

Abstract
The phytopathogenic bacterium Xylella fastidiosa is the etiological agent of various plant diseases. To survive under oxidative stress imposed by the host, microorganisms express antioxidant proteins, including cysteine-based peroxidases named peroxiredoxins. This work is a comprehensive analysis of the catalysis performed by PrxQ from X. fastidiosa (XfPrxQ) that belongs to a peroxiredoxin class still poorly characterized and previously considered as moderately reactive toward hydroperoxides. Contrary to these assumptions, our competitive kinetics studies have shown that the second-order rate constants of the peroxidase reactions of XfPrxQ with hydrogen peroxide and peroxynitrite are in the order of 107 and 106 m−1 s−1, respectively, which are as fast as the most efficient peroxidases. The XfPrxQ disulfides were only slightly reducible by dithiothreitol; therefore, the identification of a thioredoxin system as the probable biological reductant of XfPrxQ was a relevant finding. We also showed by site-specific mutagenesis and mass spectrometry that an intramolecular disulfide bond between Cys-47 and Cys-83 is generated during the catalytic cycle. Furthermore, we elucidated the crystal structure of XfPrxQ C47S in which Ser-47 and Cys-83 lie ∼12.3 Å apart. Therefore, significant conformational changes are required for disulfide bond formation. In fact, circular dichroism data indicated that there was a significant redox-dependent unfolding of α-helices, which is probably triggered by the peroxidatic cysteine oxidation. Finally, we proposed a model that takes data from this work as well data as from the literature into account.
Keywords: Enzyme Kinetics, Enzyme Mechanisms, Peroxidase, Protein Structure, Thiol, Peroxiredoxin, Thioredoxin
Introduction
The Gram-negative, xylem-limited phytopathogenic bacterium Xylella fastidiosa is the etiological agent of economically important diseases in citrus and many other plant species. The 9a5c strain of X. fastidiosa, which has been linked to citrus variegated chlorosis, was the first plant pathogenic bacterium whose genome was completely sequenced (1). Citrus variegated chlorosis is the major problem faced by the Brazilian citrus industry and is responsible for significant losses in orange production (2). X. fastidiosa also imposes severe economic damage for other countries of the American continent because other strains of this bacterium provoke Pierce disease in grapevines, phony peach disease, and leaf scorch diseases in almond and oleander (3). During infection by microorganisms, one of the first plant responses is the production of oxidants, including hydrogen peroxide, organic hydroperoxides, and peroxynitrite (4–6). To counteract the release of these oxidants, microorganisms have developed defenses such as antioxidant enzymes. In the X. fastidiosa genome, five genes encode homologous proteins involved in hydroperoxide decomposition: catalase, glutathione peroxidase (GPx),2 Ohr, AhpC, and PrxQ proteins (1). All of them except GPx protein were identified in the whole cell extract and extracellular fraction of the citrus-isolated strain 9a5c (7).
Among the antioxidant proteins of X. fastidiosa, AhpC and PrxQ are members of a very large and ubiquitous family of cysteine-based peroxidases designated as peroxiredoxin (Prx). Prx enzymes are non-heme peroxidases with a catalytic activity toward hydrogen peroxide, peroxynitrite, and various organic hydroperoxides that is endowed by reactive cysteine residues. Prxs are widely distributed and are found in the majority of the pathogenic bacteria. In Escherichia coli, Prx proteins are among the 10 most abundant cellular proteins (8), and in other bacteria, Prxs were characterized as species-specific antigens (9–11).
Prxs have been classified into two groups, 2-Cys Prx and 1-Cys Prx, based on the mechanism of catalysis. The 2-Cys Prx group can be further divided into typical 2-Cys Prx and atypical 2-Cys Prx, according to the localization of the additional cysteine involved in catalysis (12, 13). All groups share a common initial step of catalysis; the oxidation of a conserved reactive cysteine (the so-called peroxidatic cysteine) to a sulfenic acid intermediate (Cys-SOH) with the reduction of the hydroperoxide substrate to the correspondent alcohol. The fate of the sulfenic acid intermediate is distinct among the Prx groups. In typical 2-Cys Prx enzymes, the sulfenic acid reacts with a second cysteine residue located in the C terminus of the other subunit (the so-called resolving cysteine), resulting in the formation of an intermolecular disulfide bond. In contrast, the resolution reaction in atypical 2-Cys Prx enzymes occurs inside the same subunit, resulting in an intramolecular disulfide bond formation. The 1-Cys Prx enzymes do not contain a resolving cysteine, and their sulfenic acid cysteine is stabilized by the active site micro-environment of the polypeptide backbone. The catalytic cycle is completed when disulfide or sulfenic acid is reduced. For most Prxs, this reduction is carried out by Trx (14).
Besides this classification, several others were proposed based on amino acid sequence similarity (15, 16). Later on, another classification based on both amino acid sequence and structural similarities was proposed and provided insights on the evolution of proteins within the Trx superfamily, which includes Prxs (17). Among the four Prx classes, class 1 is the most ancestral from which the other three classes arose. However, they are the least characterized class. Here, we will adopt the systems proposed by Copley et al. (17), and for clarity the relationship between the various classifications is presented in Table 1. Additionally, GPx enzymes are included in Table 1 because these proteins present the Trx fold and also are cysteine-based thiol-dependent peroxidases (18, 19).
TABLE 1.
Classifications for Prxs
| Subfamilies of Prxsa |
Mechanism of catalysis | Representative proteinse | ||
|---|---|---|---|---|
| Copley et al. (17)b | Trivelli et al. (16)c | Hofmann et al. (15)d | ||
| Class 1 Prx | C-type | V | Atypical 2-Cys Prx | Bcp/PrxQ/Dot5 |
| Class 2 Prx | E-type | IV | Atypical 2-Cys Prx | TPx |
| Class 3 Prx | D-type | III | All of them | Prdx5/Pmp20/Ahp1 |
| Class 4 Prx | A-type | I | Typical 2-Cys Prx | Prdx1–4/Tsa1/Tsa2 |
| B-type | II | 1-Cys Prx | Prdx6/Prx1 | |
| Class 5 Prxf | Atypical 2-Cys Prx | GPx | ||
a First line, in boldface type, is the subfamily in which XfPrxQ is included.
b Data are according to both motifs and structural elements.
c Data are according to conserved residues around the active site: A-type Prx (YPXDF(T/S)]FVCPP(T/S)E(I/L/V)); B-type Prx (HPXDFTPVCPTTE(L/F)); C-type Prx (YPX(A/D)XTP(G/V)CPTX(Q/E)XCX(F/L)); D-type Prx (XP(G/A)A(F/Y)(T/S)(P/G)XCP(S/T)XXHXP); and E-type Prx (XP(D/S)DTXVCPXX(Q/S)X(K/R)). D-type Prx also includes the so-called type II Prxs.
d Data are according to the five major molecular clades of a dendrogram of the Prx family.
e Bcp and TPx are bacterial Prx designations; PrxQ is a bacterial or plant Prx designation; Dot5, Pmp20, Ahp1, Tsa1, and Tsa2 are fungi Prx designations; Prdx1–6 is a mammalian Prx designation; GPx is the glutathione peroxidase designation.
Historically, all classes of Prxs have been considered only moderately reactive because their catalytic efficiencies (kcat/Km) toward hydroperoxides as determined by steady-state kinetics were in the 105 m−1 s−1 range. In contrast, selenocysteine-containing GPx (∼108 m−1 s−1) and heme-containing catalases (∼106 m−1 s−1) presented considerably higher values (20). More recently with the development of new assays, Prx enzymes were considered as reactive as selenium- and heme-containing proteins (21–23). However, it should be mentioned that only class 3 Prx and class 4 Prx (composed mostly of typical 2-Cys Prx but also 1-Cys Prx proteins) were analyzed by these assays, and consequently, the catalytic efficiencies for enzymes of the other Prx classes remain to be determined.
Class 1 Prx (Table 1) include, among other proteins, Bcp and PrxQ proteins with homologs present in a wide range of organisms from bacteria to unicellular eukaryotes and plants (24–28). Among all Prxs, proteins belonging to class 1 can be considered the least reactive Prxs toward hydroperoxides (23, 29). Despite this, the biological importance of these proteins comes from the observations that bcp deletion in E. coli rendered cells hypersensitive to hydrogen peroxide, TBHP, and linoleic acid hydroperoxide (24), and expression of prxq in Arabidopsis thaliana was stimulated by hydrogen peroxide, TBHP, and diamide (30). Interestingly, bcp expression was induced in the bacterium Frankia sp. during the formation of symbiosis with the plant, Alnus glutinosa (31). Recently, PrxQ proteins were further categorized into two subfamilies as follows: PrxQα proteins that have the two catalytic cysteine residues (peroxidatic and resolving cysteines) with four-residue spacing, and PrxQβ proteins that do not have a cysteine vicinal to the peroxidatic cysteine (25).
