

Abstract
N-terminal processing of proteins is a process affecting a large part of the eukaryotic proteome. Although N-terminal processing is an essential process, not many large inventories are available, in particular not for human proteins. Here we show that by using dedicated mass spectrometry-based proteomics techniques it is possible to unravel N-terminal processing in a semicomprehensive way. Our multiprotease approach led to the identification of 1391 acetylated human protein N termini in HEK293 cells and revealed that the role of the penultimate position on the cleavage efficiency by the methionine aminopeptidases is essentially conserved from Escherichia coli to human. Sequence analysis and comparisons of amino acid frequencies in the data sets of experimentally derived N-acetylated peptides from Drosophila melanogaster, Saccharomyces cerevisiae, and Halobacterium salinarum showed an exceptionally higher frequency of alanine residues at the penultimate position of human proteins, whereas the penultimate position in S. cerevisiae and H. salinarum is predominantly a serine. Genome-wide comparisons revealed that this effect is not related to protein N-terminal processing but can be traced back to characteristics of the genome.
Protein Nα-terminal acetylation (i.e. N-acetylation) in which an acetyl group is transferred from acetyl-coenzyme A to the α-amino group of the N-terminal residue of a protein is one of the most common covalent modifications of proteins. This modification can occur on the ultimate methionine residue, which forms the main target of acetylation, or after the cleavage of the N-terminal methionine residue. Together, these modifications occur on the vast majority of eukaryotic proteins (1, 2). For mammalian systems, it has been suggested that up to 90% of the proteins can be N-acetylated (3, 4). Cleavage of N-terminal methionine residues and N-acetylation occurs co-translationally on nascent polypeptide chains as they leave the ribosome. Protein N-terminal methionine excision is performed by the ubiquitous, essential methionine aminopeptidase enzymes. The ability of these enzymes to cleave a methionine residue is dependent on the penultimate residue according to experimental evidence and predictions based on both in vivo and in vitro data (2, 5). In general, methionine residues are removed more efficiently if the penultimate residue has a small radius of gyration (i.e. a small side chain). The preferred residues can be approximately placed in the order of glycine, alanine, serine, cysteine, threonine, proline, and valine (3).
Nearly all N-acetylations are accomplished by N-terminal acetyltransferase (NAT)1 complexes of which some are known to associate with the ribosome complexes (6). Attempts have been made to predict the likelihood of N-terminal acetylation based on the properties of the N-terminal amino acid residue, but such methods are still largely ineffective (7–10). Most of the current insights into sequence specificity for N-acetylation comes from studies using yeast strains in which specific NAT genes were deleted. In these studies the substrate specificities for the yeast acetyltransferases (Ard1p, Nat3p, and Mak3p) were deduced from the lack of acetylation of protein subsets in the different yeast knock-out strains. The corresponding substrate proteins were classified as either NatA (Ard1p), NatB (Nat3p), or NatC (Mak3p) substrates. Proteins with Ser, Ala, Gly, Thr, Cys, and Val N termini are most likely substrates of NatA. Proteins with Met-Glu or Met-Asp termini and subclasses of proteins with Met-Asn and Met-Met termini are potential substrates of NatB. Subclasses of proteins with Met-Ile, Met-Leu, Met-Trp, or Met-Phe termini are considered putative NatC substrates. However, despite a substantial amount of data, in most cases, the efficiency of N-acetylation on a given protein cannot be accurately predicted solely from its primary amino acid sequence (10).
NatA is the acetyltransferase responsible for most of the protein acetylation observed in yeast. Based on the existence of homologous N-acetylases in many eukaryotic organisms (worms, flies, plants up to human) it has been suggested that yeast and more complex eukaryotic systems have a similar set of machinery for N-terminal acetylation (3, 4). However, it cannot be excluded that unrecognized NATs may exists because there are proteins with unusual and rare N-terminal sequences that are not substrates for the known transferases. For example, Cys-Asp actin in yeast is not, as expected, a NatA substrate (3). The situation for human cells is most likely even more incomplete. For instance, the human genome encodes two orthologues for both Ard1p (hArd1p and hArd2p) and Nat1p (hNat1p and hNat2p), the two components of the yeast NatA complex. It was recently shown that hArd2p had acetyltransferase activity, but the expression levels of hARD2 (and hNat2) appear to be quite low in most tissues, and therefore the exact contribution of these proteins to N-acetylation remains unclear. Recent large scale proteomics studies on a yeast strain expressing the human NatA demonstrated that hNatA acts on almost the same set of (yeast) proteins as yNatA, indicating that NatA complexes of humans and yeast have nearly identical specificities (11). Nevertheless, only a part (57%) of all protein substrates in yeast are N-acetylated, whereas almost all (84%) protein substrates in HeLa cells are N-acetylated.
The large scale experimental identification of N-acetylated protein termini is still somewhat in its infancy, although it has seen a rapid growth in the last decade corresponding to the development of high throughput proteomics methods. Several groups have applied two-dimensional gel electrophoresis to identify N-acetylated proteins because they show markedly different electrophoretic behavior compared with their non-acetylated form (9, 12). More recently, peptide-centric approaches have been introduced for the analysis of protein N-terminal peptides. In a typical peptide-centric experiment, proteins are first digested by the protease trypsin, separated based on their biophysical properties (e.g. charge or hydrophobicity), and identified using tandem mass spectrometry. Because trypsin cleaves proteins adjacent to basic amino acids, the resulting peptides sequester typically two charges (one at their N terminus and one at the C-terminal basic residue). N-Acetylated peptides originating from the original protein N terminus do not have such a free amine group at the N terminus and therefore generally acquire one charge less. This biophysical feature can be exploited in strong cation exchange chromatography, which can separate peptides according to their charge. Several studies have already shown that SCX, when performed at acidic pH, can be used to more or less enrich for N-acetylated peptides (13, 14).
Another successful method that has been used to map N-acetylated protein termini is the so-called combined fractional diagonal chromatography (COFRADIC) technology (15–18). Diagonal peptide chromatography consists of two consecutive, identical peptide separations that contain an enzymatic reaction or chemical labeling step in between that alter the chromatographic properties of only a subset of the peptides. Such altered peptides can therefore be distinguished from non-altered peptides in a series of secondary peptide separation steps. Gevaert et al. (16) introduced and exploited this procedure for the sorting of protein N-terminal peptides in protease degradome and xenoproteome studies. Recently, a more refined COFRADIC technique was described that combined SCX separation with an enzymatic step liberating pyroglutamyl peptides for 2,4,6-trinitrobenzenesulphonic acid modification to allow COFRADIC sorting (18). Using this procedure, close to 95% of all COFRADIC-sorted peptides were found to α-acetylated. As a recent example, Arnesen et al. (11) reported on the use of COFRADIC to isolate N-terminal peptides and characterize the N-terminal acetylation of 742 proteins from human HeLa cells and 379 protein from yeast. Aivaliotis et al. (19) charted the N-acetylated terminal proteome from the two prokaryotes Halobacterium salinarum and Natronomonas pharaonis combining data from COFRADIC- and SCX-based approaches, which led to about 600 and 300 N-terminal peptides of the two organisms, respectively. Their data revealed that, perhaps surprisingly, in archaea ∼60% of the proteins undergo methionine cleavage and 13–18% of the proteins become Nα-acetylated. Most recently, Goetze et al. (20) revealed, by combining data of SCX, COFRADIC, and multiple multidimensional protein identification technology experiments, a first glimpse of the N-terminal proteome in Drosophila melanogaster Kc167 cells, reporting 900 in vivo acetylated N-terminal peptides.
