

Abstract
Benzodiazepines are widely used in clinics and for recreational purposes, but will lead to addiction in vulnerable individuals. Addictive drugs increase the levels of dopamine and also trigger long-lasting synaptic adaptations in the mesolimbic reward system that ultimately may induce the pathological behavior. The neural basis for the addictive nature of benzodiazepines however remains elusive. Here we show that benzodiazepines increase firing of dopamine neurons of the ventral tegmental area through the positive modulation of GABAA receptors in nearby interneurons. Such disinhibition, which relies on α1-containing GABAARs expressed in these cells, triggers drug-evoked synaptic plasticity in excitatory afferents onto dopamine neurons and underlies drug reinforcement. Taken together, our data provide evidence that benzodiazepines share defining pharmacological features of addictive drugs through cell type-specific expression of α1-containing GABAARs in the ventral tegmental area. The data also suggest that subunitselective benzodiazepines sparing α1 may be devoid of addiction liability.
Addictive drugs can be classified into three groups, according to the cellular mechanism through which they increase mesolimbic dopamine (DA)1. Opioids, cannabinoids and the club drug γ-hydroxy butyrate reduce release from inhibitory afferents onto DA neurons, through their respective G-protein coupled receptors on GABA neurons. These substances activate pre- and postsynaptic receptors, indirectly increasing the firing rate of DA neurons, a mechanism defined as disinhibition. Nicotine, as a member of the second group, directly depolarizes DA neurons by activating α4β2-containing acetylcholine receptors, while the third group targets DA transporters (e.g. cocaine and amphetamines). It remains unclear whether these mechanisms can account for the addiction liability of benzodiazepines (BDZs)2, which are positive modulators of GABAAR function.
In addition to increasing mesolimbic DA, another common feature to all addictive drugs studied so far is that they trigger adaptive synaptic plasticity in the ventral tregmental area (VTA)3. Hours after the initial exposure, excitatory afferents onto DA neurons of the VTA are strengthened, in part by the insertion of GluR2-lacking AMPARs4,5 6. To test whether a similar mechanism is elicited by BDZs, we examined whether a single injection of a BDZ would, in addition to an increase of AMPA/NMDA ratio7, also cause a change in slope of the current-voltage (iv)-curve of evoked excitatory postsynaptic currents (EPSCs). Such rectification reflects the presence of GluR2-lacking AMPARs, which are calcium permeable and blocked by polyamines at positive potentials.
BDZ-evoked plasticity in dopamine neurons
In slices obtained 24 h after the i.p. injection of midazolam (MDZ), diazepam (DZ), or flunitrazepam (FZ), the rectification index (RI = EPSC−65 mV/EPSC+35 mV) was significantly higher than in slices from saline-injected controls (Fig. 1a and Supplementary Fig. 2). Similar rectification was measured after an injection of morphine (Mor), a member of the class of drugs that cause disinhibition of DA neurons8. The BDZ antagonist flumazenil (Flu) blocked rectification when co-injected with MDZ but was without effect when co-injected with a control saline solution (Fig. 2 and Supplementary Fig. 2). The adaptive plasticity induced by systemic BDZs was also observed 24 h after local application of MDZ into the VTA by stereotactic injection (0.5 μl of a 8 mg/ml solution over 10 minutes; Fig. 1b). Thus, BDZ-dependent effects on VTA circuitry are sufficient to induce this cellular hallmark of addictive drugs.
Figure 1. BDZ-evoked synaptic plasticity is abolished in α1(H101R) mutant mice.

a, Top panel; normalized AMPAR-EPSCs obtained at -65, 0 and +35 mV in slices from WT mice i.p. injected with saline, MDZ (0.5 mg/kg) or Mor (15 mg/kg) 24 h prior to sacrifice. Middle panel; corresponding iv-curves. Bottom panel; bar graphs represent group data for the RI. F(2;21) = 9.08. b, AMPAR-EPSCs, iv-curves and RI (top, middle and bottom panel, respectively) observed when ACSF or MDZ were injected into the VTA in WT mice. t(11) = 5.43. c, Similar experiments performed with α1(H101R) mice. Note that Mor induces a rectification that is similar in WT and mutant mice. F(2;16) = 17.88. d, Similar experiments performed with α1(H101R) mice when MDZ was injected intra-VTA. n = 6-10.
Figure 2. Synaptic plasticity evoked by α1-subunit selective compounds.