Here, we structurally and functionally characterized a class 1 Prx enzyme from X. fastidiosa named here as XfPrxQ, including the evaluation of its kinetic properties by means of new methodologies currently available. This study represents the first determination of the second-order rate constants for the reaction of a class 1 Prx enzyme with hydrogen peroxide and peroxynitrite. Through this analysis, it was possible to observe that XfPrxQ reactivity toward hydroperoxides is comparable with the most reactive enzymes (class 3 Prx, class 4 Prx, GPx, and catalases). Furthermore, the catalytic cycle of XfPrxQ was elucidated by multiple approaches such as x-ray crystallography, CD, site-directed mutagenesis, biochemical assays, and mass spectrometry. Moreover, by the identification of Trx, among other thiols, as the reducing substrate, we were able to reconstitute in vitro the pathway involved in hydroperoxide decomposition. Finally, we propose a model for the redox-dependent structural changes in PrxQβ proteins that is consistent with all of the data presented here as well as information from the literature.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of PrxQ and Its Mutants from X. fastidiosa
The prxq gene (GenBankTM accession number AAF83771, originally annotated as bcp gene) was amplified by PCR using genomic DNA of the X. fastidiosa 9a5c strain as template and the following combination of primers: forward 5′-GGAATTCCATATGAACATCGGCGACACC-3′ (NdeI) and reverse 5′-CGCGGATCCTTATTGCTCGGCATG-3′ (BamHI). The restriction sites shown in parentheses are underlined in the sequences. The amplified DNA fragment was digested with NdeI and BamHI and cloned into an NdeI-BamHI-digested pET15b vector (Novagen EMD Biosciences, Inc., Merck). The sequence of the inserted DNA was confirmed by sequencing, and the obtained expression plasmid was named pET15b-PrxQ. Expression plasmids for four XfPrxQ mutant proteins, C23S, C47S, C83S, and C101S, in which cysteines were replaced individually by serines, were generated following the protocol of the QuikChange site-directed mutagenesis kit (Agilent Technologies, Inc., Stratagene Products Division, La Jolla, CA). We used four pairs of complementary mutagenic primers (C23S, 5′-GGCAGCACCAGCAAAACACTGAG-3′; C47S, 5′-CACGCCAGGCTCTAGCACAGAAG-3′; C83S, 5′-CGACAGCTTCTCTGCCAAACAGGG-3′; and C101S, 5′-GTGACGCAATACTGTCTAAAGCATTTGACG-3′; the respective mutated codons are underlined in the sequences) and the pET15b-PrxQ as template. Resulting plasmids were used to transform E. coli BL21(DE3) (Novagen EMD Biosciences, Inc.) by electroporation, and a single colony of each generated strain was inoculated in LB medium (50 ml) containing 0.1 mg of ampicillin/ml and grown overnight at 37 °C. The culture was then transferred to 1 liter of fresh LB medium containing ampicillin and cultured further until the A600 reached 0.6–0.8. The expression of the recombinant protein was then induced by the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and growth was maintained at 20 °C. After overnight incubation, cells were harvested by centrifugation and stored at −20 °C. Frozen cells were suspended in start buffer (20 mm sodium phosphate buffer (pH 7.4), containing 0.5 m sodium chloride, 20 mm imidazole, and 2 mm phenylmethyl sulfonyl fluoride) and disrupted by sonication. The cell extracts were kept in ice during 1% streptomycin sulfate treatment for 20 min. The supernatants clarified by centrifugation were loaded onto a nickel affinity column (HisTrap FF crude from GE Healthcare) that had been equilibrated with start buffer. The conditions of His-tagged protein purification were optimized according to the manufacturer's instructions. Excess imidazole was removed from purified proteins by gel filtration using HiTrap desalting columns (GE Healthcare). Purified proteins were stored in 50 mm sodium phosphate buffer (pH 7.4) and 50 mm sodium chloride. Protein concentrations were determined spectrophotometrically at 280 nm. The extinction coefficients for reduced XfPrxQ proteins (ϵ280 = 16,960 m−1 cm−1) were calculated using the ProtParam tool available on line (32). When required, XfPrxQ proteins were reduced with 5 mm TCEP for 30 min at room temperature. Excess TCEP was removed by gel filtration using HiTrap desalting columns (GE Healthcare).
Determination of Hydroperoxide Concentration
Peroxynitrite concentrations were measured spectrophotometrically at 302 nm (ϵ302 = 1,670 m−1 cm−1). Hydrogen peroxide concentrations were measured at 240 nm (ϵ240 = 43.6 m−1 cm−1). The concentration of TBHP and CHP stock solutions was determined by an enzymatic coupled assay as described previously (33) at 340 nm (ϵ340 = 6,200 m−1 cm−1) following NADPH oxidation.
Determination of DTT-dependent Peroxidase Activity and XfPrxQ Inactivation by Dimedone Treatment
The concentration of remaining hydroperoxide was determined according to the procedure described previously (34). The peroxidase reactions of XfPrxQ wild-type and mutant proteins were started by the addition of hydroperoxide (concentration as indicated in the figure legends) into a 0.1-ml reaction mixture containing 50 mm HEPES-HCl (pH 7.4), 1 mm azide, 0.1 mm diethylenetriaminepentaacetic acid, and XfPrxQ wild-type or mutant protein (concentration as indicated in the figure legends) and then incubated at 37 °C. At the appropriate time, the reaction was terminated by the addition of 0.9 ml of ferrous xylenol orange solution (25 mm Fe(NH4)2(SO4)2, 0.1 mm xylenol orange, 4 mm butylated hydroxytoluene, and 0.25 m sulfuric acid (pH ∼1.6)) and then incubated at 37 °C for 30 min. The concentration of remaining hydroperoxide was determined by measuring the absorbance at 560 nm. Hydroperoxide consumption due to the enzymatic reaction was determined by subtracting the nonenzymatic reaction that occurs in the absence of protein from the reaction that occurs in the presence of protein. To identify sulfenic acid-containing protein by inactivation with dimedone (35), XfPrxQ wild-type and mutant proteins were incubated with 1.1 eq of hydrogen peroxide in the absence or presence of 1,000 eq of dimedone at room temperature for 30 min prior to the initiation of the peroxidase reaction.
Determination of Trx-dependent Peroxidase Activity of XfPrxQ
The peroxidase activity linked to NADPH by the Trx system was determined by monitoring the decrease of absorbance at 340 nm as described previously (36). For this purpose, XfTsnC (Trx from X. fastidiosa, GenBankTM accession number AAF85495) and XfTrxR (TrxR from X. fastidiosa, GenBankTM accession number AAF84257) were cloned and expressed in E. coli, as described in the supplemental Fig. S1. The reaction mixture contained 50 mm sodium phosphate buffer (pH 7.4), 0.3 mm NADPH, and concentrations of Trx, TrxR, and XfPrxQ wild-type or mutant protein, as indicated in the figure legends. The reaction was started by the addition of various concentrations of hydroperoxide and was carried out at 37 °C. As a control, XfPrxQ proteins were omitted from the assay mixture. The concentration of NADPH was determined spectrophotometrically at 340 nm (ϵ340 = 6,200 m−1 cm−1).
Bisubstrate Steady-state Kinetic Analysis of XfPrxQ
The peroxidase assays monitoring the decrease of absorbance at 340 nm due to NADPH oxidation were carried out as described above. The reaction mixture contained 50 mm sodium phosphate buffer (pH 7.4), 0.2 mm NADPH, 5.0 μm XfTrxR, 0.2 μm XfPrxQ, various concentrations of XfTsnC (1–4 μm), and various concentrations of either hydrogen peroxide (50–200 μm), TBHP (75–300 μm), or CHP (50–200 μm). The reaction was started by the addition of hydroperoxide and was carried out at 37 °C. As a control, XfPrxQ proteins were omitted from the assay mixture. All kinetic data were analyzed and fitted with Michaelis-Menten equation using GraphPad Prism 4 software (GraphPad Software, Inc., San Diego) and following kinetics equations described previously (37).
Competitive Kinetic Studies with HRP
The second-order rate constants of the reaction of XfPrxQ with hydrogen peroxide and peroxynitrite were determined by competition with HRP as described previously (22). In a reaction mixture containing 0.1 m sodium phosphate buffer (pH 7.4), 0.1 mm diethylenetriaminepentaacetic acid, 8.0 μm HRP, and various concentrations of reduced XfPrxQ (as indicated in the legend or abscissa axis of the figures), either 4.0 μm hydrogen peroxide or 8.0 μm peroxynitrite was mixed at 37 °C. The extent of conversion to compound I was determined by measuring the absorbance at 403 nm (ϵ403 = 102,000 m−1 cm−1) before and 2 min after the addition of hydroperoxide. The percentage of inhibition of HRP oxidation (F/1 − F) was plotted against XfPrxQ concentrations that caused it to obtain the corresponding second-order rate constant (kXfPrxQ), as calculated by Equation 1 below. The second-order rate constants of the reaction of HRP with hydrogen peroxide (kHRP = 1.7 × 107 m−1 s−1) and peroxynitrite (kHRP = 1.02 × 106 m−1 s−1) have been previously determined (22).
To determine the pKa of the peroxidatic cysteine, the second-order rate constants of the reaction of XfPrxQ with hydrogen peroxide were also determined at different pH values by the experiment described above.
HPLC Separation of Tryptic Peptides and Mass Spectrometry
To identify the disulfide-containing peptide, XfPrxQ was submitted to chemical modification and tryptic digestion prior to HPLC-ESI-MS analyses. Reduced XfPrxQ was incubated with 1.1 eq of hydrogen peroxide at room temperature for 30 min to generate the disulfide form. Excess hydrogen peroxide was removed by gel filtration using HiTrap desalting columns (GE Healthcare) with 5 mm sodium citrate buffer (pH 5.0). Disulfide-containing XfPrxQ was then treated with 100-fold excess of NEM at room temperature for 1 h to prevent nonspecific disulfide bond formation. The NEM-alkylated disulfide-containing XfPrxQ was resuspended in 6 m guanidine solution. Dilutions with 0.1 m Tris-HCl (pH 8.0) were made to reach 1 m guanidine, and the sample was incubated overnight at 37 °C after addition of Trypsin Gold (Mass Spectrometry Grade, Promega Corp., Madison, WI) to a final trypsin:XfPrxQ ratio of 1:20 (w/w). Approximately 6 μg of protein was injected onto a reverse phase C18 column (150 × 3.0 mm, 5 μm, ACE 5 C-18 HL, Advanced Chromatography Technologies, Aberdeen, Scotland, UK) connected to a Micromass Quattro II triple quadrupole mass spectrometer (Waters Corp.) equipped with an electrospray ionization source operating in positive ion mode. After a 5-min washing with 22.5% acetonitrile (in 0.1% trifluoroacetic acid), peptides were eluted with a linear gradient from 22.5 to 60.0% acetonitrile (in 0.1% trifluoroacetic acid) over 25 min at 150 μl/min (obtained by flow-splitting). Peaks were also detected at 214 nm. Mass spectra were acquired and analyzed using MassLynx software (version 3.2, Waters Corp.). To comparatively identify the disulfide-containing peptide, an aliquot of tryptic digested protein was incubated with 10 mm TCEP at room temperature for 30 min, and ∼6 μg of protein were also analyzed by HPLC-ESI-MS.