Recently, we refined an SCX-based peptide separation method to achieve higher resolution in the separation of singly charged peptides (21). We showed that using this SCX approach we could base-line resolve and thus separate singly charged N-acetylated peptides from singly charged phosphorylated peptides. In previous reports, these latter two peptide categories were found to largely co-elute, hampering their targeted analysis significantly (22, 23). Here, we exploited this improved separation power in a targeted analysis of N-acetylated peptide termini from human HEK293 cells. Additionally, we took advantage of the complementarity of the proteases Lys-N, Lys-C, and trypsin to identify a total of 1391 non-redundant acetylated protein N termini with a false discovery rate of <1% from approximately 1 mg of protein, the largest data set of human acetylated protein N termini to date. We analyzed the presence of consensus sequence motifs in the experimentally observed peptides and observed several remarkable sequence features, especially in the putative hNatA substrates. Additionally, we compared our data with other reported data sets on acetylated protein N termini from D. melanogaster Kc167 cells, Saccharomyces cerevisiae, H. salinarum, and human HeLa cells (11), revealing similar characteristics but also striking differences between N-terminally acetylated proteins from different organisms. Most notably our data reveal that the cleavage efficiency by methionine aminopeptidases is conserved from E. coli to human. However, human N-acetylated peptides showed an exceptionally higher frequency of alanine residues at the penultimate position, whereas the penultimate position is predominantly a serine in S. cerevisiae and H. salinarum. Genome-wide comparisons revealed that this effect is not related to protein N-terminal processing but can be traced back to genome characteristics.
MATERIALS AND METHODS
Ammonium bicarbonate, sodium phosphate, potassium fluoride, potassium chloride, sodium orthovanadate, acetic acid, and formic acid were purchased from Sigma. The proteolytic enzymes trypsin and Lys-C were obtained from Roche Diagnostics, and Lys-N was from Seikagaku Corp. (Tokyo, Japan). Aqua C18, 5-μm, 200-Å resin and ReproSil-Pur C18-AQ, 3-μm 120-Å resin were purchased from Phenomenex (Torrance, CA) and Dr. Maisch GmbH (Ammerbuch, Germany), respectively. Fused silica capillaries (50- and 100-μm inner diameter, 375-μm outer diameter) were obtained from Bester (Amstelveen, The Netherlands), and the PolySULFOETHYL column (200 × 2.1 mm, pore diameter of 200 Å) was purchased from PolyLC Inc. (Columbia, MD). Opti-Lynx C18 cartridges from Optimize Technologies (Oregon City, OR) were used for on-line trapping and desalting of peptides. The HPLC grade acetonitrile was purchased from Biosolve (Valkenswaard, The Netherlands), and potassium silicate (KASIL 1624) was from PQ Europa (Winschoten, The Netherlands).
Preparation of HEK293 Lysate
HEK293 cells were grown in plates until confluence as described previously (21). The cells were harvested by abrasion and lysed by resuspension in lysis buffer (50 mm ammonium bicarbonate, pH 8, 8 m urea, EDTA-free protease inhibitor mixture, 1 mm potassium fluoride, 1 mm sodium orthovanadate, 5 mm potassium phosphate). The lysate was vortexed and incubated on ice for 20 min. Any remaining cells and debris were removed by centrifugation at 1000 × g for 10 min at 4 °C. The final protein concentration of the sample was determined using the 2DQuant kit (GE Healthcare).
Proteolytic Cleavage
Four 1-mg aliquots of the HEK293 lysate were resuspended in 8 m urea, 50 mm NH4HCO3, pH 8 and reduced and alkylated with 45 mm DTT (50 °C, 15 min) and 100 mm iodoacetamide (dark, room temperature, 15 min). Two aliquots were diluted to 2 m urea, 50 mm NH4HCO3 urea and digested with trypsin (1:50, w/w) overnight at 37 °C followed by dilution to 1 m urea, 50 mm NH4HCO3 and an additional digestion with trypsin (1:50, w/w) for 4 h. The other two aliquots were independently digested with Lys-N (1:85, w/w) or Lys-C (1:50, w/w) overnight at 37 °C and diluted to 1 m urea, 50 mm NH4HCO3, and a second digestion for 4 h was performed with either Lys-N (1:85, w/w) or Lys-C (1:50, w/w) (24). All digests were desalted using Sep-Pak 50-mg C18 cartridges (Waters Corp.) and reconstituted in 10% formic acid (FA) for further analysis (21).
Strong Cation Exchange
Each of the peptide mixtures was loaded onto two C18 Opti-Lynx cartridges using an Agilent 1100 HPLC system at a flow rate of 100 μl/min in 0.05% FA essentially as described previously (21, 25). Elution from the trapping cartridges was achieved using 80% acetonitrile, 0.05% FA, and the eluted sample was loaded onto a 200 × 2.1-mm PolySULFOETHYL A column (PolyLC Inc.) for 10 min at the same flow rate. The different peptide populations were separated using a non-linear 65-min gradient at 200 μl/min solvent A (5 mm KH2PO4, 30% Acetonitrile, 0.05% FA) and solvent B (5 mm KH2PO4, 30% acetonitrile, 0.05% FA, 350 mm KCl). From 0 to 10 min isocratic flow of 100% solvent A was performed, and from 10 to 15 min a linear gradient up to 26% solvent B, from 15 to 40 min a linear gradient to 35% solvent B, and from 40 to 45 min a linear gradient to 60% solvent reaching 100% solvent B at 49 min were performed. The column was then washed for 6 min with 100% solvent B and finally equilibrated with 100% solvent A for 9 min. Fractions were collected at 1-min intervals for 40 min, dried, and resuspended in 60 μl of 10% formic acid. Twenty microliters of each fraction was used for further analysis.
Mass Spectrometry
The nano-LC-MS/MS analysis was performed using an LTQ-Orbitrap (Thermo, San Jose, CA) and an Agilent 1200 series LC system equipped with a 20-mm Aqua C18 trapping column (packed in house; inner diameter, 100 μm; resin, 5 μm) and a 400-mm ReproSil-Pur C18-AQ analytical column (packed in house; inner diameter, 50 μm; resin, 3 μm). Trapping was performed at 5 μl/min for 10 min in solvent A (0.1 m acetic acid in water), and elution was achieved with a linear gradient of 10–35% B (0.1 m acetic acid in 80:20 acetonitrile/water) for 90 min with a total analysis time of 120 min. The flow rate was passively split to 100 nl/min during the gradient analysis. Nanospray was achieved using a distally coated fused silica emitter (New Objective, Cambridge, MA) (outer diameter, 360 μm; inner diameter, 20 μm; tip inner diameter, 10 μm) biased to 1.7 kV. A 33-megaohm resistor was introduced between the high voltage supply and the electrospray needle to reduce the ion current.