a, Normalized AMPAR-EPSCs obtained at -65, 0 and +35 mV in slices from WT mice injected with ZOL (5 mg/kg i.p.), L-838 417 (10 mg/kg i.p.) and MDZ together with Flu (5 mg/kg), 24 h prior to sacrifice. b, Corresponding iv-curves. c, Bar graphs representing group data for the RI. F(2,19) = 28.97. n = 6-8.
BDZs bind to GABAARs at the interface between α and γ subunits9 in a subunit-dependent manner. GABA neurons in many parts of the brain express the α1 subunit isoform10, while midbrain DA neurons lack α1 but express α2, α3, and α4 subunit isoforms11. Thus, the addictive potential of BDZs might rely on the potentiation of α1-containing GABAARs, which would selectively inhibit GABA neurons and lead to disinhibition of DA neurons. To test this idea, we examined whether MDZ (i.e. a rapidly acting, non-selective BDZ with a very strong brain uptake12) has an effect in mice with a point mutation (H101R) in the α1 subunit that disrupts the BDZ binding site13. In α1(H101R) mice, MDZ no longer had an effect on the RI of AMPAR EPSCs in DA neurons (Fig. 1c). This was not due to a general loss of adaptive plasticity, as morphine still caused a strong rectification. Stereotactic injections of MDZ into the VTA also failed to elicit rectifying AMPAR-mediated EPSCs in α1(H101R) mice (Fig. 1d). Furthermore, MDZ increased the AMPA/NMDA ratio, while control injections of ACSF were without effect in either genotype (Supplementary Fig. 3).
We next used pharmacological tools to confirm the involvement of α1. Zolpidem (ZOL) is a non-classical BDZs selective for α1-containing GABAARs14 while the experimental compound L-838 417 does not modulate receptors that contain α115. We therefore tested whether ZOL and L-838 417 could evoke synaptic plasticity in DA neurons. We found that a single injection of ZOL led to rectifying AMPAR-mediated EPSCs, while L-838 417 did not affect the iv-curve (Fig. 2). Taken together with the results in α1(H101R) mice described above, we conclude that BDZ-evoked synaptic plasticity depends on α1-containing GABAARs within the VTA.
Cell type-specific expression of α1
To identify α1-expressing cells in the VTA, we next carried out immunohistochemical staining for tyrosine hydroxylase (TH) and the α1 subunit isoform in GAD-67 GFP mice (Fig. 3a). These experiments confirmed that α1 was expressed mainly in GFP-positive neurons, but not in TH-positive DA neurons. Quantifications revealed that 81% of the GABA neurons contained the α1 subunit isoform, while this was the case only in 7% of the DA neurons (Inset Fig. 3a). We also observed α1-staining that could neither be associated to TH-positive nor GAD67-GFP-expressing cells. This may reflect the pool of the so-called tertiary cells that are neither DA- nor GABA-neurons16,17 or be due to detectability limits in fine processes.
Figure 3. α1 is selectively expressed in GABA neurons of the VTA.
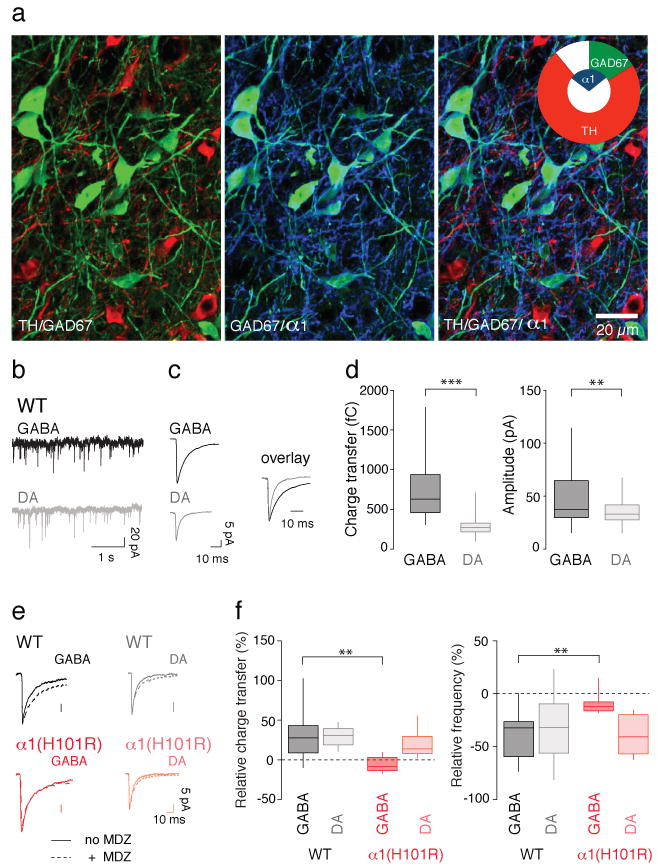
a, Immunohistochemical staining for tyrosine hydroxylase (TH, red) and ༟1 (blue) in VTA slices of GAD67-GFP (green) knock-in mice. Concentric pie charts represent the fraction of α1-positive cells (inner segment), and quantification of the two cell types (outer segment, n = 4 mice). Overlap between inner and outer segments represents colocalization. b, Example trace of mIPSCs recordings in GABA and DA neurons obtained in slices from WT mice. c, Representative averaged mIPSC trace from a GABA and a DA neuron. The overlay shows the difference in kinetics when the two currents are normalized to the average mIPSC peak amplitude. d, Box-plots represent group data for charge transfer and amplitude of mIPSCs obtained from GABA and DA neurons in slices from WT mice. t(75) = 7.55 and t(75) = 3.16, respectively. (n = 25-48). e, Representative average traces of mIPSCs before (solid line) and after (dotted line) application of MDZ (100 nM) in slices from WT and α1(H101R) mice. f, Corresponding box-plots representing group data for relative increase in charge transfer and frequency after MDZ bath-application. t(14) = 3.06 and t(14) = 3.23. n = 6-10.