Crystallization, X-ray Diffraction Data Collection, Molecular Replacement, and Refinement of XfPrxQ C47S
For crystallization assays, XfPrxQ C47S protein was concentrated to 5–15 mg/ml in 5 mm sodium citrate buffer (pH 5.0). Crystallization trials were performed using the sitting-drop vapor diffusion method, and crystals suitable to x-ray diffraction experiments were obtained from the protein after reduction with 10 mm DTT. The optimal condition was obtained with the reservoir solution composed of 0.1 m HEPES-HCl (pH 7.5) and 20% PEG 8000 at 293 K. The crystals were soaked in a solution of 2 μl of the reservoir solution containing 20% glycerol (v/v) and then flash-frozen in a nitrogen gas stream. Crystal x-ray diffraction data were collected at 110 K using synchrotron radiation at the protein crystallography beamline W01B-MX2 of the Brazilian Synchrotron Light Laboratory, LNLS. Diffraction data were recorded on a MarMosaic 225 detector. Data sets were indexed and integrated with MOSFLM (38) and scaled and merged using SCALA (39, 40) from the CCP4 package (41). The crystal structure of XfPrxQ C47S was determined by molecular replacement methods using the PHASER program (42) and coordinates of tryparedoxin peroxidase from Crithidia fasciculata (PDB code 1E2Y, 30% sequence identity) (43). Crystallographic refinement was carried out using the REFMAC 5.0 program (44) from the CCP4 package, alternating with visual inspection of the electron density maps and manual model rebuilding with the Coot program (version 0.5.2) (45). Structural representations were generated using PyMOL (version 0.99rc6, DeLano Scientific LLC, San Carlos, CA). The coordinates have been deposited in the PDB under accession code 3IXR.
CD Spectroscopy of XfPrxQ
XfPrxQ wild-type and mutant proteins in the reduced form were analyzed before and after treatment with 1.2 eq of hydrogen peroxide at room temperature for 10 min. The CD spectra were generated using a 0.1-cm path length cuvette containing 10 μm of protein in 20 mm sodium phosphate buffer (pH 7.4). The assays were carried out at 20 °C in a Jasco J-720 spectropolarimeter (Jasco, Easton, MD). Spectra were presented as an average of eight scans recorded from 195 to 245 nm.
RESULTS
Prxs from X. fastidiosa
In the genome of X. fastidiosa 9a5c, there are two open reading frames for Prx-like proteins with accession numbers AAF83771 and AAF84339, which were annotated in the X. fastidiosa Comparative Genome Project as Bcp and AhpC, respectively (1). Following the suggestion of Kong et al. (27), we designated Bcp protein from X. fastidiosa as a PrxQ protein. The prxq gene (located in coordinates 920,666–921,145, complementary strand) codes for a predicted 159-amino acid sequence with an estimated molecular mass of 17,813 Da, and ahpc gene (GenBankTM accession number AAF84339; located in coordinates 1,487,808–1,488,368) codes for a predicted 186 amino acid sequence with an estimated molecular mass of 20,606 Da. The XfAhpC (where XfAhpC is subunit C of alkyl hydroperoxide reductase from X. fastidiosa) protein displays high amino acid sequence similarity with proteins from other bacteria (69% amino acid identity with AhpC protein from E. coli). In contrast, XfPrxQ only shares 34% sequence identity with E. coli Bcp protein.
Because proteins belonging to the Prx1 class are scarcely studied, we chose to characterize XfPrxQ. Therefore, recombinant XfPrxQ was obtained, and its purity was ascertained by nonreducing SDS-PAGE (see supplemental Fig. S1).
Thiol-dependent Peroxidase Activity of XfPrxQ
Recombinant XfPrxQ was active as initially attested by its ability to protect glutamine synthetase from oxidative inactivation by a metal-catalyzed thiol system (DTT/Fe3+/O2) but not by a non-thiol system (ascorbate/Fe3+/O2), confirming that XfPrxQ is a thiol-specific antioxidant protein (see supplemental Fig. S2). Therefore, XfPrxQ is re-reduced by DTT but not by ascorbate.
Interestingly, DTT-dependent peroxidase activity of XfPrxQ was only observed when the dithiol compound DTT was employed at high concentrations. At a 2 mm concentration of DTT, the specific activity of XfPrxQ was 0.22 ± 0.01 and 0.13 ± 0.01 μm/min/μm, respectively, when CHP and TBHP were the substrates. Because DTT consumed hydrogen peroxide nonenzymatically under these conditions, determination of hydrogen peroxide elimination through XfPrxQ catalysis was impracticable. In contrast, the other Prx from X. fastidiosa (XfAhpC) required lower concentrations of DTT. At 1.5 mm concentration of DTT, the specific activity of XfAhpC was 1.17 ± 0.03, 1.02 ± 0.01, and 0.84 ± 0.02 μm/min/μm, respectively, when hydrogen peroxide, CHP, and TBHP were the substrates.
Because XfPrxQ is only slightly reducible by DTT, it was relevant to identify the biological reducing system for this enzyme. DTT-dependent peroxidase activity of XfPrxQ was greatly stimulated by addition of Trx from X. fastidiosa (XfTsnC) into the reaction mixture (Fig. 1A), suggesting XfTsnC as a possible XfPrxQ biological electron donor. In the presence of XfTsnC, the specific activity of XfPrxQ was 1.76 ± 0.09, 1.49 ± 0.07, and 0.67 ± 0.04 μm/min/μm, respectively, when hydrogen peroxide, CHP, and TBHP were the substrates (Fig. 1B).
FIGURE 1.

Peroxidase activity of XfPrxQ. A, removal of CHP was determined by DTT-dependent peroxidase assay in the presence of 5 μm XfPrxQ (closed squares), 10 μm XfTsnC (open triangles), and 5 μm XfPrxQ plus 10 μm XfTsnC (open squares). Peroxidase reactions were carried out in a reaction mixture containing 200 μm CHP and 0.5 mm DTT, at 37 °C. B, removal of hydrogen peroxide (open squares), TBHP (open triangles), and CHP (closed triangles) by XfPrxQ in DTT-dependent peroxidase assays. Peroxidase reactions were carried out in a reaction mixture containing 200 μm hydroperoxide, 0.5 mm DTT, 5 μm XfPrxQ, and 7 μm XfTsnC at 37 °C. At the indicated times, the remaining hydroperoxides were quantified in triplicate, and results are the mean ± S.D. (error bars). C, NADPH oxidation coupled to hydrogen peroxide reduction in the presence of 1 μm XfPrxQ (closed squares); 0.8 μm XfTsnC and 0.3 μm XfTrxR (open triangles); 1 μm XfPrxQ, 0.8 μm XfTsnC, and 0.3 μm XfTrxR (closed triangles); and 1 μm XfAhpC, 0.8 μm XfTsnC, and 0.3 μm XfTrxR (open squares). NADPH oxidation was measured at 340 nm in a reaction mixture containing 250 μm NADPH and 500 μm hydrogen peroxide at 37 °C. D, wild-type XfPrxQ was subjected to nonreducing SDS-PAGE under reducing conditions (lane 2), treated with 1 eq of hydrogen peroxide (lane 3) and treated with 5 eq of hydrogen peroxide (lane 4). Lane 1 shows molecular mass standards (BenchMark Protein Ladder, Invitrogen). Reduced XfPrxQ protein was treated with hydrogen peroxide at room temperature for 30 min. Approximately 4 μg of protein was loaded by lane.
Trx-dependent Peroxidase Activity of XfPrxQ
To better characterize the enzymatic properties of XfPrxQ, the Trx-linked peroxidase assay was carried out. In the presence of XfTsnC and XfTrxR, no NADPH oxidation was observed when XfPrxQ was omitted from the reaction mixture. The addition of XfPrxQ resulted in significant oxidation of NADPH (Fig. 1C). A heterologous Trx system, composed by E. coli proteins, TrxA (Trx) and TrxB (TrxR), also supported the peroxidase activity of XfPrxQ (data not shown). In contrast, the Trx system of X. fastidiosa did not support the peroxidase activity of XfAhpC (Fig. 1C).
In the genome of X. fastidiosa, there is an ORF predicted to encode an AhpF homolog, just downstream of the ahpc gene (1). In bacteria, the AhpF enzyme acts as a dedicated AhpC reductase, directing the transfer of electrons from NADH to AhpC (46). Corroborating this AhpC dedicated activity, recombinant AhpF from X. fastidiosa (XfAhpF, accession number AAF84340) did not support the peroxidase activity of XfPrxQ (see supplemental Fig. S3).
Because some Prxs display peroxidase activity supported by glutathione or glutaredoxin (47, 48), these processes were also investigated. However, neither glutathione nor glutaredoxin 1 from E. coli supported peroxidase activity (see supplemental Fig. S4). These data indicated that XfPrxQ is a Trx-dependent peroxidase.
Functional Cysteine Residues of XfPrxQ
Several Prxs similar to E. coli Bcp have two conserved cysteines with four-residue spacing (CXXXXC motif) that are involved in the intramolecular disulfide bond formation during the catalytic cycle (24, 25, 27, 28). XfPrxQ has four cysteines, and its N-terminal cysteine residue (Cys-47) is conserved in all proteins of the Bcp/PrxQ family (class 1 Prx enzymes) and is predicted to be the peroxidatic cysteine (13). However, XfPrxQ lacks the putative resolving cysteine residue at that C-terminal position of the CXXXXC motif. This motif is a feature shared by proteins from the PrxQα subfamily (Fig. 2, upper section). On the other hand, Cys-83 of XfPrxQ is also conserved in some proteins of the Bcp/PrxQ family (class 1 Prx enzymes), as is Cys-101 (Fig. 2). Moreover, Cys-23 is the unique residue that is only present in the XfPrxQ protein. Therefore, Cys-83 and Cys-101 are strong candidates for participating in an intramolecular disulfide bond in XfPrxQ.
FIGURE 2.

Analysis of the primary structures of PrxQ proteins. Amino acid sequence alignments of various PrxQ from PrxQα and PrxQβ subfamilies. The red line separates PrxQ proteins into PrxQα (upper section) and Prxβ (lower section). Conserved peroxidatic and resolving cysteine residues are highlighted in blue. Conserved residues of the active site are highlighted in orange. XfPrxQ Cys-23 and Cys-101 are highlighted in green and magenta, respectively. PrxQα subfamily is represented by proteins as follows: ScDot5, S. cerevisiae DOT5 (GI: 731778); AtPrxQ, A. thaliana PrxQ (GI: 9279611); PoPrxQ, Poplar PrxQ (Populus balsamifera subsp. trichocarpa × Populus deltoides; GI: 42795441); SlPrxQ, Sedum lineare PrxQ (GI: 75336180); CdBcp, Clostridium difficile Bcp (GI: 126699430); EcBcp, E. coli Bcp (GI: 1788825); HiBcp, Haemophilus influenzae Bcp (GI: 1573220); KpBcp, K. pneumoniae Bcp (GI: 152971345); MtBcp, Mycobacterium tuberculosis Bcp (GI: 2791423); and StBcp, S. typhimurium Bcp (GI: 16765811). PrxQβ subfamily is represented by proteins as follows: RsPrxQ, Rhodobacter sphaeroides PrxQ (GI: 77387949); XcBcp, X. campestris Bcp (GI: 66768804); AvBcp, Anabaena variabilis Bcp (GI: 75906708); SyBcp, Synechocystis sp. (GI: 16329318); TeBcp, Thermosynechococcus elongatus Bcp (GI: 22298737); and XfPrxQ, Xylella fastidiosa PrxQ (GI: 9105889). The secondary structure of XfPrxQ C47S, obtained by PROCHECK (52), is shown below its sequence (green arrows representing β-sheets and red rectangles for α-helices).