The LTQ-Orbitrap mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS. Full-scan MS spectra (300–1500 m/z) were acquired with a resolution of 60,000 at 400 m/z and accumulation to a target value of 500,000. The five most intense peaks above a threshold of 500 were selected for collision-induced dissociation in the linear ion trap at a normalized collision energy of 35 after accumulation to a target value of 30,000.
Data Analysis
All MS/MS spectra for each SCX fraction were converted to DTA files using Bioworks 3.3.1 (Thermo) with default settings, combined in a single file, and searched using the Mascot search engine (Matrix Science, London, UK; version 2.2.01) against a concatenated Swiss-Prot human database containing an equivalent size decoy protein set (version 56.2; 40,656 sequences; 22,416,002 residues) with cysteine carbamidomethylation as a fixed modification. Methionine oxidation, peptide N-terminal acetylation, and phosphorylation were chosen as variable modifications. A peptide mass tolerance of 10 ppm and fragment mass tolerance of 0.6 Da were selected. Trypsin, Lys-N, and Lys-C were chosen appropriately as the proteolytic enzymes, allowing one missed cleavage. Additionally, database searches selecting semitrypsin, semi-Lys-N, and semi-Lys-C as the proteolytic enzyme were performed. SCX fractions known to be enriched for N-terminally acetylated peptides were searched and processed separately from the other SCX fractions. For this targeted analysis, we excluded missed cleavages for fractions containing peptides with no or one basic residue and allowed one missed cleavage for fractions containing N-terminally acetylated peptides with two basic residues. The remaining SCX fractions, which were not particularly enriched for N-terminally acetylated peptides, were searched with one missed cleavage. A Mascot cutoff score corresponding to a false discovery rate of less than 1%, according to the number of decoy identifications, was selected and applied as threshold for each of the search approaches (regular enzyme search, semienzyme search targeting the fractions rich in N-terminally acetylated peptides, and the non-targeted semienzyme search). This resulted in a minimum Mascot score of 32 for peptides from trypsin- or Lys-C-generated peptides and a minimum score of 28 for Lys-N peptides. All mass spectrometry data were loaded into Scaffold v.2 (Proteome Software, Portland, OR) and can be retrieved through the Tranche repository using the following web link: https://proteomecommons.org/tranche/data-downloader.jsp?fileName=UVN8tcfpHyBzWDY4QL1nsqePFioGA58KgsNA50wGDttFe1gvnyTvMyjQmQpY1bLtHaIS1UXGpfPAlEVpw7Wy8t5fcDsAAAAAAAAFPQ%3D%3D; pass phrase: hek293acetylation.
Finally, all identifications were combined, and redundancies were eliminated from the data sets (peptides that identified the same protein N terminus were considered redundant). Amino acid frequency analysis of N-terminal peptide sequences were calculated using Weblogo.
Statistical Analysis of Amino Acid Frequencies of Protein N Termini
To determine amino acid frequency distributions from full proteins and protein N termini, the Swiss-Prot v56.2 fasta database containing protein sequences for a large variety of species was taken from the European Bioinformatics Institute/Swiss Institute of Bioinformatics repository. This fasta database was then filtered to obtain protein sets of selected species only, namely Homo sapiens, D. melanogaster, S. cerevisiae, and H. salinarum, for which experimental data on N-acetylated termini are available. Only protein entries that start with an N-terminal methionine (i.e. more than 90% of the entries) were used for this analysis. Each protein set was subsequently parsed into four subsets to obtain (i) the penultimate amino acids following the N-terminal methionine (i.e. the X in MX), (ii) amino acids 3–7 (i.e. the Ys in MXYYYYY) from the N terminus, (iii) amino acids 3–30 from the N terminus, and (iv) the full-length proteins. All redundant peptides and proteins were removed from these data sets. For each subset, the amino acid frequency was calculated. The following species were analyzed with the number of unique (non-redundant) entries in the Swiss-Prot v56.2 fasta database given in parentheses: H. sapiens (n = 18,821), D. melanogaster (n = 2789), S. cerevisiae (n = 6551), and H. salinarum (n = 443).
RESULTS
Low pH SCX has been proven to enrich for N-acetylated peptides from a pool of “regular” tryptic peptides, exploiting the fact that N-acetylated peptides have one less positive charge in solution due to the blocked N terminus (13, 26–28). However, phosphopeptides and most peptides derived from the C terminus of proteins also have a single charge. Consequently, a mixture of these three types of peptides is often obtained, hampering an analysis that is focused solely on one of three peptide types. This is reflected by the common strategy in large scale phosphoproteomics that utilizes additional enrichment steps such as IMAC and/or TiO2 (13, 29–31). These strategies often discard the pools of N-acetylated and C-terminal peptides. Recently, we demonstrated that SCX can resolve these peptide populations with identical nominal net charges, allowing a clear separation of phosphopeptides from N-acetylated peptides (21, 24). Although we initially demonstrated this resolving power for peptides created by the Lys-N protease (25), similar results could be obtained with either trypsin or Lys-C (21). Considering the data we acquired for phosphopeptides, we set out to investigate here the possibility that these three proteases could provide access to different pools of N-acetylated peptides. Fig. 1 provides a schematic overview of the experimental design. After digesting 1 mg of human HEK293 cell lysate with either trypsin (two aliquots), Lys-N, or Lys-C, we performed SCX fractionation. Subsequently, one-third of each fraction was subjected to nano-reverse phase LC-MS/MS. All resulting MS/MS spectra were searched against all human proteins in the Swiss-Prot database. Furthermore, additional searches were performed taking into account the possibility of the activity of other enzymes (described under “Materials and Methods”). The general performance of the SCX separation for different classes of peptides has been described earlier (21, 25) and is summarized in Fig. 2. As expected, the bulk of the “normal” doubly charged peptides eluted in the later SCX fractions, starting around fraction 25 (Fig. 2, bottom graph). Phosphorylated peptides were found to be clustered in two distinct regions. Doubly phosphorylated peptides were found between fractions 5 and 9, and singly phosphorylated peptides were present in fractions 15–24 (Fig. 2, bottom graph). The N-acetylated peptides were observed in three separate populations (Fig. 2, top graph, and Fig. 3). To start with, N-acetylated peptides were present in the very first fractions of the SCX run, primarily originating from Lys-N-generated N-acetylated peptides that do not contain any basic residue. These peptides bind weakly to the SCX column due to their ability to coordinate protons via the peptide backbone. N-Acetylated peptides that contain a single basic residue are clustered in SCX fractions 8–14, and the last distinct population of N-acetylated peptides eluted in SCX fractions 24–28, originating from N-acetylated peptides that contain two basic residues. This last population, which has a net charge of 2+, co-elutes with the huge population of normal doubly charged peptides. However, both of the other clusters elute largely separated from any other class of peptides. The three clusters observed for N-acetylated peptides seem to be independent of the protease used (see Fig. 2, top graph, and Fig. 3). However, it is apparent that Lys-N generates the most “zero-charged” N-acetylated peptides due to the fact that it cleaves on the N-terminal side of lysine residues. Although some variation was observed between the two trypsin replicates, their overall appearance is very similar, showing the reproducibility of the SCX separation (Fig. 3). This is also illustrated by the number of non-redundant N-acetylated protein termini identified in each independent SCX analysis, which was quite similar (between 600 and 700). For instance, the analysis of the first tryptic digest led to the identification of 666 non-redundant in vivo N-acetylated peptides with a false discovery rate below 1%. A list of the assigned N-acetylated peptides, the proteins they originate from, and additional details are provided for the experiments with trypsin (n = 666), the trypsin replicate (n = 618), Lys-C (n = 577), and Lys-N (n = 701) in supplemental Tables 1, 2, 3, and 4, respectively. It should be noted that these lists were filtered for redundancies, meaning that when multiple N-acetylated peptides were detected for the same protein terminus (due to miscleavages or additional modifications), only the most confidently identified (i.e. the highest scoring) peptide was included in the list.
