

Abstract
Endometrial carcinoma is the most common gynecological malignancy in the United States. Although most women present with early disease confined to the uterus, the majority of persistent or recurrent tumors are refractory to current chemotherapies. We have identified a total of 11 different FGFR2 mutations in 3/10 (30%) of endometrial cell lines and 19/187 (10%) of primary uterine tumors. Mutations were seen primarily in tumors of the endometrioid histologic subtype (18/115 cases investigated, 16%). The majority of the somatic mutations identified were identical to germline activating mutations in FGFR2 and FGFR3 that cause Apert Syndrome, Beare–Stevenson Syndrome, hypochondroplasia, achondroplasia and SADDAN syndrome. The two most common somatic mutations identified were S252W (in eight tumors) and N550K (in five samples). Four novel mutations were identified, three of which are also likely to result in receptor gain-of-function. Extensive functional analyses have already been performed on many of these mutations, demonstrating they result in receptor activation through a variety of mechanisms. The discovery of activating FGFR2 mutations in endometrial carcinoma raises the possibility of employing anti-FGFR molecularly targeted therapies in patients with advanced or recurrent endometrial carcinoma.
Keywords: endometrial cancer, mutation, FGFR2, apert syndrome
Endometrial cancer is the most commonly diagnosed malignancy of the female reproductive tract in the United States. It was predicted that in 2006, approximately 41 200 new cases of cancer of the uterine corpus would be diagnosed and 7350 women would die of this disease (Jemal et al., 2006).
The FGF family comprises 18 ligands (FGF1–FGF10 and FGF16–FGF23), which signal through four transmembrane receptor tyrosine kinases (FGFR1–FGFR4) and their alternatively spliced isoforms (Ornitz and Itoh, 2001). FGF signaling has been shown to play a crucial role in many physiological and pathological processes including embryogenesis, organogenesis, angiogenesis, wound healing and tumorigenesis. Alternative splicing in the third immunoglobulin domain (D3) of FGFR is the primary determinant of both the patterns of redundancy and specificity in FGF/FGFR binding and signaling. This splicing event is tissue specific and gives rise to the IIIb and IIIc receptor isoforms for FGFR1–FGFR3, which possess distinct ligand specificities (Ibrahimi et al., 2004; Mohammadi et al., 2005; Zhang et al., 2006). For FGFR2, cells of an epithelial lineage only express the ‘IIIb’ isoform encoded by exon 8 (FGFR2b) while mesenchymally derived cells only express the ‘IIIc’ isoform utilizing exon 9 (FGFR2c). Specificity of signaling is also provided by tissue-specific expression of receptors, ligands and heparan sulfate proteoglycans.
Germline gain-of-function mutations in FGFR1, 2 and 3 have been reported in a variety of craniosynostosis syndromes and chondrodysplasia syndromes. The genotype/phenotype correlations in these disorders are complex, with over 14 distinct clinical syndromes associated with mutations in one of the three receptors and several clinical syndromes, for example, Pfeiffer and Crouzon Syndrome associated with mutations in different receptors (Passos-Bueno et al., 1999; Wilkie, 2005).
Several of the FGFRs have been implicated in cancer through chromosomal translocations, activating mutations and aberrant splicing. Recent analyses of mutations from kinome screens performed in several cancer types further implicate the FGF signaling pathway in tumorigenesis (Greenman et al., 2007). The Catalog of Somatic Mutations in Cancer (COSMIC) provides a repository of all somatic changes reported to date in this receptor family (http://www.sanger.ac.uk/genetics/CGP/cosmic/).