Recombinant XfPrxQ was obtained at the molecular mass expected for a monomer (20.0 kDa, including the 2.2-kDa His tag) by nonreducing SDS-PAGE, regardless of its oxidative state (Fig. 1D), indicating that an intermolecular disulfide bond was not formed throughout its catalytic cycle. In agreement with the formation of an intramolecular disulfide bond during the catalytic cycle, thiol titrations of XfPrxQ by 5,5′-dithiobis(2-nitrobenzoic acid) under denaturing conditions indicated the presence of four thiols per molecule for the reduced form of protein, whereas two thiols were detected in the oxidized form. About three thiols per molecule were detected in reduced XfPrxQ under native conditions, indicating the presence of one solvent inaccessible thiol (see supplemental Table SI).
To identify the location of the putative intramolecular disulfide bond and to characterize the catalytic cycle of XfPrxQ, a series of biochemical assays were conducted on wild-type and mutant proteins in which each cysteine residue was individually replaced by serine. In the thiol titration assay, XfPrxQ C47S was the only mutant protein presenting the same number of thiol groups (three per molecule) under oxidizing and reducing conditions, indicating the absence of thiol oxidation (see supplemental Table SI), which is consistent with the prediction that Cys-47 is the peroxidatic cysteine of XfPrxQ. The other mutant proteins presented differences higher than 1.6 thiols per molecule between reduced and oxidized forms, suggesting that disulfides were formed in the oxidized state. Therefore, thiol titration assay was unable to identify the resolving cysteine. Interestingly, hydrogen peroxide-treated XfPrxQ C83S exhibited several bands on nonreducing SDS-PAGE, which is distinctly divergent from the other mutant proteins. This result suggests nonspecific disulfide bond formation consistent with the lack of the resolving cysteine in the XfPrxQ C83S mutant protein (see supplemental Fig. S5). As mentioned above, thiol titrations under native conditions of wild-type protein in the reduced form indicated that one cysteine residue was inaccessible for reaction with 5,5′-dithiobis(2-nitrobenzoic acid). Because the number of thiol groups between denatured and native forms of the C101S mutant was about the same, it is possible that Cys-101 is buried in the polypeptide backbone.
As the peroxidase activity of XfPrxQ was abolished by mutation of Cys-47 (Fig. 3, A and B), this residue is the peroxidatic cysteine as predicted by amino acid alignment and thiol titration. Moreover, XfPrxQ C23S and XfPrxQ C101S retained substantial activity in comparison with the wild-type protein. However, the C83S mutant protein had divergent effects on the enzymatic activity when its DTT- or Trx-dependent peroxidase activity was analyzed. Although XfPrxQ C83S appeared to be gradually inactivated by the hydroperoxide during the time course of the Trx-linked peroxidase assay (Fig. 3B), its DTT-dependent peroxidase activity was about 2-fold increased when compared with the wild-type protein (Fig. 3A).
FIGURE 3.

Effects of the Cys → Ser mutations on XfPrxQ disulfide bond formation and peroxidase activity. A, removal of CHP in DTT-dependent peroxidase assay by XfPrxQ proteins as follows: wild-type (gray squares), XfPrxQ C23S (black squares), XfPrxQ C47S (gray triangles), XfPrxQ C83S (black triangles), and XfPrxQ C101S (open triangles). Peroxidase reactions were carried out in a reaction mixture containing 200 μm CHP, 2 mm DTT, and 25 μm XfPrxQ proteins at 37 °C. As a negative control, a peroxidase reaction was performed without XfPrxQ protein (open squares). At the indicated times, the remaining CHP was quantified in triplicate, and results are the mean ± S.D. (error bars). B, NADPH oxidation coupled to CHP reduction in the presence of XfPrxQ proteins as follows: wild-type (gray squares), XfPrxQ C23S (black squares), XfPrxQ C47S (gray triangles), XfPrxQ C83S (black triangles), and XfPrxQ C101S (open triangles). Assays were carried out in a reaction mixture containing 250 μm CHP, 200 μm NADPH, 1 μm XfPrxQ proteins, 1 μm E. coli Trx, and 0.1 μm E. coli TrxR at 37 °C. As a negative control, peroxidase reactions were performed without XfPrxQ protein (open squares). C, dimedone inactivation of XfPrxQ wild-type and C83S proteins. Prior to the DTT-dependent peroxidase assay, XfPrxQ proteins were incubated with 1.1 eq of hydrogen peroxide in the absence (control, black bars) or presence of 1,000 eq of dimedone (white bars) at room temperature for 30 min. Peroxidase reactions were carried out in a mixture containing 300 μm TBHP, 1 mm DTT, 1 μm E. coli Trx, and 10 μm XfPrxQ proteins at 37 °C. After 10 min, the remaining TBHP was quantified in triplicate, and results are the mean ± S.D. (error bars). Peroxidase activity was considered 100% when wild-type XfPrxQ was assayed in the absence of dimedone.
It is likely that Trx may not be efficient at reducing sulfenic acids generated in XfPrxQ C83S and thereby Cys-47 could be overoxidized to either sulfinic or sulfonic acids. It is well known that overoxidation of Prx provokes their inactivation (13, 20, 49). Inactivation may not have occurred in the DTT-dependent peroxidase assay (Fig. 3A) because sulfenic acid in Cys-47 could be more readily reduced by DTT than by Trx.
To test if sulfenic acid was generated and persisted for a longer time in the absence of the putative resolving cysteine, XfPrxQ wild-type and C83S proteins were preincubated with dimedone, a sulfenic acid-modifying reagent (50), in the presence or absence of hydrogen peroxide. Excess reagents were removed before assaying peroxidase activity. Dimedone was not able to inactivate wild-type XfPrxQ probably because the attack of resolving cysteine to the Cys-47-SOH intermediate outcompetes its alkylation by dimedone (Fig. 3C). However, dimedone likely inactivated the XfPrxQ C83S protein because sulfenic acids persist long enough to react with dimedone only in the absence of Cys-83. Dimedone also protected XfPrxQ C83S from precipitation by the hydrogen peroxide treatment (see supplemental Fig. S6), probably by preventing the reaction of sulfenic acids with other XfPrxQ cysteine residues after protein denaturation in nonreducing SDS-PAGE. These data strongly suggest Cys-83 as the resolving cysteine of XfPrxQ.
Unequivocal localization of peroxidatic and resolving cysteines of XfPrxQ was obtained by HPLC-ESI-MS analyses. NEM-alkylated disulfide-containing wild-type XfPrxQ was trypsin-digested under denaturing conditions and resolved on a C-18 column connected to an ESI-MS. The chromatogram of the NEM-alkylated disulfide-containing peptide mixture (Fig. 4A) was compared with that of the TCEP-treated mixture (Fig. 4B). A well resolved peak was detected in the TCEP-treated sample at 16.97 min (peak D in Fig. 4B). The mass of this species corresponded to the peptide containing the Cys-47 residue (Fig. 4D). In this reduced sample, we were unable to find the other peptide of the disulfide-containing peptide. However, we detected the original disulfide-containing peptide in the NEM-alkylated disulfide-containing peptide mixture (peak C in Fig. 4A). The mass of this species corresponded to the expected peptide formed by a disulfide bond between Cys-47 and Cys-83 (Fig. 4C). In addition, we detected two peptides that corresponded to the peptide containing the Cys-23 residue and the peptide containing the Cys-101 residue, both conjugated with NEM (data not shown). These two peptides were detected in the NEM-alkylated disulfide-containing peptide mixture and the TCEP-treated mixture. Thus, the hydrogen peroxide treatment of XfPrxQ resulted in the formation of an intramolecular disulfide bond between Cys-47 and Cys-83.
FIGURE 4.

Localization of peroxidatic and resolving cysteines of XfPrxQ by HPLC-ESI-MS. Disulfide-containing XfPrxQ was submitted to NEM alkylation and tryptic digestion prior to HPLC-ESI-MS analyses, as described under “Experimental Procedures.” A, C-18 HPLC c of the NEM-alkylated disulfide-containing peptide mixture. The arrow indicates the retention time of the peptide scanned in C (15.66 min). B, chromatogram of the TCEP-treated mixture. The arrow indicates the retention time of the peptide scanned in D (16.97 min). Peptide elution was monitored at 214 nm. C, ion scan at 15.66 min retention time of the chromatogram showed in A. Two protonated parent ions could be used to assign a peptide formed by a disulfide bond between Cys-47 and Cys-83 (4353.07 atomic mass units). D, ion scan at 16.97 min retention time of the chromatogram showed in B. Three protonated parent ions could be used to assign a peptide containing the Cys-47 residue (3461.71 atomic mass units).
Another mass spectrometry experiment was performed by the Proteomics Platform (Eastern Quebec Genomics Center, Quebec, Canada) and confirmed the results presented above. Briefly, NEM-alkylated disulfide-containing XfPrxQ was reduced by DTT, alkylated with iodoacetamide, and finally digested by trypsin. Trypsinized protein was then analyzed by HPLC-C18-ESI-MS/MS and again indicated a disulfide bond between Cys-47 and Cys-83. In this experiment, XfPrxQ peptides containing Cys-47 and Cys-83 were preferentially alkylated by iodoacetamide, whereas peptides containing Cys-23 and Cys-101 were preferentially alkylated by NEM (see supplemental Table SII).