Fig. 1.
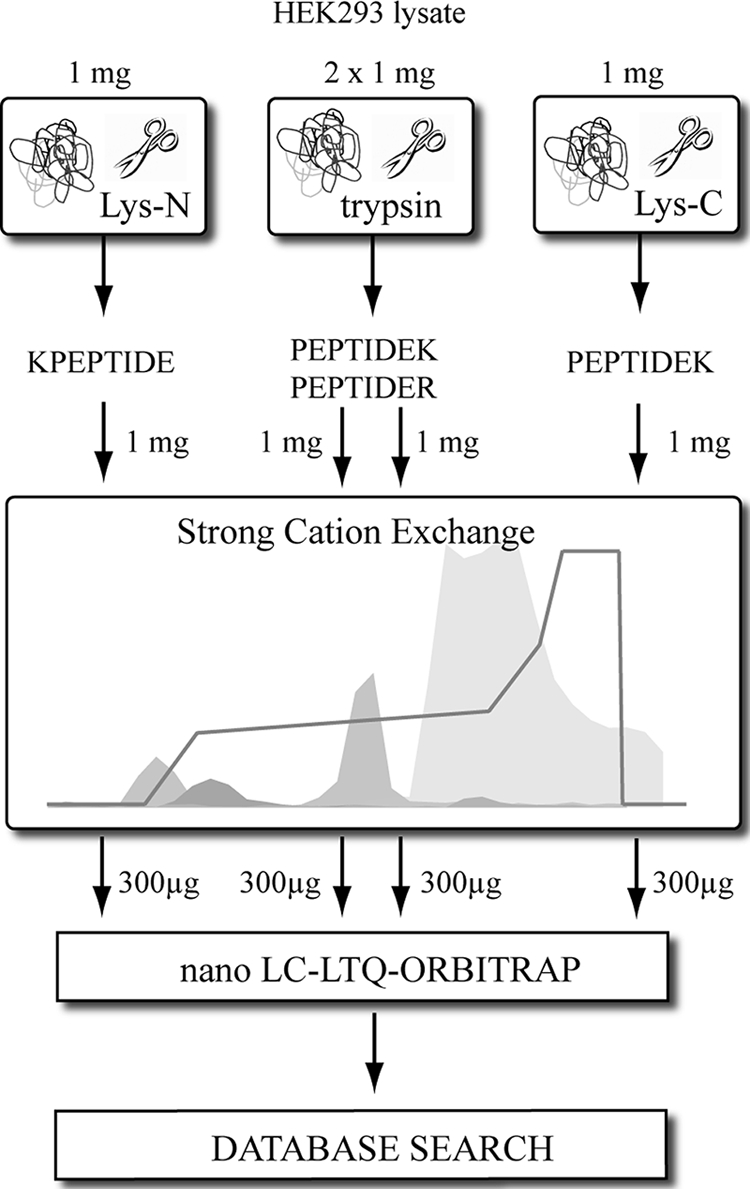
Scheme of experimental approach for analysis of N-acetylated protein N termini in human HEK293 cells. Four individual sets of peptides, generated by trypsin (twice), Lys-C, and Lys-N, each originating from 1 mg of HEK293 lysate, were subjected to SCX separation, LC-MS/MS, and database searches.
Fig. 2.

Schematic of SCX fractionation of different classes of proteolytic peptides. The bottom graph displays the observed elution patterns of unmodified doubly charged peptides, typically very abundant in (tryptic) digests, in dark gray, whereas the elution of doubly and singly phosphorylated peptides is displayed in light grey. In the top diagram, the experimentally derived elution profiles of the number of N-acetylated peptides are displayed in a cumulative manner for the four experiments using trypsin, Lys-C, and Lys-N. Characteristic patterns are observed for N-acetylated peptides containing zero, one, or two basic residues whereby the first two categories can be nearly base-line resolved from the other classes of peptides.
Fig. 3.

Overview of number of non-redundant N-acetylated peptides observed per SCX fraction. The graphs display the observed elution patterns of N-acetylated peptides for the four experiments (trypsin (twice), Lys-C, and Lys-N). Although there is some variation, they all possess similar elution patterns and number of identified N-acetylated peptides. Shown in black are the number of peptides originating from N termini predicted by the annotated genome (starting at either position 1 or 2 in the protein), whereas displayed in gray are peptides with a start residue at a different position in the proteins.
Next, we evaluated the redundancy, reproducibility, and complementarity of the multienzyme approach with the additional aim to generate an overall non-redundant data set of in vivo N-acetylated protein termini of human HEK293 cells. Initially, undersampling and SCX reproducibility were evaluated by comparing the overlap of trypsin replicate experiments (Fig. 4). A total of 422 N-acetylated peptides were detected in both experiments, implying a 49% overlap. To assess the complementarity between the trypsin-based experiments and the Lys-C and Lys-N experiments, all non-redundant N-terminally acetylated sequences were compared based on protein accession and starting position. The overlap between the N-acetylated peptides identified with trypsin compared with either Lys-C or Lys-N was somewhat lower (about 30%) when compared with the trypsin replicate experiments (Fig. 4). The lower level of overlap suggests a significant degree of complementarity; nevertheless, the substantial overlap allows a large portion of N-acetylated protein termini to be validated. Whenever a particular N-acetylated termini peptide was observed multiple times (in any one of the four digest experiments), only the most confident peptide identification was kept for the non-redundant list. Combining data from all four experiments (a redundant list of 2562 N-terminally acetylated peptides) and filtering led to the experimental identification of a total of 1391 non-redundant in vivo N-acetylated protein termini listed in supplemental Table 5. These results provide currently the most extensive data set of human in vivo N-acetylated protein termini.
Fig. 4.

Redundancy, reproducibility, and complementarity of multienzyme approach and non-redundant data set of in vivo N-acetylated protein termini in human HEK293 cells. Each of the Venn diagrams displays the overlap in identified acetylated protein N termini between a single trypsin data set and each of the other data sets. As expected, the overlap between the trypsin replicates is larger than between trypsin and Lys-C or Lys-N. After filtering for redundant protein N termini, we obtained experimental identification of a total of 1391 in vivo N-acetylated protein termini. FDR, false discovery rate.