As part of the Cancer Genome Project, Cancer Cell Line Project (http://www.sanger.ac.uk/genetics/CGP/CellLines/), 3/10 uterine cancer cell lines were found to harbor FGFR2 variants. Exon 8 is three nucleotides longer than exon 9, making the FGFR2b isoform one codon longer than the FGFR2c isoform. All mutations are numbered in the text relative to the epithelially expressed FGFR2b isoform (NP_075259.2), however, for ease of comparison to the majority of previously published reports, the equivalent mutation numbered relative to the FGFR2c isoform (NP_000132.1) is also provided in Table 2. The N550K variant identified in two of the endometrial cell lines was likely to result in receptor activation as an identical mutation in FGFR3 occurs in patients with hypochondroplasia (Passos-Bueno et al., 1999; Figure 1). These preliminary data suggested that activation of FGFR2 might play a role in endometrial tumorigenesis. Next, we sought to determine the spectrum and frequency of activating FGFR2 mutations in primary uterine cancers. Direct sequencing of the exons in which germline activating mutations in FGFR2 and FGFR3 had previously been identified (exons 7, 8, 9, 10, 13 and 15) was performed for 187 primary uterine cancers, representing all grades and stages of tumors and the major histologic subtypes of endometrial carcinoma (Table 1). See Supplementary methods for details. For a subset of tumors (32 endometrioid endometrial cancers plus 17 carcinosarcomas) exons 5–18 encompassing the second and third immunoglobulin domains (hereafter referred to as D2 and D3), transmembrane domain and the entire kinase domain were sequenced to determine the relative occurrence of novel somatic mutations. In addition to the mutations found in exons 7, 10, 13 and 15, one additional mutation was identified in this more extensive mutational screen, a 2 bp deletion in exon 18. Mutations were identified in 19 cases (10%). Eighteen of 115 endometrioid endometrial cancers (16%) had mutations and a single serous carcinoma (1/45, 2%) harbored a mutation. No mutations were seen in carcinosarcomas or clear cell cancers. For all mutations, constitutional DNA was sequenced to confirm that the mutation arose somatically. Among the endometrioid cases, 11/49 tumors with defective DNA mismatch repair deficiency (22%) had FGFR2 mutations and 6/61 (10%) with normal mismatchhad mutations (P = 0.10). It should be noted that microsatellite instability (MSI) status was not determined for five tumors. We did not include the 2 bp deletion in an MSI-positive case, as it is unlikely to be activating and thus may represent a bystander mutation. Although there was a higher frequency of mutations in tumors demonstrating MSI, we would argue that these mutations in FGFR2 are pathogenic due to the fact that the same mutations are observed in both MSI positive and microsatellite stable (MSS) primary tumors and that the majority of the mutations are identical to those activating mutations identified in the germline, a coincidence one would not expect if they were bystander mutations associated with MSI. Of the 12 different mutations we identified, seven had previously been reported associated with craniosynostosis or skeletal dysplasia syndromes, one (A315T) occurred at a FGFR2c residue at which a similar missense mutation had been reported (A315S) and four mutations were novel (Figure 1).
Table 2.