To assess the functional consequences of this cell type-specific isoform expression for inhibitory transmission, we characterized miniature inhibitory postsynaptic currents (mIPSCs) in the presence of the glutamate receptor blocker kynurenic acid to isolate GABAAR-mediated currents (Fig. 3b, c). On average mIPSCs in GABA neurons were slower and bigger than those in DA neurons, leading to a significantly larger charge transfer in the former (Fig. 3d). This difference was of similar magnitude in WT and α1(H101R) mice (Supplementary Fig. 4), in line with previous reports13 that baseline transmission in mutant mice is normal. Moreover, the frequency of mIPSCs as well as the multiplicity factor (See methods for detailed description, supplementary Fig. 4c) were similar in GABA and DA neurons in both genotypes. Although this approach has its limitations18, it suggests that the numbers of inhibitory synapses are in the same range in the two cell types. To further confirm that synapses on GABA neurons express α1-containing GABAARs, we tested for effects of MDZ on charge transfer and frequency of mIPSCs in WT and α1(H101R) mice. In DA neurons, MDZ significantly increased the charge transfer and decreased the mIPSC frequency in both genotypes. In GABA neurons, MDZ increased the charge transfer and decreased mIPSC frequency in slices from WT mice, but was without effect on mIPSCs recorded in slices from α1(H101R) mice (Fig. 3e, f and supplementary Fig. 5a). As expected, MDZ had no effect on mIPSC amplitude in either cell type or genotype19 (Supplementary Fig. 5b). The observation that the mIPSC frequency is reduced by BDZs except in GABA neurons of α1(H101) mice is surprising at first, but could reflect presynaptic GABAARs. In fact such receptors have been described in the VTA, which, upon activation reduce the release probability20.
Since DA neurons express a set of many subunits11 the identification of the molecular composition of the GABAAR is difficult. Most DA neurons actually express the α3 subunit isoform (96%, supplementary Fig. 6). Importantly the majority of GABA neurons do not express the α3 subunit isoform (70%) even though significant heterogeneity was observed. In heterologous expressed systems, currents of α1-containing receptors are smaller than those of α3-containing ones21. This however does not apply to DA neurons in the VTA since in α3 KO mice currents are reduced only by 50%22. Our results establish that in α1(H101R) mice endogenous GABAA-mediated synaptic transmission is normal, while the positive modulation of MDZ was abolished in GABA neurons, because the α1 subunit isoform is selectively expressed in these cells.
Cellular determinants of disinhibition
In WT mice, mIPSCs in both GABA and DA neurons were enhanced by BDZs. However, when BDZs are administered whilst transmission is intact, the extent of current amplification in DA neurons depends on the frequency of synaptic events, which originate in the interneurons upstream. We therefore monitored the effect of MDZ on spike-driven, spontaneous IPSCs (sIPSC) in DA neurons (Fig. 4). Although, the charge transfer of sIPSCs on average increased after MDZ (in line with the mIPSC data), there was a strong reduction of the frequency of spike-driven events in DA neurons (Supplementary Fig. 7). As a result, when we integrated the charge transfer of sIPSCs over time before and after application of MDZ (relative total current), we found a significant decrease (Fig. 4b). Because interneurons are efficiently inhibited by MDZ, fewer spikes are generated, strongly decreasing the number of sIPSC, an effect that predominates over the MDZ amplification of the individual event. In α1(H101R) mice, in contrast, we observed an increased total current in DA neurons because the GABA neurons were insensitive to MDZ. In summary, in the WT mice, MDZ led to a net decrease of the total inhibitory current in DA neurons, which could be sufficient to cause their disinhibition (see supplementary Fig. 1 for schematics).