Kinetic Parameters of XfPrxQ
Because XfPrxQ has two substrates, hydroperoxide and reduced Trx, the more appropriate way to kinetically characterize XfPrxQ is by steady-state bisubstrate kinetic analyses (37), during which the initial rate of hydroperoxide consumption by XfPrxQ was indirectly measured by the NADPH oxidation with varying concentrations of hydroperoxide and XfTsnC in the presence of a molar excess of both XfTrxR and NADPH. Results obeyed the Michaelis-Menten equation with saturation by both substrates (hydroperoxide and XfTsnC), making it possible to determine the enzymatic parameters, kcat and Km. The reciprocal initial rates plotted against the reciprocal concentrations of the reducing substrate (XfTsnC) resulted in straight and parallel lines for different concentrations of hydroperoxide (Fig. 5). These data suggested that the catalysis by XfPrxQ proceeds by a ping pong bi bi mechanism that can be described as a sequence of two bimolecular reactions (Reactions 1 and 2) occurring consecutively and independently of each other,
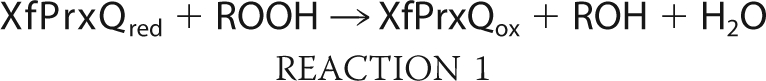 |
 |
where ROH is hydroperoxide-derived alcohol and ROOH is hydroperoxide. The ordinate intercepts (1/Vmax(app)) were replotted against 1/[hydroperoxide], revealing an enzyme substitution mechanism with saturation kinetics (37). Assuming a ping pong mechanism for all equations, the y intercept of the secondary plot was equivalent to 1/Vmax, and the slope was equivalent to Km(hydroperoxide)/Vmax, whereas the slope of the primary plot was equivalent to Km(XfTsnC)/Vmax. XfPrxQ presented slightly higher specificity toward hydrogen peroxide and CHP (kcat/Km = 3.35 × 104 and 5.34 × 104 m−1 s−1, respectively) compared with TBHP (kcat/Km = 0.56 × 104 m−1 s−1) (Table 2). The catalytic efficiency determined for XfPrxQ in relation to XfTsnC was in the 105 m−1 s−1 range, and no significant difference was observed for the three hydroperoxide substrates investigated. Therefore, taking into account this steady-state kinetics analysis, it can be assumed that XfPrxQ is moderately reactive toward hydroperoxides. It is important to mention that cleavage of the His tag from recombinant fusion proteins did not change considerably their activities (data not shown).
FIGURE 5.

Steady-state bisubstrate kinetics analyses of XfPrxQ. NADPH oxidation was measured at 340 nm at 37 °C in a reaction mixture containing 0.2 mm NADPH, 5.0 μm XfTrxR, 0.2 μm XfPrxQ, various concentrations of XfTsnC (1–4 μm), and various concentrations of hydroperoxide. The data were fitted using GraphPad Prism 4. On the left side, primary plot of 1/(initial rate) versus 1/(XfTsnC concentration) at various concentrations of hydroperoxide is shown. Concentrations of hydrogen peroxide and CHP are as follows: 50 μm (open squares), 100 μm (closed squares), and 200 μm (open triangles). Concentrations of TBHP are as follows: 75 μm (open squares), 150 μm (closed squares), and 300 μm (open triangles). Each hydroperoxide concentration was analyzed in triplicate, and initial rates are the means ± S.D. (error bars). On the right side, replot of the y-intercept of primary plot versus 1/(hydroperoxide concentration).
TABLE 2.
Steady-state bisubstrate kinetic parameters of XfPrxQ
| Substrate | Kinetic parameters of XfPrxQa |
|||
|---|---|---|---|---|
| Vmaxb | kcat | Kmc | kcat/Km | |
| μm s−1 | s−1 | μm | m−1s−1 | |
| Hydrogen peroxide | 0.24 ± 0.02 | 1.10 ± 0.07 | 33 ± 19 | 3.35 × 104 |
| XfTsnC | 6.8 ± 1.3 | 1.62 × 105 | ||
| TBHP | 0.09 ± 0.04 | 0.41 ± 0.18 | 72 ± 41 | 0.56 × 104 |
| XfTsnC | 3.0 ± 1.9 | 1.34 × 105 | ||
| CHP | 0.44 ± 0.16 | 2.00 ± 0.73 | 37 ± 19 | 5.34 × 104 |
| XfTsnC | 12.2 ± 5.5 | 1.64 × 105 | ||
a Steady-state bisubstrate kinetic analyses of XfPrxQ were performed as described in Fig. 5.
b These values are the 1/(y intercept ± S.D.) of the replot of the y intercept of primary plot versus 1/hydroperoxide concentration in Fig. 5 (right plots).
c For hydroperoxide substrates, these values are the average ± S.D. of Km obtained from the nonlinear regression analysis of initial rate versus hydroperoxide concentration, using GraphPad Prism 4. For XfTsnC, these values were obtained from (slope ± S.D.) of the replot of the y intercept of the primary plot versus 1/(hydroperoxide concentration) in Fig. 5 (right plots) multiplied by the Vmax obtained here (slope = Km(XfTsnC)/Vmax).
New kinetic approaches have revealed that class 3 Prx and class 4 Prx enzymes present high reactivity toward hydroperoxides (21–23, 51), changing the view that Prx enzymes are only moderately reactive. Because this kind of analysis had not yet been performed for class 1 Prx enzymes, the second-order rate constant of the reaction between reduced XfPrxQ and hydrogen peroxide was measured by competitive assay (22). Hydrogen peroxide-mediated oxidation of HRP to compound I was inhibited by XfPrxQ in a concentration-dependent manner. The plot of (F/(1 − F))·kHRP·[HRP] against XfPrxQ concentration was linear (Fig. 6A) and allowed the determination of the second-order rate constant of the reaction between XfPrxQ and hydrogen peroxide as (4.53 ± 0.39) × 107 m−1 s−1 (pH 7.4) and 37 °C. The second-order rate constant of XfPrxQ C83S was also determined in the same range (107 m−1 s−1) (pH 7.4) and 37 °C (Fig. 6B), indicating that condensation between the sulfenic acid derivative of peroxidatic cysteine and the resolving cysteine did not interfere in the analysis.
FIGURE 6.

Rate constant determinations of the reactions between XfPrxQ and either hydrogen peroxide or peroxynitrite by kinetic competitive approach with HRP. The reaction mixtures containing 8 μm HRP and reduced XfPrxQ in the specified concentrations were incubated with 8 μm hydrogen peroxide or peroxynitrite. After a 2-min incubation at 37 °C, formation of HRP compound I was measured at 403 nm. Each XfPrxQ protein concentration was analyzed in triplicate, and values are the means ± S.D. (error bars). A, rate constant of the reaction between wild-type (WT) XfPrxQ and hydrogen peroxide. Slope = kWT XfPrxQ = (4.53 ± 0.39) × 107 m−1 s−1. B, rate constant of the reaction between XfPrxQ C83S and hydrogen peroxide. Slope = kXfPrxQ C83S = (1.06 ± 0.07) × 107 m−1 s−1. C, variation of the second-order rate constant of the reaction between wild-type XfPrxQ and hydrogen peroxide as a function of the pH. D, Rate constant of the reaction between wild-type XfPrxQ and peroxynitrite. Slope = kWT XfPrxQ = (1.04 ± 0.03) × 106 m−1 s−1.
The kinetic competitive approach with HRP was also used to determine the pKa of the XfPrxQ-reactive cysteine. The second-order rate constants of the reaction of XfPrxQ with hydrogen peroxide were determined at different pH values in the range of 4.5 to 7.5, and the experimental pH profiles showed a single pKa of 6.2 (Fig. 6C). This pKa value corresponds to the pKa of the peroxidatic cysteine residue of XfPrxQ (22).
The second-order rate constant of the reaction between reduced XfPrxQ and peroxynitrite can also be determined by the kinetic competitive approach with HRP (22). Once again, XfPrxQ competed with HRP for peroxynitrite in a concentration-dependent manner (Fig. 6D). By the linear plot of (F/(1 − F))·kHRP·[HRP] against XfPrxQ concentration, we calculated the second-order rate constant of the reaction of XfPrxQ and peroxynitrite to be (1.04 ± 0.03) × 106 m−1 s−1 (pH 7.4) and 37 °C. The result is in the range of 105 to 107 m−1 s−1 rate constants reported so far (23).
Crystal Structure of XfPrxQ C47S
Once several biochemical properties of XfPrxQ were characterized, we attempted to obtain its structure in the reduced and oxidized forms to perform structural and functional relationship experiments. Despite the efforts to obtain structures for the wild-type protein, we could only obtain crystals of XfPrxQ C47S, which is probably representative of the reduced enzyme state of the wild-type protein, as discussed below and assumed by researchers in the field. XfPrxQ C47S crystallized in space group P212121 (orthorhombic) with one molecule per asymmetric unit. In the final model, which includes 156 of 159 residues of XfPrxQ, the missing residues are in a large loop located between the β6 and β7 (Gly-114 and Arg-115) and in the C-terminal amino acid (Gln-159). Nine residues were marked as alanine due to the lack of electronic density in the amino acid side chains (Lys-41, Lys-110, Thr-111, Met-112, Tyr-113, Gln-116, Ile-118, Lys-141, and Glu-158). Because XfPrxQ C47S was expressed as an N-terminal His-tagged recombinant protein, three amino acids residues of the tag were assigned as −2 (Gly), −1 (Ser), and 0 (His). All non-glycine and non-proline residues fall in the most favored or additionally allowed regions of the Ramachandran plot, as defined by the PROCHECK program (version 3.4.4) (52). Additionally, 172 water molecules were assigned in the final model. The structure was refined to a final crystallographic R-factor of 19.8% and R-free of 24.6% at 1.60 Å of resolution. Crystallization data and refinement statistics are presented in the supplemental Table SIII. The structural analysis, performed using the PISA program (53) from the CCP4 package, suggested that XfPrxQ is a monomer in solution with a surface area of 7691.4 Å2.
As expected, the structure of XfPrxQ C47S includes a canonical Trx fold (20) with insertions and extensions that form a five-stranded mixed β-sheet (in the order β3↑-β4↑-β5↑-β8↓-β9↑) surrounded by six α-helices and four additional β-strands in the α1-β1-β2-α2-β3-α3-β4-α4-β5-α5-β6-β7-β8-β9-α6 arrangement of secondary structure elements. According to structural comparisons with known Prx structures and based on the classification suggested by Copley et al. (17), we confirmed that XfPrxQ is a class 1 Prx (Table 1).
A small number of structural coordinates of class 1 Prx enzymes were only very recently made available in the RCSB Protein Data Bank. Of seven structures available, three of them are structures of the same protein, Bcp from Xanthomonas campestris (54), which is a member of the PrxQβ subfamily that shares 66% of amino acid sequence identity with XfPrxQ. The atom coordinates of these structures (54) were deposited during the development of this manuscript. Furthermore, the four other structures refer to members of the PrxQα subfamily (PDB codes 2CX3, 2CX4, 2YWN, and 3DRN); however, only 3DRN has been published (55).