Spectrum of FGFR2 mutations in primary endometrial cancers
| Case ID | FGFR2b nucleotide a | FGFR2b codon b | FGFR2c codon c | Histotype | Stage | Grade | MSI status |
|---|---|---|---|---|---|---|---|
| AN3 CAd | A929G | K310R | K310R | Endometrioid | Positive | ||
| AN3 CAd | T1650G | N550K | N549K | Endometrioid | Positive | ||
| MFE296 | T1650G | N550K | N549K | Endometrioid | Negative | ||
| ESS-1 | G943Ae | A315Tf | Stromal sarcoma | Negative | |||
| 1359 | C755G | S252W | S252W | Endometrioid | I | 2 | Positive |
| 1574 | C755G | S252W | S252W | Endometrioid | I | 2 | Positive |
| 1492d | C755G | S252W | S252W | Endometrioid | I | 1 | Negative |
| 1484 | C755G | S252W | S252W | Endometrioid | III | 3 | Negative |
| 1316 | C755G | S252W | S252W | Endometrioid | III | 1 | Negative |
| 1792 | C755G | S252W | S252W | Endometrioid | III | 1 | Positive |
| 1438 | C755G | S252W | S252W | Serous | IV | 3 | Negative |
| 1482 | C755G | S252W | S252W | Endometrioid | IV | 2 | Positive |
| 1267 | T1650A | N550K | N549K | Endometrioid | II | 2 | Positive |
| 1391 | T1650A | N550K | N549K | Endometrioid | III | 2 | Positive |
| 1528 | T1650G | N550K | N549K | Endometrioid | IV | 2 | Negative |
| 1655 | A1127G | Y376C | Y375C | Endometrioid | III | 2 | Positive |
| 1492d | A1127G | Y376C | Y375C | Endometrioid | I | 1 | Negative |
| 1684 | C1118G | S373C | S372C | Endometrioid | I | 1 | Positive |
| 1094 | T1147C | C383R | C382R | Endometrioid | I | 1 | Positive |
| 1361 | T1175G | M392R | M391R | Endometrioid | I | 1 | Positive |
| 1744 | A1642G | I548V | I547V | Endometrioid | III | 2 | Positive |
| 1717 | A1978G | K660E | K659E | Endometrioid | I | 2 | Negative |
| 1272 | Intron10 A>C+2 | Endometrioid | I | 1 | Negative | ||
| 1289 | 2290-91 del CT | Frameshift | Frameshift | Endometrioid | I | 3 | Positive |
Abbreviation: MSI, microsatellite instability.
Numbering relative to NM_022970.2.
Numbering relative to NP_075259.2.
Numbering relative to NP_000132.1.
Two mutations in one sample.
Numbering according to the NM_000141 as ESS-1 was derived from a stromal sarcoma expressing the FGFR2c isoform.
There is no alanine residue at the equivalent position in FGFR2b.
Figure 1.
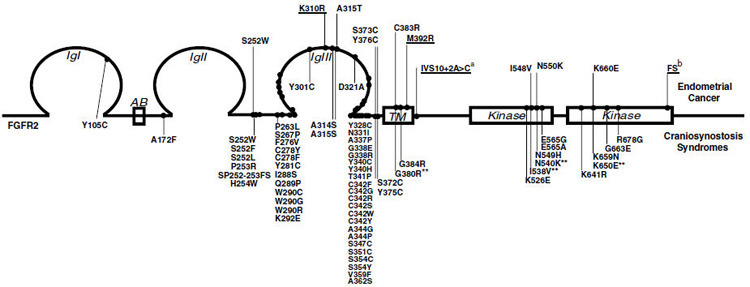
Schematic representation of FGFR2 mutations. FGFR2 mutations mapped to functional domains. Somatic mutations identified in primary endometrial cancers and cell lines are presented above the schematic representation of the protein and are numbered relative to FGFR2b (NP_075259.2). Germline mutations associated witha variety of craniosynostosis syndromes and numbered relative to FGFR2c (NP_000132.1) are presented below (http://www.hgmd.cf.ac.uk/ac/index.php). (Passos-Bueno et al., 1999; Wilkie, 2005) Four somatic FGFR2 endometrial mutations, while not previously reported in the germline, have an identical missense change reported in the paralogous position in FGFR3c in a skeletal chondrodysplasia (indicated with **) (http://www.hgmd.cf.ac.uk/ac/index.php). Novel mutations are underlined. aThe IVS10 + 2A > C mutation is hypothesized to result in a relative increase in the proportion of the + VT spliceform (see text for details). bFS refers to a 2 bp deletion 2290–91 (CT) resulting in a frameshift and premature truncation.
Table 1.