Figure 4. The total current generated by sIPSC in DA neurons is decreased by MDZ.
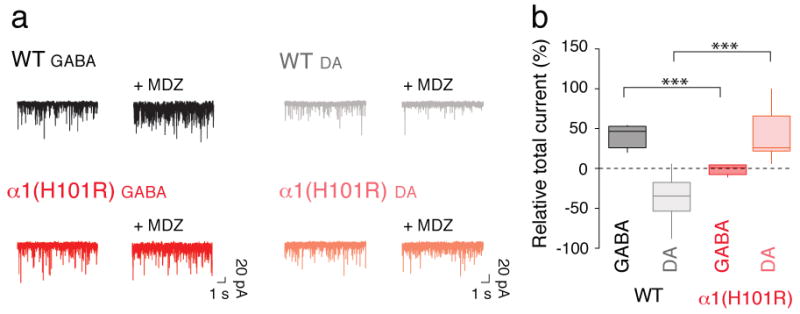
a, Example trace of sIPSCs recordings in GABA and DA neurons obtained before and after application of MDZ in slices from WT and α1(H101R) mice. sIPSCs were abolished with picrotoxin (PTX, 100 μM, not shown). b, Group data for the relative increase in the overall charge transfer (1 min) after MDZ bath-application. Note that in WT mice the total current in DA neurons decreases with MDZ application while in α1(H101R) mice there is an increase. GABA/WT vs GABA/α1(H101R) t(9) = 6.39, DA/WT vs DA/α1(H101R) t(15) = 5.50. n = 6-7.
We therefore tested the effect of MDZ on the firing rate of DA neurons in the VTA by performing extracellular single unit recordings in vivo. When the drug was injected into the tail vein of WT mice, we recorded a significant increase of the firing rate that was reversed by Flu (Fig. 5a, e, g). In stark contrast, no such disinhibition could be observed in α1(H101R) mice (Fig. 5b, e, g). In line with a disinhibition scenario, the data in the DA neurons were mirrored by the observations in GABA neurons. MDZ caused an inhibition of the spontaneous firing rates, at times leading to complete spike suppression (Fig. 5c, f, g). In α1(H101R) mice MDZ did not significantly affect firing in GABA neurons (Fig. 5d, f, g). The specificity of these findings are further demonstrated by the observation that, in mice where a different α subunit isoform had been mutated (α3(H126R) mice)23, MDZ caused an increase in the firing rate of DA neurons comparable to WT mice (Supplementary Fig. 8). The magnitude of increase in the firing rate was inversely related to the basal firing rate, which further suggests a disinhibition scenario (Fig. 5e). Moreover, in α1(H101R) mice disinhibition of DA neurons was observed with Mor, an effect that was also inversely correlated to the basal firing rate (Fig. 5h). Although anesthesia may modify the overall distribution of firing rates and therefore the magnitude of the disinhibition, the mean basal firing rates observed here were comparable to values recorded in freely moving animals24,25.
Figure 5. Opposing effects of MDZ on in vivo firing rates of DA and GABA neurons.