DISCUSSION
In this work we used SCX chromatography to chart the N-acetylated protein termini present in the proteome of human HEK293 cells. Although SCX has been used before for a targeted analysis of N-acetylated terminal peptides originating from protein termini (11, 14, 19), we were able to achieve nearly base-line separation of phosphopeptides from N-acetylated peptides, improving the targeted analysis of both subclasses. By using this two-dimensional approach, we were able to identify over 600 N-acetylated peptides from a tryptic digest of approximately 300 μg of human HEK293 cells. We implemented a multienzyme approach (32, 33) to cover a larger section of the N-terminal proteome using trypsin, Lys-C, and a relative new player in the field, Lys-N (24, 25). Each of these experiments performed quite similarly when evaluated by the number of non-redundant N-acetylated peptides identified (i.e. 600–700 per experiment). After removing overlap, we exposed 1391 unique non-redundant N-acetylated protein N termini.
N-Acetylated N-terminal Proteome of Human Cells
The here reported data set of unique non-redundant N-acetylated human protein N termini is the largest reported to date. Previous work by Polevoda and Sherman (3) listed over 450 yeast N-acetylated proteins and 300 N-acetylated mammalian proteins. Frottin et al. (5) gathered a data set from the literature consisting of 832 protein N termini in E. coli, and Aivaliotis et al. (19) identified close to 600 N termini in the archaeum H. salinarum. Arnesen et al. (11) applied COFRADIC in combination with SCX to isolate N-terminal peptides and thus determined the N termini of 742 human HeLa cells and 379 S. cerevisiae protein N termini. Of these reported N termini, 632 and 241 were identified as in vivo N-acetylated peptides. The remainder were unmodified N termini that were in vitro acetylated to allow isolation and identification (18). Most recently, Goetze et al. (20) described, by combining data of SCX, COFRADIC, and multiple multidimensional protein identification technology experiments, slightly over 900 in vivo acetylated N-terminal peptides in D. melanogaster Kc167 cells (20). Arnesen et al. (11) applied a similar methodological strategy on a human cell line (i.e. HeLa) to generate their data, providing an ideal reference for evaluation and comparison with our data set. Initially, we evaluated the overlap between the set of in vivo N-acetylated peptides reported by Arnesen et al. (11) in HeLa cells, and our data set, which is derived from HEK293 cells (Fig. 5A). Of the 1391 N-acetylated peptides detected in our study, 299 were also reported by Arnesen et al. (11), whereas they identified 333 in vivo N-acetylated peptides not present in our data set. Similar small overlaps were observed when we compared our data set with the smaller data set obtained in our laboratory by Dormeyer et al. (14) extracted from a crude membrane fraction of human embryonic carcinoma cells (Fig. 5A). We note that all these studies have been based on using transformed human cell lines and, therefore, may potentially not accurately represent the N-terminal proteome of primary human cells or specific human tissue. Still, the relatively small overlaps observed are likely due to differences in the (sub)proteome of these different cell lines, “undersampling” of the full proteome, and the different methods used to enrich for N-acetylated peptides. Combining the results of these three studies provides a list of 1861 non-redundant acetylated N-terminal peptides (supplemental Table 6).
Fig. 5.

Characteristics of experimentally measured N-terminal proteome. A, three-way comparison of N-acetylated peptides identified by studies performed on human teratocarcinoma Nt2/d1 cells (14), HeLa cells (11), and HEK293 cells (our present data) revealing a small overlap of 14% between the HeLa and HEK293 study. Combining the results of these three studies provides a list of 1861 non-redundant acetylated N-terminal peptides (supplemental Table 6). B, frequency distribution of acetylated N-terminal amino acid residues. Results are summarized for data on HEK293 cells (this work), HeLa cells (11), D. melanogaster (20), H. salinarum (19), and S. cerevisiae (11). The data on the two human cell lines are very similar. N-terminal acetylation on serine residues is frequently observed in organisms such as H. salinarum and S. cerevisiae, whereas acetylation on alanine is more abundant in human HEK293 and HeLa cells. C, bar chart illustrating the effect of the penultimate amino acid residue on the efficiency of methionine cleavage. If, for example, an alanine is in the second position, the N-terminal methionine is cleaved off in nearly 100% of the cases; however, when a valine is in this position, only 50% of the N-terminal peptides undergo methionine cleavage.
Furthermore, we evaluated the nature of the experimentally observed ultimate N-terminal residue of the acetylated protein termini (Fig. 5B). We found that this residue was an alanine residue in almost half of the detected protein N termini (47%) with additional abundant residues being methionine (24%), serine (18%), threonine (4%), and glycine (2%). The severe dominance of the alanine residue is quite apparent but is also present in the data set of Arnesen et al. (11) from human HeLa cells (n = 632) in which the most abundant N-terminal residues are alanine (45%), methionine (31%), and serine (17%). However, in contrast to this observation, a very different occurrence of primary residues is observed in S. cerevisiae and H. salinarum with serine being the most prominent N-terminal residue (43 and 84%, respectively) followed by methionine (20 and 5%, respectively) and alanine (15 and 9%, respectively). Although the S. cerevisiae and H. salinarum data sets are smaller (n = 267 and n = 88, respectively) than the human data sets, our analysis suggests that these differences are significant. Finally, the experimental data available for D. melanogaster reveals an acetylated N-terminal proteome state somewhat in between human and S. cerevisiae with the most abundant N-terminal residues being methionine (39%), serine (29%), and alanine (23%). Below we will discuss whether this behavior is related to specific N-terminal processing or characteristics of the whole genome.
Efficiency of Methionine Processing
Our data also provide a resource for the qualitative assessment of the in vivo probability/efficiency of N-terminal methionine cleavage in the human proteome. Although we do not have, in contrast to the COFRADIC experiments, data on the concomitant non-acetylated peptide counterparts, our data allow us to qualitatively assess the methionine cleavage efficiency by comparing the number of observed N-acetylated peptides with a specific N-terminal amino acid residue with the number of peptides that have that same amino acid in the penultimate position next to an acetylated methionine. Hereby, we assume that the acetylation efficiency of Ala and Met-Ala are in vivo similar. For instance, we detected 651 peptides N-acetylated at an alanine residue but only four N-acetylated peptides starting with Met-Ala, indicating that more than 99% of the observed proteins that have an alanine residue in the penultimate position have their methionine readily cleaved in human cells. In contrast, we detected 11 N-acetylated peptides starting with valine and 13 peptides initiated by Met-Val, indicating a lower methionine cleavage efficiency (46%). This finding could be further substantiated by the fact that four of the 13 N-acetylated Met-Val peptides were also identified in a form that lacked the methionine and were instead acetylated on the valine residue. Fig. 5C summarizes the qualitative efficiency of methionine cleavage for all penultimate amino acid residues based on our experimental data set. Considering only penultimate residues for which we detected at least 10 peptides, the cleavage probability was found to be highest for alanine (99%, 90%) and serine (99%, 84%) followed by glycine (90%, 97%), threonine (90%, 90%), valine (46%, 84%), glutamine (40%), asparagine (30%, 16%), leucine (17%, 16%), glutamic acid, (10%) and aspartic acid (10%, 16%). With a few exceptions, our values agree very well with those (given in italics) compiled by Frottin et al. (5) that were based on data for 862 E. coli proteins and/or on in vitro peptide assays and also agree with the values extracted from the work of Polevoda and Sherman (3). Our human proteome data confirm that the penultimate residue plays an important role in the cleavage efficiency of the methionine aminopeptidases and that in general methionine residues are removed more efficiently if the penultimate residue has a small radius of gyration. This rule seems to be highly conserved from E. coli to human. We performed a sequence alignment of several methionine amino peptidases that revealed, in line with these findings, high homology (data not shown).