The Ser-47, which replaces the peroxidatic cysteine in XfPrxQ C47S, is located at the N terminus of helix α3 (Fig. 7A). Electron density maps of Ser-47 indicated that the side chain can adopt two distinct conformations. Interactions between amino acids reflect the configuration that usually stabilizes reactive cysteine in the thiolate form (R-S−) within the active site of Prxs in only one of the conformations (Fig. 7B, the other conformation is represented in supplemental Fig. S7) (12, 20). In this conformation, Oγ of Ser-47 (Cys-47 in the wild-type protein) can make hydrogen bonds with O of Lys-41 (3.17 Å), Oγ of Thr-44 (3.19 Å), and Nη1 of Arg-122 (3.22 Å). One water molecule (H2O-290) interacts with Oγ of Ser-47, with O and N of Lys-41, and with Oγ of Thr-44. Furthermore, Oϵ1 of Glu-50 makes a polar interaction with Nη1 of Arg-122 (2.90 Å) that appears to orient the Arg-122 and thereby allows the interaction between Arg-122 and Ser-47. Residues conserved to Arg-122 are postulated to affect the pKa of the peroxidatic cysteine and consequently influences its nucleophilicity (17). Thus, the pKa of 6.2 for the peroxidatic cysteine residue of XfPrxQ determined here (Fig. 6C) could be attributed (at least partially) to the nearby guanidino group of Arg-122. Indeed, mutation of this Arg in other Prxs abolishes or diminishes peroxidase activity (56). The stabilization of this negative charge on Cys-47 could also arise from the influence of electrostatic interactions between the thiolate anion and other positively charged residues in the active site region (57). Finally, the positive electrostatic potential of the XfPrxQ surface, where the peroxidatic cysteine is located (Fig. 7C), and the localization of the peroxidatic cysteine in the N terminus of helix α2 (58) can also be correlated with the low pKa value of Cys-47.
FIGURE 7.

Structural features of the XfPrxQ C47S protein. A, schematic representation of the XfPrxQ C47S structure. The five-stranded mixed β-sheet of the Trx fold is colored in orange. Some elements of secondary structure are labeled, and the N and C termini are indicated. Side chains of Ser-47 and Cys-83 are shown in ball and stick mode. B, interaction network in the active site of XfPrxQ C47S. Residues are represented as balls and sticks, and atoms are colored following the CPK color scheme (carbon, green; oxygen, red; nitrogen, blue; and sulfur, yellow). The magenta ball represents water-290. Hydrogen bonds and salt bridges are indicated by dashed lines. C, XfPrxQ C47S mapped by electrostatic surface potentials (red, negatively charged; blue, positively charged), as calculated with the APBS program (65). The white circle demarcates the surface in the vicinity of the active site.
In only the conformation described in Fig. 7B, Ser-47 is also exposed to the surface of the protein, allowing interaction with substrate. The Ser-47 side chain is located in a shallow cavity with a surface that is made up of the side chains of Pro-40, Lys-41, Thr-44, Gly-46, and Arg-122. In this cavity, the Ser-47 side chain occupies 0.3 Å2 of solvent-accessible surface, as calculated using the ArealMol program from the CCP4 package (41) and using a radius of 1.4 Å for probe solvent molecules. The shape of this cavity suggests that it could accommodate substrates of various forms and sizes, which is in agreement with the low specificity of XfPrxQ toward hydroperoxides (Table 2).
Electron density maps of the resolving cysteine (Cys-83) indicated that the side chain can also adopt two conformations. Interestingly, Cys-83 is located at the middle of helix α4 and is 12.26–14.07 Å from the peroxidatic Cys-47. Therefore, to produce distances within 2 Å (the idealized bonding distance between Sγ atoms in disulfide bonds), substantial backbone conformational changes in XfPrxQ would be required to form the intramolecular disulfide bond observed through mass spectrometry experiments (Fig. 4 and supplemental Table SII).
Because we expected that some unfolding of α-helices containing active cysteine residues (helix α3 and/or helix α4) should take place to allow disulfide bond formation between Cys-47 and Cys-83, we analyzed XfPrxQ in different oxidation states by CD spectroscopy. In fact, the CD spectrum of oxidized wild-type XfPrxQ in the far-UV region displayed a decrease in the signal intensity at 222 nm relative to the reduced XfPrxQ, which is consistent with a smaller α-helical content (Fig. 8A). A similar structural change due to oxidation occurred in the XfPrxQ C83S protein (Fig. 8C) but not in the XfPrxQ C47S protein (Fig. 8B). Estimating the α-helical content of these enzymes by the molar ellipticity at 222 nm as described previously (59), we found that oxidation of XfPrxQ wild-type and C83S proteins resulted in a 4.7 and 4.0% reduction in α-helical content, respectively. Because helices α3 and/or α4 are 6.7 and 6.1% of the XfPrxQ α-helical content, respectively, CD estimation indicates that at least part of helices α3 and α4 underwent a structural unfolding due to the redox state change of XfPrxQ during its catalytic cycle. Taking all of the experimental data presented here as well as information from the literature into account, a model for the catalytic mechanism for class 1 Prx is proposed below.
FIGURE 8.

Conformational analysis of XfPrxQ. CD spectra in the reduced form (solid line) and treated with 1.2 eq of hydrogen peroxide (dashed line) of wild-type XfPrxQ (A), XfPrxQ C47S (B), and XfPrxQ C83S (C) proteins. Spectra were recorded at 20 °C using a protein concentration of 10 μm in 20 mm sodium phosphate buffer (pH 7.4).
DISCUSSION
Class 1 Prx enzymes (Table 1) are poorly characterized, with crystal structures being elucidated only very recently. This study represents a comprehensive structural and functional analysis of XfPrxQ as a representative enzyme of class 1 Prx. Of the four cysteine residues of XfPrxQ (Cys-23, Cys-47, Cys-83, and Cys-101), three of them are conserved among PrxQβ orthologs (Cys-47, Cys-83, and Cys-101). Here, we unequivocally established that peroxidatic Cys-47 and resolving Cys-83 form an intramolecular disulfide bond in the oxidized state.
Functional studies with XfPrxQ mutant proteins indicated the role of Cys-47 as the peroxidatic cysteine. Additional approaches were necessary to identify Cys-83 as the resolving cysteine of XfPrxQ, including mass spectrometry approaches (Fig. 4 and supplemental Table SII). This study is the first experimental demonstration of intramolecular disulfide bond formation in solution by a protein member of the PrxQβ subfamily, providing unequivocal evidence that XfPrxQ follows the atypical 2-Cys Prx mechanism.
Compared with class 4 Prx from X. fastidiosa (XfAhpC), high concentrations of thiol compound (2 mm) were required to detect DTT-dependent peroxidase activity of XfPrxQ, suggesting that disulfides in class 1 Prx are very stable. Again, this finding is consistent with the proposal that the recycling step of Prx reduction limits the catalytic cycle (21–23, 51). This result can explain why DTT-dependent peroxidase activity of XfPrxQ C83S was higher than wild-type XfPrxQ activity (Fig. 3A) even though they have similar second-order rate constants of the reaction with hydrogen peroxide (Fig. 6, A and B). It could also explain, for example, why DOT5 (nuclear thiol peroxidase from Saccharomyces cerevisiae), a class 1 Prx protein, has a markedly lower DTT-dependent antioxidant activity of glutamine synthetase protection than those of the other yeast isoenzymes belonging to class 3 Prx and 4 Prx (29).
Therefore, it was relevant to identify in this work that the Trx system of X. fastidiosa composed by XfTsnC and XfTrxR, but not other thiol-reducing systems, is the biological reducing system of XfPrxQ (Fig. 3 and supplemental Figs. S3 and S4). In agreement with this finding, DTT-dependent peroxidase activities of other class 1 Prx enzymes were also greatly stimulated by Trx proteins (25, 28, 56, 60, 61). Furthermore, the midpoint potential (−325 mV) of plant PrxQ is in agreement with the necessity of a protein with high reducing power, such as thioredoxin, to support the peroxidase activity of PrxQ enzymes (28).
Bisubstrate steady-state kinetics analysis of XfPrxQ resulted in catalytic efficiencies in the range of 103 to 104 m−1 s−1 (Table 2), which would indicate that this peroxidase is only moderately reactive when compared with GPx and catalases (20). Recent studies indicated that class 3 Prx and class 4 Prx proteins (Table 1) are also highly reactive toward hydroperoxides, and the apparent ambiguity between these studies was resolved with the proposal that reduction of Prxs is the rate-limiting step (21–23, 51). Similarly, by competitive kinetic studies with HRP, the second-order rate constants of XfPrxQ were shown to be high for hydrogen peroxide (in the 107 m−1 s−1 range) and for peroxynitrite (106 m−1 s−1 range). These are the first determinations of second-order rate constants of the reaction of a reduced class 1 Prx enzyme and hydroperoxides, and the results have changed the paradigm that class 1 Prx enzymes present moderate reactivities within the Prx family of enzymes. Instead, these values demonstrate that class 1 Prx proteins are as reactive toward hydroperoxides as class 3 Prx, class 4 Prx, GPx, and catalases (21–23, 51), which is consistent with the severe phenotypes derived from the deletion of their genes (24, 30, 31). The second-order rate constant of XfPrxQ reacting with peroxynitrite is similar to the rate constant values of class 4 Prx from bacteria and tryparedoxin peroxidases from trypanosomatids (62, 63) and is even higher than the rate constant values of class 4 Prx from yeast (k ∼105 m−1 s−1) (22), although it is smaller than the respective parameters for class 3 Prx from mammals (k ∼107 m−1 s−1) (51).
The pKa value of the peroxidatic cysteine residue of XfPrxQ (pKa = 6.2) determined here (Fig. 6C) is in agreement with the low pKa thiols of Prxs characterized so far, conferring high nucleophilicity to its sulfur atom. Nevertheless, the second-order rate constant determined for the protonated thiol form of Cys-47 is also very high (in the range of 106 m−1 s−1) (Fig. 6C). Thus, stabilization of thiolate alone is not sufficient to confer reactivity. Several aspects of Prx reactivity remain elusive, such as the identification of residues responsible for the removal of the proton from the sulfur atom of the peroxidatic cysteine and/or protonation of the RO− leaving group (20, 51). Although it has been assumed that crystal structures of Prxs with a serine in place of the peroxidatic cysteine mimic the reduced form, the hydroxyl proton cannot be abstracted from the oxygen atom of a serine residue in the same way as in the peroxidatic cysteine. Therefore, residues surrounding Ser-47 are expected to accommodate it in a different position in comparison with the thiolate in the wild-type protein. Nevertheless, most of the available crystal structures of Prxs described so far in the reduced form have serine in place of the peroxidatic cysteine.