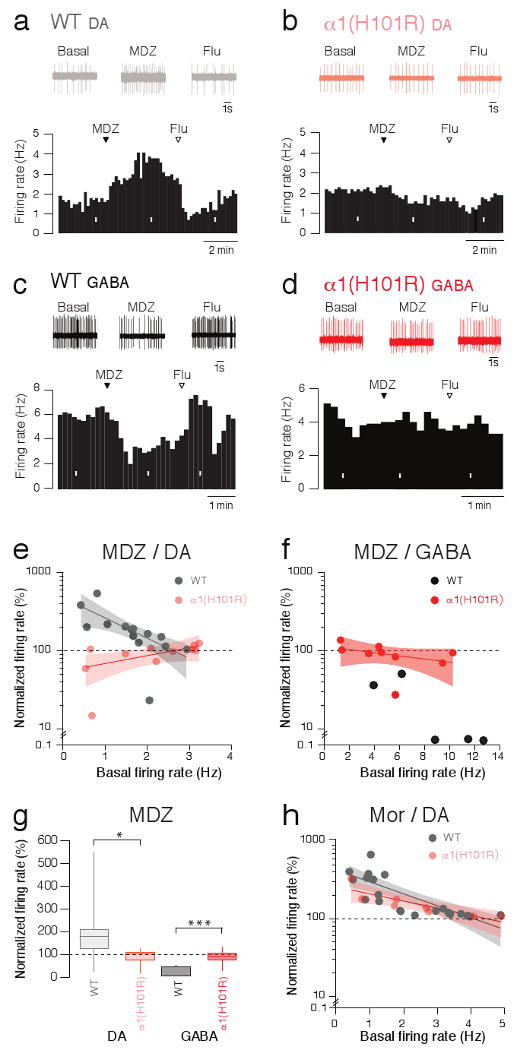
a, Representative extracellular single unit recording of a DA neuron during the i.v. injection of MDZ (0.5 mg/kg) in WT mice. Corresponding firing frequency plot (lower panel; Flu 1 mg/kg). b, Same experiment in α1(H101R) mice. c, Same experiment as in a) while monitoring a GABA neuron. d, Response of a GABA neuron to MDZ in an α1(H101R) mouse. White bars indicate time windows of traces shown above. e, Normalized firing rate of DA neurons in response to MDZ as a function of the basal activity in WT and α1(H101R) mice. WT/ α1(H101R) : F(2;23) = 10.63. f, Corresponding plot with the results obtained in GABA neurons. Notice that 3 out of 5 neurons were completely silenced, which precluded fitting. g, Box-plots representing group data for relative change in firing rate. WT DA/ α1(H101R) DA: t(23) = 2.70, WT GABA/ α1(H101R) GABA t(12) = 4.60. h, Normalized firing rate in response to i.v. injection of Mor (5 mg/kg) as a function of the basal activity in WT and α1(H101R) mice. Solid lines: regression curves; shaded area: 95% confidence intervals. n = 5-15.
Self-administration of midazolam
The results above demonstrate that α1-containing GABAAR mediate the increase of mesolimbic DA in response to BDZs. Furthermore DA antagonists can reduce self-administration of and preference to these drugs26,27. We therefore tested the impact of the α1 subunit isoform on oral self-administration of MDZ, by offering the mice a free choice of two drinking solutions (Fig. 6a). During the first three days the two bottles contained water. Sucrose was then added in both bottles to mask any bitter taste. This led to an increase of the overall consumption, but no particular preference. Finally, MDZ was added to one of the two bottles. During the test period with MDZ, the total consumption did not change in either genotype (Fig 6b). A preference for the MDZ solution developed rapidly in WT mice, but not α1(H101R) mice (Fig. 6c, d). WT mice drank between 0.8-1.1 mg/kg/24h of MDZ, which corresponds to a pharmacological dose. Two control experiments were carried out using a similar protocol. First, we offered α1(H101R) mice a choice between water and a sucrose solution. Both WT and mutant mice developed a strong preference for sucrose, indicating that α1(H101R) mice are not generally deficient in reward reinforcement (Supplementary Fig. 9). We also tested whether α3(H126R) mice, where MDZ caused a normal disinhibition of DA neurons (Supplementary Fig. 8) would develop a preference for MDZ, which was indeed the case (Supplementary Fig. 10). Although BDZs, particularly MDZ, may enhance taste perception28, this is unlikely to influence the interpretation of these data, as several studies have shown that BDZ-mediated taste enhancement is independent of α1-containing GABAAR29,30.
Figure 6. Oral self-administration of MDZ.