Dissimilarity in N-terminal Sequences between Proteomes
Several approaches have been used to predict the nature and frequency of N-acetylation for protein N termini, suggesting that the presence of certain amino acids close to the N terminus of the protein may play an important role. To analyze the presence of potential motifs in our data set, we visualized specific (sub)sets of the observed protein N termini using Weblogo, a program that generates sequence logos from multiple sequence alignments. To include also the shortest detected N-acetylated N-terminal peptides in our alignments, we only considered the first six amino acids of the identified peptides. The top row in Fig. 6 shows the sequence logos obtained from our data set of 1391 N-acetylated peptides and those generated from the in vivo N-acetylated peptide data sets from HeLa and S. cerevisiae reported by Arnesen et al. (11). These sequence logos reiterate that the relative frequency of the terminal amino acid residues is very comparable between our study on HEK293 cells and the HeLa cells of Arnesen et al. (11), whereas very different relative abundances are observed for the N-terminal residues of S. cerevisiae. Moreover, the relative frequency of specific amino acids over the first six-residue stretch is quite similar between the human HEK293 and HeLa cells. In the human cells, the relative high frequency of Glu, Ala, Asp, Ser, Thr, and Gly at the second position is quite evident. Strikingly, at positions 3–6, Ala, Ser, Glu, and Gly also seem more abundant. In the S. cerevisiae data set, however, this preference in amino acid composition is much less evident. Notably in S. cerevisiae, lysine seems to be more present throughout the analyzed sequences.
Fig. 6.

Sequence logos illustrating frequency of amino acid residue occurrence in primary N-terminal stretch of proteins. The amino acid position (1 marking the ultimate N-terminal residue) is indicated below each sequence logo. The top row shows the logos obtained from our data set of 1391 N-acetylated peptides detected in HEK293 cells and those generated from the in vivo N-acetylated peptide data sets from HeLa cells, S. cerevisiae (11), D. melanogaster (20), and H. salinarum (19). These sequence logos reveal that the relative frequency of the terminal amino acid residues is very comparable between the HEK293 and HeLa cells, whereas different relative abundances are observed for the N-terminal residues of S. cerevisiae. The second and third rows reveal subsets of the experimentally measured HEK293 N-acetylated peptides, dividing them in classes of peptides starting with an alanine, serine, threonine, methionine, glycine, and valine. Similarly, the fourth row contains sequence logos for peptides starting with an Ala-Ala, Ala-(Asp/Glu), or Ala-Ser stretch. The fifth row displays for comparison the sequence logos for all proteins in the human genome with a (Met)-Ala-Ala sequence (from the Swiss-Prot database) and shows amino acid frequency plots of N-terminally acetylated peptides from D. melanogaster and H. salinarum.
To explore these phenomena in more detail, we generated sequence logos for subsets of proteins, dividing our data set of 1391 N-acetylated peptides in classes of peptides starting with an alanine, serine, and threonine (potential NatA substrates) presented in the second row of Fig. 6. Sequence motifs for peptide subsets starting with a methionine (likely NatB substrates), glycine, and valine are presented in the third row of Fig. 6. In our set of experimentally observed N-acetylated peptides in the subset of N-acetylated peptides starting with a methionine, the penultimate residue is in most cases an aspartic or glutamic acid, which is also a feature of peptides starting with a valine and to a lesser extent for those starting with a glycine. Although for N-acetylated peptides starting with an alanine, serine, or threonine, also aspartic and glutamic acid residues are observed at the second position, but they are less frequent. This observation is probably related to the specificity in efficiency of methionine processing that has been described above. As methionine cleavage is less efficient for termini with an aspartic or glutamic acid at the penultimate position (Fig. 5C), it is not surprising to find that for the majority of N-acetylated methionine peptides the penultimate residue is an aspartic or glutamic acid. Another interesting feature is the relative high similarities observed between sequence stretches for peptides starting with a methionine and those staring with a valine, although the latter supposedly are largely NatA substrates, whereas methionine-starting peptides are NatB substrates.
Another observation for the group of peptides displayed in the second row of Fig. 6 is that for the N-acetylated peptides starting with an alanine and to a lesser extent serine and threonine there is the apparent repetitive occurrence of the starting amino acid. For instance, N-acetylated peptides starting with an alanine are enriched further in the N-terminal stretch of six amino acid residues for alanine. For N-acetylated serine peptides, a small further enrichment is observed for serine residues, and for N-acetylated peptides starting with a threonine especially at the adjacent subsequent position, frequently a threonine is observed. This “self-repetitive” behavior is notably absent for the peptides starting with a Met, Val, or Gly. The fourth row contains sequence logos for peptides starting with Ala-Ala, Ala-(Asp/Glu), and Ala-Ser, which further iterates the self-repetitive behavior in the peptides starting with Ala-Ala, whereas this behavior is absent in the peptides starting with Ala-(Asp/Glu). Notably, in the peptides staring with Ala-Ser, the dominance of alanine in the later part of the sequence is diminished in favor of serine (and glycine).
To test whether these self-repetitive patterns have their origins in the selectivity of N-acetyltransferases or other N-terminal protein processing mechanisms or whether specific frequency patterns are already present throughout the proteome, we retrieved all predicted N termini from the Swiss-Prot database. From these, we selected protein termini that had an alanine at the second and third amino acid residues, resulting in a full set of 1085 proteins. The sequence logo obtained for the first six of the amino acid residues of these proteins is shown in the fifth row of Fig. 6. This logo resembles the logo obtained from our experimental data set of N-acetylated peptides starting with Ala-Ala, indicating that this self-repetitive behavior has its unknown origin at the genome level and is most likely not a specific feature of N-terminal processing. It has been stated previously, in agreement with our data, that the protein termini of proteins of mammalian systems are enriched for alanine residues (34).