Uterine cancer patient demographics, disease characteristics and FGFR2 mutation status
|
Cohort n = 187 |
||
|---|---|---|
|
|
||
|
Entire uterine cancer cohort n (%) |
Cases with FGFR2 mutations n (%) |
|
| Age (years) | 66.5±11.1a | |
| Race | ||
| Caucasian | 150 (80) | 18 (12) |
| African-American | 33 (18) | 1 (3) |
| Other or not specified | 4 (2) | 0 (0) |
| Histology | ||
| Endometrioid | 115 (61) | 18 (16) |
| Serous or mixed serous/ endometrioid |
45 (24) | 1 (2) |
| Clear cell | 8 (4) | 0 (0) |
| Adenocarcinoma not otherwise specified |
1 (<1) | 0 (0) |
| Carcinosarcoma | 17 (9) | 0 (0) |
| Uterine stromal sarcoma |
1 (<1) | 0 (0) |
| Stage | ||
| I | 79 (42) | 9 (11) |
| II | 16 (9) | 1 (6) |
| III | 67 (36) | 6 (9) |
| IV | 25 (13) | 3 (12) |
| FIGO grade | ||
| 1 | 49 (26) | 7 (14) |
| 2 | 39 (21) | 9 (23) |
| 3b | 99 (53) | 3 (3) |
Mean±standard deviation.
All carcinomas withserous and clear cell features along with carcinosarcomas and sarcoma classified as grade 3.
The distribution of mutations according to tumor histotype, along with the grade and stage of tumors harboring FGFR2 mutations are summarized in Table 2. The S252W mutation was the most common mutation identified, seen in eight independent tumors. This mutation occurs in the linker region between D2 and D3, which provides key contacts with the FGF ligand. The S252W and the adjacent P253R mutations cause Apert syndrome, the most severe of the craniosynostosis syndromes characterized by craniosynostosis as well as severe syndactyly of the hands and feet (Wilkie et al., 2002). Structural, biological and extensive in vitro affinity studies have been performed with either the S252W FGFR2c and/or S252W FGFR2b mutant receptors showing that this mutation increases the binding affinity of the receptor for multiple FGFs from two- to eight-fold, in addition to violating the ligand-binding specificities attributed to the alternatively spliced isoforms (Yu et al., 2000; Ibrahimi et al., 2001, 2004).
The prevalence of the S252W mutation in this panel of tumors suggests positive selection for this mutant in endometrioid endometrial cancers. Although the expression of all FGF receptor and ligands has not been examined in normal cycling endometrium and endometrial cancers, there are several studies reporting the expression of FGFR2 and FGF2 predominantly in the basal part of luminal and glandular epithelium (Sangha et al., 1997; Moller et al., 2001). Several studies have also shown an increase in FGF2 expression in the glandular epithelia associated with complex hyperplasia and adenocarcinoma (Gold et al., 1994). These endometrial epithelial cells express only the FGFR2b isoform, which cannot bind FGF2. However, the acquisition of the S252W mutation in these cells would be anticipated to result in autocrine activation of the S252W FGFR2b receptor. The S252W mutation also enables the mutant receptor to bind FGF9, which is highly expressed in the endometrial stroma (Tsai et al., 2002). The prevalence of the S252W mutation suggests that the different FGFR2 isoforms play important roles in mediating directional epithelial-mesenchymal signaling in the endometrium. To the best of our knowledge, there have been no reports on endometrial cancers in patients with Apert syndrome, however our data might suggest an increased risk for endometrial cancer in female patients with Apert syndrome.
Four additional extracellular domain mutations were identified, K310R and A315T in the cell lines, and S373C and Y376C in primary tumors, the latter mutation seen in two independent tumors (Figure 1, Table 2). Functional studies performed on those extracellular mutations in FGFR2c (or the paralogous FGFR3) resulting in either the loss or gain of an additional cysteine residue have demonstrated that these missense changes result in constitutive receptor dimerization due to the formation of inter- rather than intramolecular disulphide bonds (Wilkie et al., 2002). In the germline, the extracellular juxtamembrane FGFR2c mutations S372C and Y375C have been reported in several individuals with Beare–Stevenson cutis gyrata syndrome, a craniosynostosis syndrome witha broad range of additional clinical features (Passos-Bueno et al., 1999). The paralogous mutations in FGFR3c (G370C and Y373C) are also associated with a severe chondrohyperplasia, thanatophoric dysplasia type I (Passos-Bueno et al., 1999). Similar to the A315S mutation, the A315T mutation is likely to confer upon FGFR2c the ability to bind FGF10 illegitimately (Ibrahimi et al., 2004).