a, Protocol for behavioral experiment. b, Total consumption successively with water, sucrose, and MDZ (0.005 mg/ml) + sucrose (4 %) in WT mice (black) and α1(H101R) mice (red). Note that WT and α1(H101R) mice drink similar amounts of liquids. c, Relative MDZ consumption in WT and α1(H101R) mice. d, Corresponding box plots for relative average consumption of MDZ at days indicated. n = 12-18 mice in 4-6 cages. F(3;16) = 5.39
Discussion
Based on our data, we propose that BDZs increase DA levels through disinhibition, similar to opioids, cannabinoids, and GHB. This disinhibition is dependent on the BDZ binding site on α1-containing GABAARs in the VTA. The net effect of BDZs on the VTA circuit is dominated by the role of α1-containing GABAARs, which is supported by the following three observations. First, GABAARs mediated quantal transmission is stronger in GABA neurons than in DA neurons, as evidenced by the larger charge transfer of mIPSCs (Fig. 3d). Second, GABA neurons have a higher input resistance than DA neurons17, allowing the same charge transfer to more effectively change the membrane potential of GABA neurons than DA neurons. Finally, the BDZ-dependent enhancement of each IPSC on DA neurons causes little inhibition of DA neuron activity because GABA neurons fall silent and no longer generate those IPSCs. Our model could also apply to earlier work probing the effect of the GABAAR agonist muscimol31. When administered directly into the VTA, muscimol causes an increase of DA levels in the nucleus accumbens32. This effect only occurs at low doses, which led to the conclusion that the effect is mediated indirectly on non-DA neurons33,34. This inverse dose-dependence may be due to the fact that muscimol, unlike BDZs, is not a positive modulator but an agonist. In line with this interpretation, muscimol at high concentrations in fact inhibits DA neurons35.
The implication of α1 in the addictive effect of BDZs is surprising since the clinically available compound ZOL is selective for this subunit and has been claimed to carry a low risk for addiction36. However this optimistic view contrasts with the observation that ZOL is readily self-administered37 and the clinical reality. Our data with the subunit isoform-selective compounds also show that ZOL triggers drug-evoked plasticity and suggest that α1-sparing compounds may be promising candidates in the search for BDZs devoid of addiction liability. Since α1-containing GABAA receptors outside the VTA mediate additional effects such as seizure control, sedation and anterograde amnesia38, α1-sparing compounds will certainly not be suitable for all indications. The dissociation between anxiolysis, mainly α2-mediated23, and addiction however seems possible in principle. This is particularly appealing since high anxiety levels suggest increased vulnerability for addiction39.
In conclusion, our work unravels the molecular basis of the defining pharmacological features that BDZs share with addictive drugs, which we believe will be key for designing new BDZs with lower addiction liability. However, we note that increased levels of mesolimbic dopamine are necessary for addiction, but not sufficient on their own. Recent studies suggest that early drug-evoked plasticity in the VTA may facilitate addiction by gating more enduring forms of adaptations in target regions of the mesolimbic system, which would represent the eventual locus underlying long-term addictive behaviors40,41. Coinciding factors of vulnerability, either in the initial events in the VTA or subsequent events in mesolimbic targets, may ultimately explain individual variations in susceptibility to addiction, both for BDZs and for other drugs42.
Methods Summary
Horizontal slices of the midbrain (250 μm) were prepared as previously described43 from C57BL/6 mice, Pitx3-GFP knock-in mice44, GAD67-GFP Δneo mice45 and α1(H101R) knock-in mice13 24 h after i.p. or intra-VTA (ML ±0.8, AP -2.4, DV -4.4 mm from Bregma) injections of different BDZs. AMPAR-mediated EPSCs were recorded in presence of d-APV and picrotoxin. mIPSCs were recorded in presence of kynurenic acid (2 mM) and tetrodotoxin (TTX, 500 nM). In vivo extracellular single unit recordings of DA neurons in the VTA (ML: -1.2, AP: -3.2, DV -4 to 4.5 mm from the bregma) were carried out in WT, α1(H101R) and α3(H126R)23 knock-in mice. Drugs were delivered through the tail vein. Immunofluorescence with a guinea pig antibody against the α1 or α3 subunit, a mouse antibody against TH, and a rabbit antibody against eGFP was performed as previously described10 in GAD67-GFP Δneo mice. For the oral self-administration of MDZ, mice were housed with free access to two bottles containing either MDZ in sucrose or sucrose alone. Grouped data are expressed as means ± SEM. For statistical comparisons the one-way Anova, Bonferroni matched or the paired Student's t-tests were used. The levels of significance are indicated as follows: * P<0.05, ** P<0.01, *** P<0.001.
Supplementary Material
Acknowledgments
We thank members of the Lüscher lab as well as Matthew Frerking, Mauro Serafin and Hanns Möhler for critical reading of the manuscript. Yuchio Yanagawa provided the GAD-67 GFP Δneo mouse line and Klaus A. Miczek and Hanns Ulrich Zeilhofer for help with the GABAA mutant mouse lines. This work is supported by the National Institute on Drug Abuse (DA019022; PS), the Swiss National Science Foundation and the European Neuroscience Institute Network. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIDA or the National Institutes of Health.