Establishing that this observed behavior is already present at the genome level, we next determined amino acid frequency distributions from full proteins and protein N termini as present in the Swiss-Prot database for H. sapiens, D. melanogaster, S. cerevisiae, and H. salinarum. For all protein entries starting with an N-terminal methionine, we calculated the amino acid frequency for (a) the penultimate amino acids following the N-terminal methionine (i.e. the X in MX), (b) the amino acid stretch from 3 to 7 from the N terminus, (c) the amino acid stretch from 3 to 30 from the N terminus, and (d) the full-length proteins. The frequency plots of the last three categories were found to be highly similar (supplemental Table 7). However, in comparing the amino acid frequency profiles between the penultimate amino acids and the full-length proteins, substantial differences between genomes were observed. In Fig. 7, the amino acid frequency profiles of the penultimate amino acids and the full-length proteins are compared for H. sapiens, D. melanogaster, S. cerevisiae, and H. salinarum. Most strikingly, in the human genome the occurrence of a penultimate alanine is about 3-fold enriched when compared with the whole proteome. This enrichment is still about 2-fold in D. melanogaster but nearly absent in S. cerevisiae and H. salinarum. In contrast, in the S. cerevisiae and H. salinarum genomes, the occurrence of a penultimate serine is about 3–5-fold enriched when compared with the whole proteome. This enrichment is still about 2-fold in D. melanogaster but absent in H. sapiens. Other trends can be observed, such as leucine not often being present at the penultimate position of proteins, but most of them are less striking. Our analysis indicates that there is a clear bias around the translation start of proteins, albeit that this bias is most evident for the penultimate amino acid residue and strikingly different for genomes from different branches of the tree of life. Although a serine bias has been suggested for S. cerevisiae by analyzing the context around the AUG start codons, these phenomena have been less described from a comparative genomics point of view (35, 36). This bias at the genome level is experimentally verified in the analysis of N-acetylated protein N termini of the four mentioned organisms discussed above. Overall, our data indicate that the mechanisms for N-terminal processing of proteins are preserved throughout the tree of life, whereas genome-defined differences do exist in the N-terminal proteome of these species.
Fig. 7.

Proteome-wide amino acid frequency distributions. The frequency of occurrence for the penultimate amino acid (i.e. Met-X) residue of protein N termini is given in black solid bars, and for comparison, the frequency of occurrence over all intact proteins present in the proteome is give by the striped bars (data were taken from the Swiss-Prot v56.2 database for H. sapiens (n = 18,821), D. melanogaster (n = 2789), S. cerevisiae (n = 6551), and H. salinarum (n = 443)).
Non-predicted Protein N Termini and Protein Isoforms
Of the 1391 N-acetylated peptides, 1192 (86%) start at the predicted ultimate or at the penultimate amino acid residue (Fig. 3, in black). This leaves about 200 primarily N-acetylated peptides that do not correspond to the predicted start amino residue (Fig. 3, in gray). These peptides contain potentially interesting information about these proteins. For instance, we found confident evidence for two variants of the polypyrimidine tract-binding protein 1 (PTBP1). These two proteins were found to be N-acetylated (underlined) either at the N-terminal Met amino acid or at the Ser residue at position 32 (MDGIVPDIAVGTKRGSDELFSTCVTNGPFIMSSNSASAAN). Another interesting example is the Aurora B kinase of which we very confidently detected the N-acetylated peptide SRSNVQ starting at residue 43. This start site is especially intriguing because this would represent a truncated form of the kinase that lacks the N-terminal part of the protein containing the “A-box” motif. It has been stated that Aurora A and B variants missing this protein domain become much more stable as they are not as readily degraded by the proteasome (37). Increased stability of Aurora can lead to uninhibited cell growth, which is further substantiated by the fact that other variant forms of Aurora B have been linked to tumorigenesis and cancer (38, 39). As our data are from an immortalized HEK293 cell line, the observation of this Aurora B truncation site is conceivable. Another example is RUSC1, a protein putatively involved in regulation of nerve growth factor-dependent neurite outgrowth. For this protein, we detected the N-acetylated peptide AEAQSG, starting at residue 471. In the UniProt database, a second isoform of this protein has been predicted that misses the first 469 residues with residue 470 being a Met. Our data confirm this prediction. Because we did not detect an N-acetylated peptide for RUSC1 starting at the predicted N terminus, we cannot distinguish whether the observed N-acetylated terminus at amino acid residue 471 is indeed an isoform or whether the genome annotation is simply incorrect. As a final example, several isoforms of the SCOC protein are described in the UniProt database with different deletions in the N-terminal region. In our data set, we detected the N-acetylated peptide MMNADM, which is in good agreement with the predicted isoform 4 (Q9UIL1-4) missing amino acids 1–77. Such examples show that our data are a valuable source to verify predicted proteins or discover new isoforms of known proteins.
In conclusion, we applied a straightforward but refined proteomics strategy to identify almost 1400 N-terminally acetylated peptides in a human cell line using SCX in combination with a multiprotease protein digestion approach. These data represent the largest inventory of human acetylated protein N termini to date, and we report on novel protein isoforms for the Aurora B kinase and the RUSC protein for example. In conjunction with extensive bioinformatics analysis of annotated proteomes from different species and comparisons with other data sets on acetylated protein N termini, our data provide new insights into N-terminal processing and characteristics of the N-terminal proteome. The mechanisms for N-terminal processing seem to be largely conserved between organisms as varied as H. sapiens, D. melanogaster, S. cerevisiae, and H. salinarum whereby in all these species methionine cleavage of protein N termini is clearly dependent on the penultimate amino acid. Our experimental data suggest that proteins from more basal organisms, such as S. cerevisiae and H. salinarum, more likely have a serine residue as their penultimate acetylated residue, whereas higher organisms, such as H. sapiens, display a much higher preference for alanine. Genome-wide comparisons revealed that this effect is not related to protein N-terminal processing but can be traced back to characteristics of the whole genome with a clear bias around the translation start of proteins, which is strikingly different for genomes from different branches of the tree of life.
Supplementary Material
Acknowledgments
We thank Dr. Pantelis Hatzis for supplying the HEK293 cells.
Footnotes
* This work was supported by the Netherlands Proteomics Centre.
 This article contains supplemental Tables 1–7.
This article contains supplemental Tables 1–7.
1 The abbreviations used are:
- NAT
- N-terminal acetyltransferase
- SCX
- strong cation exchange
- COFRADIC
- combined fractional diagonal chromatography
- FA
- formic acid.