Cys-101 is the most buried cysteine residue of XfPrxQ, which is consistent with the observation that this is the cysteine residue that is more inefficiently titrated by 5,5′-dithiobis(2-nitrobenzoic acid) (supplemental Table SI). Furthermore, XfPrxQ C101S was the most unstable among the mutant proteins, easily precipitating in solution (data not shown), indicating that this residue plays some structural role. Interestingly, Cys-101 is conserved in orthologous proteins from E. coli, Salmonella typhimurium, Klebsiella pneumoniae, and X. campestris (Fig. 2) but apparently does not play a role in catalysis.
Because Ser-47 (peroxidatic Cys-47 in the wild-type protein) and Cys-83 of XfPrxQ are very distant from one another, substantial conformational changes (probably by the unfolding of α-helices where peroxidatic and resolving cysteines are located) are required to generate an intramolecular disulfide bond. Some insights on the mechanism for this conformational change might be obtained from the analysis of the recently described structures of Bcp from X. campestris (PDB codes 3GKM and 3GKK) (54) as well as structures from Aeropyrum pernix (PDB codes 2CX4 and 2CX3).
Like XfPrxQ, peroxidatic and resolving cysteines of Bcp from X. campestris are in distinct α-helices (Fig. 2). Only helix α4 that holds the resolving Cys-84 is unfolded in the disulfide-containing structure of the protein (PDB code 3GKK) (Fig. 9, A and B). In apparent contrast, when structures of reduced and oxidized forms of Bcp from A. pernix K1 are compared (PDB codes 2CX4 and 2CX3, respectively), only helix α2 (equivalent to helix α3 in X. fastidiosa and X. campestris) and not helix α3 (equivalent to helix α4 in X. fastidiosa and X. campestris) is unfolded, with an intramolecular disulfide bond in a loop (Fig. 9, C and D). Bcp from A. pernix K1 is a member of the PrxQα subfamily, and active cysteines are vicinal to each other (Cys-49 and Cys-54) and belong to the same helix α2.
FIGURE 9.

Conformational changes in Bcp from X. campestris and A. pernix K1. A, schematic representation of Bcp C48S C84S from X. campestris (PDB code 3GKM) (54). Helix α3 and α4 that suffer conformational changes are colored in cyan and orange, respectively. Peroxidatic and resolving cysteines are substituted by serines and are indicated as SerP and SerR, respectively. The root mean square deviation for the superposition of 139 corresponding Cα atoms between XfPrxQ C47S and reduced form of Bcp from X. campestris is 0.873 Å. B, schematic representation of Bcp from X. campestris in the intramolecular disulfide bond form (PDB code 3GKK) (54). C, schematic representation of Bcp from A. pernix K1 in the reduced form (PDB code 2CX4). Helix α2 that suffer conformational change is colored in magenta. Peroxidatic and resolving cysteines are indicated as CysP and CysR, respectively. The root mean square deviation for the superposition of 139 corresponding Cα atoms between XfPrxQ C47S and Bcp from A. pernix K1 is 1.785 Å. D, schematic representation of the oxidized form of Bcp from A. pernix K1 (PDB code 2CX3). Residues involved in intramolecular disulfide bond are represented as balls and sticks, and atoms are colored following CPK color scheme.
We elaborate a model in which crystal structures of PrxQβ proteins represent snapshots along the coordinate of the enzyme-catalyzed process (Fig. 10). This model also takes into account data presented here and in the literature. Because oxidation takes place in the peroxidatic cysteine (Cys-47 in XfPrxQ) of the reduced protein (Fig. 10A), structural changes in PrxQβ proteins probably begin with the oxidation to a sulfenic acid derivative by the hydroperoxide substrate. Because sulfenic acid is bulkier and more electronegative than the corresponding thiol, electrostatic interactions that stabilize thiolate in Cys-47 (Fig. 7B) might be lost, triggering the unfolding of helix α3 (Fig. 10B). This structural change due to the oxidation of peroxidatic cysteine was in agreement with the similar CD spectra of XfPrxQ wild-type and C83S proteins (Fig. 8, A and C) but not the XfPrxQ C47S enzyme (Fig. 8B).
FIGURE 10.

Structurally detailed model of conformational changes in the PrxQβ catalytic cycle. Proposed sequence of structure snapshots along the catalytic cycle of PrxQ subfamily β-proteins. Each panel represents a different species of the proposed model. A, reduced species based on the determined crystal structure of XfPrxQ C47S. B and C, these species in dashed boxes represent hypothetical conformational intermediates based on CD data presented here. D, oxidized species based on the determined crystal structure of Bcp from X. campestris (PDB code 3GKK) (54). Peroxidatic and resolving cysteines are indicated as CysP and CysR, respectively. Residues are represented as sticks, and atoms are colored following the CPK color scheme.
It is possible that a redox-dependent change in the CD spectra of XfPrxQ C83S protein has occurred due to the formation of an intermolecular disulfide bond because this mutant protein suffered oligomerization even under native conditions (see supplemental Fig. S8). In any case, intermolecular disulfide bond formation would only be possible if the sulfenic acid derivative suffered a local unfolding of helix α3, where the peroxidatic cysteine is located. There are other reports on the redox-dependent unwinding of α-helices; however, the mechanisms are quite distinct. For instance, in the disulfide form of plant GPx, α-helix where resolving cysteine is located undergoes complete unwinding (64).
Local unfolding of helix α3 might lead to the approximation of the sulfenic acid derivative to helix α4 where resolving cysteine is located (Fig. 10B) and, finally, the formation of the intramolecular disulfide bond (Fig. 10C). It is possible that the formation of the disulfide bond may provoke partial unfolding of helix α4 (Fig. 10C), followed by the full refolding of helix α3 and restructuration of the active site (Fig. 10D). Interestingly, this characteristic is unique to PrxQβ because the active site is fully unfolded in all other Prxs in the disulfide configuration. The model depicted in Fig. 10 is consistent with all available crystallographic structures of class 1 Prx enzymes and with our CD spectra (Fig. 9), mass spectra (Fig. 4 and supplemental Table SII), and other biochemical assays.
In conclusion, we show here that class 1 Prx enzymes present a distinct catalytic mechanism among cysteine-based peroxidases, although they are also very reactive toward hydroperoxides. Because these proteins are not present in mammalian hosts (Table 1), they may represent a promising target for the development of drugs against bacterial pathogens.
Supplementary Material
Acknowledgments
We thank Dr. Paolo Di Mascio for providing the facilities for mass spectrometry and Fernanda M. Prado for assistance with mass spectrometry data collection. We thank Dr. Shaker Chuck Farah for providing the facilities for CD and Diorge Paulo de Souza for assistance with CD data collection. We also thank Dr. Gisele Monteiro and Dr. Daniel C. Pimenta for helpful discussions and experimental support.
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo, Conselho Nacional de Desenvolvimento Científico e Tecnológico (as part of Projeto Milênio Redoxoma), and the Brazilian Synchrotron Light Laboratory under Proposal D03B-MX1 7568 (performed on beamline W01B-MX2).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8, Tables SI–SIII, and additional references.
- GPx
- glutathione peroxidase
- Bcp
- bacterioferritin comigratory protein
- CHP
- cumene hydroperoxide
- DTT
- 1,4-dithiothreitol
- ESI-MS
- electrospray ionization mass spectrometry
- HPLC
- high performance liquid chromatography
- HRP
- horseradish peroxidase
- NEM
- N-ethylmaleimide
- Prx
- peroxiredoxin
- TBHP
- t-butyl hydroperoxide
- TCEP
- tris(2-carboxyethyl)phosphine
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- PDB
- Protein Data Bank.