We identified two mutations, C383R and M392R within the transmembrane domain. The C383R mutation we identified is similar to a nonconservative missense mutation at the paralogous position in FGFR3 (G380R) that accounts for over 95% of patients with achondroplasia (Passos-Bueno et al., 1999). The FGFR3 G380R mutation has been reported to alter ligand mediated receptor downregulation, thus augmenting FGF signaling (Monsonego-Ornan et al., 2000; Cho et al., 2004).
In addition to the extracellular and transmembrane domain mutations, four different mutations in the FGFR2 kinase domain were identified. While two of these, N550K and K660E, have not been identified in any craniosynostosis syndromes, the similar N549H mutation in FGFR2c has been associated with Crouzon Syndrome (Wilkie, 2005) and identical mutations at the paralogous positions have been seen in FGFR3 associated with hypochondroplasia (N540K) and thanatophoric dysplasia II (K650E) (Wilkie, 2005). Crystal structures of N549H and K650N mutant FGFR2c kinases show that these mutations activate the kinase by loosening a novel autoinhibitory molecular brake at the kinase hinge region (Mohammadi, unpublished results).
The pathological consequence of the novel IVS10 + 2A > C splicing mutation is unknown, however, it is tempting to speculate that this would result in increased receptor signaling. There is alternative splicing in the intracellular juxtamembrane region in FGFR1–3 leading to the inclusion or exclusion of two amino acids, valine and threonine (VT) downstream of exon 10. The FRS2 adaptor signaling protein that links FGFRs to the MAPK and PI3K pathways binds to a sequence in the juxtamembrane domain of FGFR1 that includes the alternatively splicing VT (Burgar et al., 2002). The IVS10 + 2A > C mutation results in a GCAAGT noncanonical splice donor site. We hypothesize based on its increased frequency in the genome (Burset et al., 2000; Chong et al., 2004) that this results in a more efficient splice donor site that increases the levels of the +VT transcript, which would in turn result in increased FRS2-mediated signaling.
Two endometrial samples were shown to carry two mutations, the AN3CA MSI positive cell line which carried N550K and K310R and the MSI negative tumor 1492 which carried S252W and Y376C. The discovery of two presumably dominantly activating mutations in the same tumor is unexpected. One possible explanation is that the MSI-positive cell line is not clonal with respect to the FGFR2 mutations or, alternatively, the mutations may occur on different alleles in the same cell population. It is interesting to note that in each case there is a mutation, which is known to result in constitutive ligand-independent receptor activation, along with either the ligand-dependent S252W or the uncharacterized K310R, suggesting that additional selective pressure may exist for increased FGFR2 activation in endometrial epithelia.
Among the endometrioid cases, there was no association between FGFR2 mutation and overall or disease-free survival (data not shown). This contrasts with the presence of FGFR3 mutations in bladder cancers, which occur more frequently in superficial carcinomas than invasive carcinomas and are associated with favorable outcome (van Rhijn et al., 2001). The identification of activating mutations in FGFR2 in endometrioid endometrial cancers is of direct clinical relevance. Three cytotoxic agents, doxorubicin, cisplatin and paclitaxel, appear to be useful in the treatment of advanced or recurrent endometrial carcinoma (Obel et al., 2006). There is a clear need for additional adjuvant therapies and biologics, alone or in combination with cytotoxics, and agents with activity against activated receptor tyrosine kinases represent attractive novel therapeutics for the treatment of this common malignancy. Indeed, there is at least one FGFR inhibitor (TKI258), which is in phase I clinical trials for the treatment of multiple myeloma. Future studies will determine whether inhibition of FGFR in a subset of endometrial patients may prove to be a viable molecularly targeted therapy.