Footnotes
Author contributions: K.T. carried out all in vitro-electrophysiology experiments. M.B, G.L. and C.Y contributed equally to the in vivo-recordings. K.T. and C.C. performed the behavioral experiments. J-M.F carried out the immunohistochemistry. U.R. generated the mutant mice. C.L. designed the study and wrote the manuscript with the help of all authors.
References
- 1.Lüscher C, Ungless MA. The mechanistic classification of addictive drugs. PLoS Med. 2006;3:e437. doi: 10.1371/journal.pmed.0030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'brien CP. Benzodiazepine use, abuse, and dependence. J Clin Psychiatry. 2005;66(2):28–33. [PubMed] [Google Scholar]
- 3.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 4.Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 5.Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–9100. doi: 10.1523/JNEUROSCI.1001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mameli M, Balland B, Lujan R, Luscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- 7.Heikkinen AE, Moykkynen TP, Korpi ER. Long-lasting modulation of glutamatergic transmission in VTA dopamine neurons after a single dose of benzodiazepine agonists. Neuropsychopharmacol. 2009;34:290–298. doi: 10.1038/npp.2008.89. [DOI] [PubMed] [Google Scholar]
- 8.Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sigel E, Schaerer MT, Buhr A, Baur R. The benzodiazepine binding pocket of recombinant alpha1beta2gamma2 gamma-aminobutyric acidA receptors: relative orientation of ligands and amino acid side chains. Mol Pharmacol. 1998;54:1097–1105. doi: 10.1124/mol.54.6.1097. [DOI] [PubMed] [Google Scholar]
- 10.Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- 11.Okada H, Matsushita N, Kobayashi K, Kobayashi K. Identification of GABAA receptor subunit variants in midbrain dopaminergic neurons. J Neurochem. 2004;89:7–14. doi: 10.1111/j.1471-4159.2004.02271.x. [DOI] [PubMed] [Google Scholar]
- 12.Arendt RM, Greenblatt DJ, Liebisch DC, Luu MD, Paul SM. Determinants of benzodiazepine brain uptake: lipophilicity versus binding affinity. Psychopharmacol (Berl) 1987;93:72–76. doi: 10.1007/BF02439589. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph U, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 14.Mohler H, Benke D, Mertens S, Fritschy JM. GABAA-receptor subtypes differing in alpha-subunit composition display unique pharmacological properties. Adv Biochem Psychopharmacol. 1992;47:41–53. [PubMed] [Google Scholar]
- 15.McKernan RM, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 16.Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5- hydroxytryptamine and opioids. Neurosci. 1997;77:155–166. doi: 10.1016/s0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- 17.Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsia AY, Malenka RC, Nicoll RA. Development of excitatory circuitry in the hippocampus. J Neurophysiol. 1998;79:2013–2024. doi: 10.1152/jn.1998.79.4.2013. [DOI] [PubMed] [Google Scholar]
- 19.Poncer JC, Durr R, Gahwiler BH, Thompson SM. Modulation of synaptic GABAA receptor function by benzodiazepines in area CA3 of rat hippocampal slice cultures. Neuropharmacol. 1996;35:1169–1179. doi: 10.1016/s0028-3908(96)00055-x. [DOI] [PubMed] [Google Scholar]
- 20.Long P, et al. Nerve Terminal GABAA Receptors Activate Ca2+/Calmodulin-dependent Signaling to Inhibit Voltage-gated Ca2+ Influx and Glutamate Release. J Biol Chem. 2009;284:8726–8737. doi: 10.1074/jbc.M805322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barberis A, Mozrzymas JW, Ortinski PI, Vicini S. Desensitization and binding properties determine distinct alpha1beta2gamma2 and alpha3beta2gamma2 GABA(A) receptor-channel kinetic behavior. Eur J Neurosci. 2007;25:2726–2740. doi: 10.1111/j.1460-9568.2007.05530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yee BK, et al. A schizophrenia-related sensorimotor deficit links alpha 3-containing GABAA receptors to a dopamine hyperfunction. Proc Natl Acad Sci U S A. 2005;102:17154–17159. doi: 10.1073/pnas.0508752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Löw K, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- 24.Robinson S, Smith DM, Mizumori SJ, Palmiter RD. Firing properties of dopamine neurons in freely moving dopamine-deficient mice: effects of dopamine receptor activation and anesthesia. Proc Natl Acad Sci U S A. 2004;101:13329–13334. doi: 10.1073/pnas.0405084101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neurosci. 2002;114:475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- 26.Pilotto R, Singer G, Overstreet D. Self-injection of diazepam in naive rats: effects of dose, schedule and blockade of different receptors. Psychopharmacol (Berl) 1984;84:174–177. doi: 10.1007/BF00427442. [DOI] [PubMed] [Google Scholar]