REFERENCES
- 1.Simpson A. J., Reinach F. C., Arruda P., Abreu F. A., Acencio M., Alvarenga R., Alves L. M., Araya J. E., Baia G. S., Baptista C. S., Barros M. H., Bonaccorsi E. D., Bordin S., Bové J. M., Briones M. R., Bueno M. R., Camargo A. A., Camargo L. E., Carraro D. M., Carrer H., Colauto N. B., Colombo C., Costa F. F., Costa M. C., Costa-Neto C. M., Coutinho L. L., Cristofani M., Dias-Neto E., Docena C., El-Dorry H., Facincani A. P., Ferreira A. J., Ferreira V. C., Ferro J. A., Fraga J. S., França S. C., Franco M. C., Frohme M., Furlan L. R., Garnier M., Goldman G. H., Goldman M. H., Gomes S. L., Gruber A., Ho P. L., Hoheisel J. D., Junqueira M. L., Kemper E. L., Kitajima J. P., Krieger J. E., Kuramae E. E., Laigret F., Lambais M. R., Leite L. C., Lemos E. G., Lemos M. V., Lopes S. A., Lopes C. R., Machado J. A., Machado M. A., Madeira A. M., Madeira H. M., Marino C. L., Marques M. V., Martins E. A., Martins E. M., Matsukuma A. Y., Menck C. F., Miracca E. C., Miyaki C. Y., Monteriro-Vitorello C. B., Moon D. H., Nagai M. A., Nascimento A. L., Netto L. E., Nhani A., Jr., Nobrega F. G., Nunes L. R., Oliveira M. A., de Oliveira M. C., de Oliveira R. C., Palmieri D. A., Paris A., Peixoto B. R., Pereira G. A., Pereira H. A., Jr., Pesquero J. B., Quaggio R. B., Roberto P. G., Rodrigues V., de M, Rosa A. J., de Rosa V. E., Jr., de Sá R. G., Santelli R. V., Sawasaki H. E., da Silva A. C., da Silva A. M., da Silva F. R., da Silva W. A., Jr., da Silveira J. F., Silvestri M. L., Siqueira W. J., de Souza A. A., de Souza A. P., Terenzi M. F., Truffi D., Tsai S. M., Tsuhako M. H., Vallada H., Van Sluys M. A., Verjovski-Almeida S., Vettore A. L., Zago M. A., Zatz M., Meidanis J., Setubal J. C. (2000) Nature 406, 151–159 [DOI] [PubMed] [Google Scholar]
- 2.Amaro A. A., Maia M. L., Gonzales M. A. (1998) in Citrus Variegated Chlorosis (Donadio L. C., Moreira C. S. eds) pp. 123–139, Estação Experimental de Citricultura, Bebedouro, SP, Brazil [Google Scholar]
- 3.Hendson M., Purcell A. H., Chen D., Smart C., Guilhabert M., Kirkpatrick B. (2001) Appl. Environ. Microbiol. 67, 895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wrzaczek M., Brosché M., Kollist H., Kangasjärvi J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5412–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenhaken R., Levine A., Brisson L. F., Dixon R. A., Lamb C. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 4158–4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koszelak-Rosenblum M., Krol A. C., Simmons D. M., Goulah C. C., Wroblewski L., Malkowski M. G. (2008) J. Biol. Chem. 283, 24962–24971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smolka M. B., Martins-de-Souza D., Martins D., Winck F. V., Santoro C. E., Castellari R. R., Ferrari F., Brum I. J., Galembeck E., Della Coletta Filho H., Machado M. A., Marangoni S., Novello J. C. (2003) Proteomics 3, 224–237 [DOI] [PubMed] [Google Scholar]
- 8.Link A. J., Robison K., Church G. M. (1997) Electrophoresis 18, 1259–1313 [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi R., Matsuo K., Yamazaki A., Takahashi M., Fukasawa Y., Wada M., Abe C. (1992) Infect. Immun. 60, 1210–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torian B. E., Flores B. M., Stroeher V. L., Hagen F. S., Stamm W. E. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 6358–6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Toole P. W., Logan S. M., Kostrzynska M., Wadström T., Trust T. J. (1991) J. Bacteriol. 173, 505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Netto L. E., de Oliveira M. A., Monteiro G., Demasi A. P., Cussiol J. R., Discola K. F., Demasi M., Silva G. M., Alves S. V., Faria V. G., Horta B. B. (2007) Comp. Biochem. Physiol. C Toxicol. Pharmacol. 146, 180–193 [DOI] [PubMed] [Google Scholar]
- 13.Rhee S. G., Chae H. Z., Kim K. (2005) Free Radic. Biol. Med. 38, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 14.Poole L. B. (2007) Subcell. Biochem. 44, 61–81 [DOI] [PubMed] [Google Scholar]
- 15.Hofmann B., Hecht H. J., Flohé L. (2002) Biol. Chem. 383, 347–364 [DOI] [PubMed] [Google Scholar]
- 16.Trivelli X., Krimm I., Ebel C., Verdoucq L., Prouzet-Mauléon V., Chartier Y., Tsan P., Lauquin G., Meyer Y., Lancelin J. M. (2003) Biochemistry 42, 14139–14149 [DOI] [PubMed] [Google Scholar]
- 17.Copley S. D., Novak W. R., Babbitt P. C. (2004) Biochemistry 43, 13981–13995 [DOI] [PubMed] [Google Scholar]
- 18.Rouhier N., Jacquot J. P. (2005) Free Radic. Biol. Med. 38, 1413–1421 [DOI] [PubMed] [Google Scholar]
- 19.Herbette S., Roeckel-Drevet P., Drevet J. R. (2007) FEBS J. 274, 2163–2180 [DOI] [PubMed] [Google Scholar]
- 20.Wood Z. A., Schröder E., Robin Harris J., Poole L. B. (2003) Trends Biochem. Sci. 28, 32–40 [DOI] [PubMed] [Google Scholar]
- 21.Parsonage D., Youngblood D. S., Sarma G. N., Wood Z. A., Karplus P. A., Poole L. B. (2005) Biochemistry 44, 10583–10592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogusucu R., Rettori D., Munhoz D. C., Netto L. E., Augusto O. (2007) Free Radic. Biol. Med. 42, 326–334 [DOI] [PubMed] [Google Scholar]
- 23.Trujillo M., Ferrer-Sueta G., Thomson L., Flohé L., Radi R. (2007) Subcell. Biochem. 44, 83–113 [DOI] [PubMed] [Google Scholar]
- 24.Jeong W., Cha M. K., Kim I. H. (2000) J. Biol. Chem. 275, 2924–2930 [DOI] [PubMed] [Google Scholar]
- 25.Wakita M., Masuda S., Motohashi K., Hisabori T., Ohta H., Takamiya K. (2007) J. Biol. Chem. 282, 27792–27801 [DOI] [PubMed] [Google Scholar]
- 26.Verdoucq L., Vignols F., Jacquot J. P., Chartier Y., Meyer Y. (1999) J. Biol. Chem. 274, 19714–19722 [DOI] [PubMed] [Google Scholar]
- 27.Kong W., Shiota S., Shi Y., Nakayama H., Nakayama K. (2000) Biochem. J. 351, 107–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rouhier N., Gelhaye E., Gualberto J. M., Jordy M. N., De Fay E., Hirasawa M., Duplessis S., Lemaire S. D., Frey P., Martin F., Manieri W., Knaff D. B., Jacquot J. P. (2004) Plant Physiol. 134, 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park S. G., Cha M. K., Jeong W., Kim I. H. (2000) J. Biol. Chem. 275, 5723–5732 [DOI] [PubMed] [Google Scholar]
- 30.Horling F., Lamkemeyer P., König J., Finkemeier I., Kandlbinder A., Baier M., Dietz K. J. (2003) Plant Physiol. 131, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammad Y., Maréchal J., Cournoyer B., Normand P., Domenach A. M. (2001) Can. J. Microbiol. 47, 541–547 [DOI] [PubMed] [Google Scholar]
- 32.Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R. D., Bairoch A. (2003) Nucleic Acids Res. 31, 3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flohé L., Steinert P., Hecht H. J., Hofmann B. (2002) Methods Enzymol. 347, 244–258 [DOI] [PubMed] [Google Scholar]
- 34.Jiang Z. Y., Hunt J. V., Wolff S. P. (1992) Anal. Biochem. 202, 384–389 [DOI] [PubMed] [Google Scholar]
- 35.Monteiro G., Horta B. B., Pimenta D. C., Augusto O., Netto L. E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 4886–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chae H. Z., Chung S. J., Rhee S. G. (1994) J. Biol. Chem. 269, 27670–27678 [PubMed] [Google Scholar]
- 37.Segel I. H. (1993) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems, pp. 606–625, Wiley Interscience, New York [Google Scholar]
- 38.Leslie A. G. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography No. 26 [Google Scholar]
- 39.Kabsch W. (1988) J. Appl. Crystallogr. 21, 916–924 [Google Scholar]
- 40.Blessing R. H. (1995) Acta Crystallogr. A 51, 33–38 [DOI] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project No. 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 42.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alphey M. S., Bond C. S., Tetaud E., Fairlamb A. H., Hunter W. N. (2000) J. Mol. Biol. 300, 903–916 [DOI] [PubMed] [Google Scholar]
- 44.Pannu N. S., Murshudov G. N., Dodson E. J., Read R. J. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 45.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 46.Poole L. B. (2005) Arch. Biochem. Biophys. 433, 240–254 [DOI] [PubMed] [Google Scholar]
- 47.Rouhier N., Gelhaye E., Jacquot J. P. (2002) J. Biol. Chem. 277, 13609–13614 [DOI] [PubMed] [Google Scholar]
- 48.Bréhélin C., Meyer E. H., de Souris J. P., Bonnard G., Meyer Y. (2003) Plant Physiol. 132, 2045–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang K. S., Kang S. W., Woo H. A., Hwang S. C., Chae H. Z., Kim K., Rhee S. G. (2002) J. Biol. Chem. 277, 38029–38036 [DOI] [PubMed] [Google Scholar]
- 50.Poole L. B. (2008) Curr. Protoc. Toxicol. 38, 17.1.1–17.2.27 [Google Scholar]
- 51.Trujillo M., Clippe A., Manta B., Ferrer-Sueta G., Smeets A., Declercq J. P., Knoops B., Radi R. (2007) Arch. Biochem. Biophys. 467, 95–106 [DOI] [PubMed] [Google Scholar]
- 52.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 53.Krissinel E., Henrick K. (2007) J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 54.Liao S. J., Yang C. Y., Chin K. H., Wang A. H., Chou S. H. (2009) J. Mol. Biol. 390, 951–966 [DOI] [PubMed] [Google Scholar]
- 55.D'Ambrosio K., Limauro D., Pedone E., Galdi I., Pedone C., Bartolucci S., De Simone G. (2009) Proteins 76, 995–1006 [DOI] [PubMed] [Google Scholar]
- 56.König J., Lotte K., Plessow R., Brockhinke A., Baier M., Dietz K. J. (2003) J. Biol. Chem. 278, 24409–24420 [DOI] [PubMed] [Google Scholar]
- 57.Sun C., Berardi M. J., Bushweller J. H. (1998) J. Mol. Biol. 280, 687–701 [DOI] [PubMed] [Google Scholar]
- 58.Kortemme T., Creighton T. E. (1995) J. Mol. Biol. 253, 799–812 [DOI] [PubMed] [Google Scholar]
- 59.Morrow J. A., Segall M. L., Lund-Katz S., Phillips M. C., Knapp M., Rupp B., Weisgraber K. H. (2000) Biochemistry 39, 11657–11666 [DOI] [PubMed] [Google Scholar]
- 60.Lamkemeyer P., Laxa M., Collin V., Li W., Finkemeier I., Schöttler M. A., Holtkamp V., Tognetti V. B., Issakidis-Bourguet E., Kandlbinder A., Weis E., Miginiac-Maslow M., Dietz K. J. (2006) Plant J. 45, 968–981 [DOI] [PubMed] [Google Scholar]
- 61.Stork T., Laxa M., Dietz M. S., Dietz K. J. (2009) Arch. Microbiol. 191, 141–151 [DOI] [PubMed] [Google Scholar]
- 62.Bryk R., Griffin P., Nathan C. (2000) Nature 407, 211–215 [DOI] [PubMed] [Google Scholar]
- 63.Trujillo M., Budde H., Piñeyro M. D., Stehr M., Robello C., Flohé L., Radi R. (2004) J. Biol. Chem. 279, 34175–34182 [DOI] [PubMed] [Google Scholar]
- 64.Koh C. S., Didierjean C., Navrot N., Panjikar S., Mulliert G., Rouhier N., Jacquot J. P., Aubry A., Shawkataly O., Corbier C. (2007) J. Mol. Biol. 370, 512–529 [DOI] [PubMed] [Google Scholar]
- 65.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.