Acknowledgements
We thank the TGen Sequencing Core for their excellent work. We also thank Feng Gao and the Biostatistics Core at Siteman Cancer Center, Barnes-Jewish Hospital Washington University for assistance with the survival analyses (CA091842). Supported in part by RO1 CA71754 (PJG and MAM), Wellcome Trust (MS, AF and HD), the Melanoma Research Foundation (PMP) and R01 CA109544 (JT).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Burgar HR, Burns HD, Elsden JL, Lalioti MD, Heath JK. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. J Biol Chem. 2002;277:4018–4023. doi: 10.1074/jbc.M107785200. [DOI] [PubMed] [Google Scholar]
- Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JY, Guo C, Torello M, Lunstrum GP, Iwata T, Deng C, et al. Defective lysosomal targeting of activated fibroblast growth factor receptor 3 in achondroplasia. Proc Natl Acad Sci USA. 2004;101:609–614. doi: 10.1073/pnas.2237184100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong A, Zhang G, Bajic VB. Information for the Coordinates of Exons (ICE): a human splice sites database. Genomics. 2004;84:762–766. doi: 10.1016/j.ygeno.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Gold LI, Saxena B, Mittal KR, Marmor M, Goswami S, Nactigal L, et al. Increased expression of transforming growth factor beta isoforms and basic fibroblast growth factor in complex hyperplasia and adenocarcinoma of the endometrium: evidence for paracrine and autocrine action. Cancer Res. 1994;54:2347–2358. [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh G, Hunter C, Bignell G, et al. Patterns of Somatic mutation in cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci USA. 2001;98:7182–7187. doi: 10.1073/pnas.121183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi OA, Zhang F, Eliseenkova AV, Itoh N, Linhardt RJ, Mohammadi M. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum Mol Genet. 2004;13:2313–2324. doi: 10.1093/hmg/ddh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Moller B, Rasmussen C, Lindblom B, Olovsson M. Expression of the angiogenic growth factors VEGF, FGF-2, EGF and their receptors in normal human endometrium during the menstrual cycle. Mol Hum Reprod. 2001;7:65–72. doi: 10.1093/molehr/7.1.65. [DOI] [PubMed] [Google Scholar]
- Monsonego-Ornan E, Adar R, Feferman T, Segev O, Yayon A. The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol Cell Biol. 2000;20:516–522. doi: 10.1128/mcb.20.2.516-522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obel JC, Friberg G, Fleming GF. Chemotherapy in endometrial cancer. Clin Adv Hematol Oncol. 2006;4:459–468. [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:Reviews3005.1–reviews3005.12. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Sangha RK, Li XF, Shams M, Ahmed A. Fibroblast growth factor receptor-1 is a critical component for endometrial remodeling: localization and expression of basic fibroblast growth factor and FGF-R1 in human endometrium during the menstrual cycle and decreased FGF-R1 expression in menorrhagia. Lab Invest. 1997;77:389–402. [PubMed] [Google Scholar]
- Tsai SJ, Wu MH, Chen HM, Chuang PC, Wing LY. Fibroblast growth factor-9 is an endometrial stromal growth factor. Endocrinology. 2002;143:2715–2721. doi: 10.1210/endo.143.7.8900. [DOI] [PubMed] [Google Scholar]
- van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61:1265–1268. [PubMed] [Google Scholar]
- Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Patey SJ, Kan SH, van den Ouweland AM, Hamel BC. FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am J Med Genet. 2002;112:266–278. doi: 10.1002/ajmg.10775. [DOI] [PubMed] [Google Scholar]
- Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci USA. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]