- 27.Fuchs V, Burbes E, Coper H. The influence of haloperidol and aminooxyacetic acid on etonitazene, alcohol, diazepam and barbital consumption. Drug Alcohol Depend. 1984;14:179–186. doi: 10.1016/0376-8716(84)90043-7. [DOI] [PubMed] [Google Scholar]
- 28.Berridge KC, Pecina S. Benzodiazepines, appetite, and taste palatability. Neurosci Biobehav Rev. 1995;19:121–131. doi: 10.1016/0149-7634(94)00026-w. [DOI] [PubMed] [Google Scholar]
- 29.Yerbury RE, Cooper SJ. Novel benzodiazepine receptor ligands: palatable food intake following zolpidem, CGS 17867A, or Ro23-0364, in the rat. Pharmacol Biochem Behav. 1989;33:303–307. doi: 10.1016/0091-3057(89)90504-2. [DOI] [PubMed] [Google Scholar]
- 30.Morris HV, Nilsson S, Dixon CI, Stephens DN, Clifton PG. Alpha1- and alpha2-containing GABAA receptor modulation is not necessary for benzodiazepine-induced hyperphagia. Appetite. 2009;52:675–683. doi: 10.1016/j.appet.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Kalivas PW, Duffy P, Eberhardt H. Modulation of A10 dopamine neurons by gamma-aminobutyric acid agonists. J Pharmacol Exp Ther. 1990;253:858–866. [PubMed] [Google Scholar]
- 32.Xi ZX, Stein EA. Nucleus accumbens dopamine release modulation by mesolimbic GABAA receptors-an in vivo electrochemical study. Brain Res. 1998;798:156–165. doi: 10.1016/s0006-8993(98)00406-5. [DOI] [PubMed] [Google Scholar]
- 33.Doherty M, Gratton A. Differential involvement of ventral tegmental GABA(A) and GABA(B) receptors in the regulation of the nucleus accumbens dopamine response to stress. Brain Res. 2007;1150:62–68. doi: 10.1016/j.brainres.2007.02.081. [DOI] [PubMed] [Google Scholar]
- 34.Grace AA, Bunney BS. Paradoxical GABA excitation of nigral dopaminergic cells: indirect mediation through reticulata inhibitory neurons. Eur J Pharmacol. 1979;59:211–218. doi: 10.1016/0014-2999(79)90283-8. [DOI] [PubMed] [Google Scholar]
- 35.Klitenick MA, DeWitte P, Kalivas PW. Regulation of somatodendritic dopamine release in the ventral tegmental area by opioids and GABA: an in vivo microdialysis study. J Neurosci. 1992;12:2623–2632. doi: 10.1523/JNEUROSCI.12-07-02623.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soyka M, Bottlender R, Moller HJ. Epidemiological evidence for a low abuse potential of zolpidem. Pharmacopsych. 2000;33:138–141. doi: 10.1055/s-2000-11224. [DOI] [PubMed] [Google Scholar]
- 37.Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR. Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proc Natl Acad Sci U S A. 2005;102:915–920. doi: 10.1073/pnas.0405621102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudolph U, Mohler H. GABA-based therapeutic approaches: GABA(A) receptor subtype functions. Curr Opin Pharmacol. 2005 doi: 10.1016/j.coph.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Koob GF. Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsych. 2009;42(1):S32–41. doi: 10.1055/s-0029-1216356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lüscher C, Bellone C. Cocaine-evoked synaptic plasticity: a key to addiction? Nat Neurosci. 2008;11:737–738. doi: 10.1038/nn0708-737. [DOI] [PubMed] [Google Scholar]
- 41.Mameli M, et al. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- 42.Redish AD, Jensen S, Johnson A. A unified framework for addiction: vulnerabilities in the decision process. Behav Brain Sci. 2008;31:415–37. doi: 10.1017/S0140525X0800472X. discussion 437-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Labouebe G, et al. RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci. 2007;10:1559–1568. doi: 10.1038/nn2006. [DOI] [PubMed] [Google Scholar]
- 44.Zhao S, et al. Generation of embryonic stem cells and transgenic mice expressing green fluorescence protein in midbrain dopaminergic neurons. Eur J Neurosci. 2004;19:1133–1140. doi: 10.1111/j.1460-9568.2004.03206.x. [DOI] [PubMed] [Google Scholar]
- 45.Tamamaki N, et al. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.