REFERENCES
- 1.Bradshaw R. A., Brickey W. W., Walker K. W. (1998) N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci 23, 263–267 [DOI] [PubMed] [Google Scholar]
- 2.Polevoda B., Sherman F. (2000) Nalpha-terminal acetylation of eukaryotic proteins. J. Biol. Chem 275, 36479–36482 [DOI] [PubMed] [Google Scholar]
- 3.Polevoda B., Sherman F. (2003) N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol 325, 595–622 [DOI] [PubMed] [Google Scholar]
- 4.Polevoda B., Sherman F. (2002) The diversity of acetylated proteins. Genome Biol 3, reviews0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frottin F., Martinez A., Peynot P., Mitra S., Holz R. C., Giglione C., Meinnel T. (2006) The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteomics 5, 2336–2349 [DOI] [PubMed] [Google Scholar]
- 6.Polevoda B., Brown S., Cardillo T. S., Rigby S., Sherman F. (2008) N(alpha)-terminal acetyltransferases are associated with ribosomes. J. Cell. Biochem 103, 492–508 [DOI] [PubMed] [Google Scholar]
- 7.Liu Y., Lin Y. (2004) A novel method for N-terminal acetylation prediction. Genomics Proteomics Bioinformatics 2, 253–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiemer L., Bendtsen J. D., Blom N. (2005) NetAcet: prediction of N-terminal acetylation sites. Bioinformatics 21, 1269–1270 [DOI] [PubMed] [Google Scholar]
- 9.Perrot M., Massoni A., Boucherie H. (2008) Sequence requirements for Nalpha-terminal acetylation of yeast proteins by NatA. Yeast 25, 513–527 [DOI] [PubMed] [Google Scholar]
- 10.Martinez A., Traverso J. A., Valot B., Ferro M., Espagne C., Ephritikhine G., Zivy M., Giglione C., Meinnel T. (2008) Extent of N-terminal modifications in cytosolic proteins from eukaryotes. Proteomics 8, 2809–2831 [DOI] [PubMed] [Google Scholar]
- 11.Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Link A. J., Robison K., Church G. M. (1997) Comparing the predicted and observed properties of proteins encoded in the genome of Escherichia coli K-12. Electrophoresis 18, 1259–1313 [DOI] [PubMed] [Google Scholar]
- 13.Villén J., Gygi S. P. (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc 3, 1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dormeyer W., Mohammed S., Breukelen B., Krijgsveld J., Heck A. J. (2007) Targeted analysis of protein termini. J. Proteome Res 6, 4634–4645 [DOI] [PubMed] [Google Scholar]
- 15.Gevaert K., Goethals M., Martens L., Van Damme J., Staes A., Thomas G. R., Vandekerckhove J. (2003) Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat. Biotechnol 21, 566–569 [DOI] [PubMed] [Google Scholar]
- 16.Gevaert K., Van Damme P., Ghesquière B., Vandekerckhove J. (2006) Protein processing and other modifications analyzed by diagonal peptide chromatography. Biochim. Biophys. Acta 1764, 1801–1810 [DOI] [PubMed] [Google Scholar]
- 17.Gevaert K., Van Damme P., Martens L., Vandekerckhove J. (2005) Diagonal reverse-phase chromatography applications in peptide-centric proteomics: ahead of catalogue-omics? Anal. Biochem 345, 18–29 [DOI] [PubMed] [Google Scholar]
- 18.Staes A., Van Damme P., Helsens K., Demol H., Vandekerckhove J., Gevaert K. (2008) Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC). Proteomics 8, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 19.Aivaliotis M., Gevaert K., Falb M., Tebbe A., Konstantinidis K., Bisle B., Klein C., Martens L., Staes A., Timmerman E., Van Damme J., Siedler F., Pfeiffer F., Vandekerckhove J., Oesterhelt D. (2007) Large-scale identification of N-terminal peptides in the halophilic archaea Halobacterium salinarum and Natronomonas pharaonis. J. Proteome Res 6, 2195–2204 [DOI] [PubMed] [Google Scholar]
- 20.Goetze S., Qeli E., Mosimann C., Staes A., Gerrits B., Roschitzki B., Mohanty S., Niederer E. M., Laczko E., Timmerman E., Lange V., Hafen E., Aebersold R., Vandekerckhove J., Basler K., Ahrens C. H., Gevaert K., Brunner E. (2009) Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLoS Biol 7, e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gauci S., Helbig A. O., Slijper M., Krijgsveld J., Heck A. J., Mohammed S. (2009) Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem 81, 4493–4501 [DOI] [PubMed] [Google Scholar]
- 22.Lemeer S., Jopling C., Gouw J., Mohammed S., Heck A. J., Slijper M., den Hertog J. (2008) Comparative phosphoproteomics of zebrafish Fyn/Yes morpholino knockdown embryos. Mol. Cell. Proteomics 7, 2176–2187 [DOI] [PubMed] [Google Scholar]
- 23.Lemeer S., Pinkse M. W., Mohammed S., van Breukelen B., den Hertog J., Slijper M., Heck A. J. (2008) Online automated in vivo zebrafish phosphoproteomics: from large-scale analysis down to a single embryo. J. Proteome Res 7, 1555–1564 [DOI] [PubMed] [Google Scholar]
- 24.Taouatas N., Drugan M. M., Heck A. J., Mohammed S. (2008) Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase. Nat. Methods 5, 405–407 [DOI] [PubMed] [Google Scholar]
- 25.Taouatas N., Altelaar A. F., Drugan M. M., Helbig A. O., Mohammed S., Heck A. J. (2009) SCX-based fractionation of Lys-N generated peptides facilitates the targeted analysis of post-translational modifications. Mol. Cell. Proteomics 8, 190–200 [DOI] [PubMed] [Google Scholar]
- 26.Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinkse M. W., Mohammed S., Gouw J. W., van Breukelen B., Vos H. R., Heck A. J. (2008) Highly robust, automated, and sensitive online TiO2-based phosphoproteomics applied to study endogenous phosphorylation in Drosophila melanogaster. J. Proteome Res 7, 687–697 [DOI] [PubMed] [Google Scholar]
- 28.Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., White F. M. (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol 20, 301–305 [DOI] [PubMed] [Google Scholar]
- 30.Villén J., Beausoleil S. A., Gerber S. A., Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinkse M. W., Heck A. J. (2006) Essential enrichment strategies in phosphoproteomics. Drug Discov. Today Technol 3, 331–337 [DOI] [PubMed] [Google Scholar]
- 32.MacCoss M. J., McDonald W. H., Saraf A., Sadygov R., Clark J. M., Tasto J. J., Gould K. L., Wolters D., Washburn M., Weiss A., Clark J. I., Yates J. R., 3rd (2002) Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. U.S.A 99, 7900–7905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohammed S., Lorenzen K., Kerkhoven R., van Breukelen B., Vannini A., Cramer P., Heck A. J. (2008) Multiplexed proteomics mapping of yeast RNA polymerase II and III allows near-complete sequence coverage and reveals several novel phosphorylation sites. Anal. Chem 80, 3584–3592 [DOI] [PubMed] [Google Scholar]
- 34.Palenchar P. M. (2008) Amino acid biases in the N- and C-termini of proteins are evolutionarily conserved and are conserved between functionally related proteins. Protein J 27, 283–291 [DOI] [PubMed] [Google Scholar]
- 35.Hamilton R., Watanabe C. K., de Boer H. A. (1987) Compilation and comparison of the sequence context around the AUG start codons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res 15, 3581–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tats A., Remm M., Tenson T. (2006) Highly expressed proteins have an increased frequency of alanine in the second amino acid position. BMC Genomics 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen H. G., Chinnappan D., Urano T., Ravid K. (2005) Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol. Cell. Biol 25, 4977–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu J., Bian M., Jiang Q., Zhang C. (2007) Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 5, 1–10 [DOI] [PubMed] [Google Scholar]
- 39.Yasen M., Mizushima H., Mogushi K., Obulhasim G., Miyaguchi K., Inoue K., Nakahara I., Ohta T., Aihara A., Tanaka S., Arii S., Tanaka H. (2009) Expression of Aurora B and alternative variant forms in hepatocellular carcinoma and adjacent tissue. Cancer Sci 100, 472–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.