

INTRODUCTION
Most organisms, including all metazoans, require molecular oxygen (O2) for survival. O2 serves as the primary electron acceptor in many cellular organic and inorganic reactions and has been harnessed by mitochondria to generate ATP via oxidative phosphorylation. Low O2 levels (hypoxia) are commonly associated with pathologies such as myocardial ischemia, stroke, inflammation, and solid tumors. However, hypoxic microenvironments also occur in the developing embryo and adult and promote the differentiation of blood cells, blood vessels, the placenta, and the heart (1). Therefore, O2 can also be viewed as a signal that patterns the cardiovascular system during embryogenesis. Tissue O2 homeostasis is maintained by multiple cellular and systemic processes, and many (but not all) of the molecular mechanisms involved have now been elucidated.
RESPONSES TO CHANGES IN O2 AVAILABILITY
Hypoxia-Inducible Factor–Dependent Responses
A critical response to hypoxia is mediated through changes in gene expression regulated by the hypoxia-inducible factors (HIFs) (see Figure 1). HIFs are members of the basic helix-loop-helix (bHLH)-PAS family of environmental sensors and bind to canonical DNA sequences (hypoxia response elements, or HREs) in the promoters or enhancers of target genes (2). They consist of an alpha (HIF-1α) and a beta (HIF-1β, or ARNT) subunit and activate approximately 200 genes encoding proteins that regulate cellular metabolism, proliferation, survival, motility, and basement membrane integrity as well as angiogenesis and hematopoiesis (3). HIF activity is primarily controlled via the stability of the alpha subunit: in O2 -replete conditions (>5% for many cells), HIF-1α is extremely labile (4). As O2 levels decrease below 5%, HIF-1α becomes increasingly stabilized and dimerizes with the constitutively nuclear ARNT protein, binds HREs, and stimulates the transcription of genes such as those encoding glycolytic enzymes, vascular endothelial growth factor (VEGF), adrenomedullin, and erythropoietin. Before the circulatory system is established, mammalian embryonic development proceeds in O2 concentrations (1–3%) at which HIF is typically active (1). Using gene-targeted mice, we and others have shown that HIF is essential for hematopoietic, vascular, and cardiac development (5–7). Because global deletion of HIF activity is embryonic lethal, the generation of conditional alleles has been essential for evaluating the role of HIF in adult life. More recent studies indicate that HIF plays a critical role in cardiovascular physiology, promoting normal function of the heart, lungs, and vascular networks (8–11).
Figure 1.
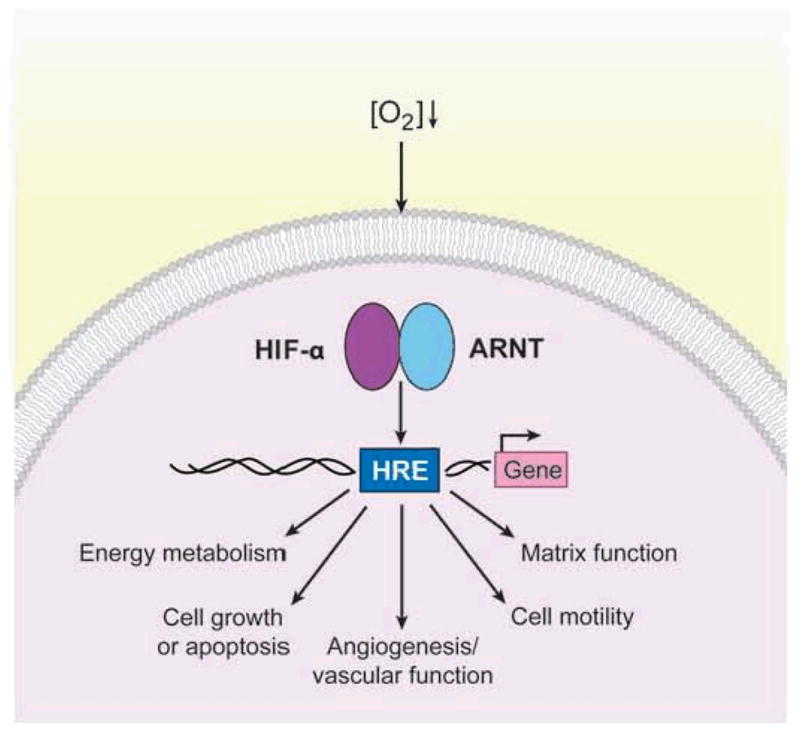
Hypoxia-inducible factor (HIF)-dependent transcriptional responses to hypoxia. In response to O2 deprivation, HIFs, consisting of an alpha and a beta (ARNT) subunit, are activated and bind to hypoxia response elements (HREs) scattered throughout the genome. HIFs stimulate the expression of approximately 200 target genes that mediate hypoxic adaptations and are involved in energy metabolism, cell growth or apoptosis, angiogenesis, cell motility, and the establishment of the extracellular matrix.
Hypoxia-Inducible Factor–Independent Responses
In mammals, hypoxia results in both acute and chronic adaptations. Rapid and reversible effects on cell signaling, contractility, ion flux, and redox state balance energy supply and demand in the face of reduced capacity for oxidative metabolism (12, 13). Whereas HIFs enable long-term cellular survival and vascular remodeling, a variety of HIF-independent pathways promote ATP conservation by limiting energy-consuming processes such as cell division, ribosome biogenesis, mRNA translation, and ion channel activity. A rapid inhibition of protein synthesis is essential for cell survival during conditions of extreme hypoxia (< 0.02 %) (14). Two central mechanisms for regulating translation initiation are the assembly of active eukaryotic initiation factor (eIF) 4F and eIF2-GTP-Met-tRNA ternary complexes (see below). The mammalian target of rapamycin (mTOR), a highly conserved serine/threonine kinase, integrates multiple environmental cues to regulate translation, metabolism, and actin organization in response to nutrient, growth factor, and O2 availability (15). mTOR sustains translation by promoting eIF4F activity. As discussed in the next section, mTOR exists in two distinct intracellular complexes known as mTORC1 and mTORC2. mTORC1 lies upstream of eIF4F and translation initiation. In contrast, ternary complex activity is largely regulated by endoplasmic reticulum (ER)-resident kinases, such as PERK, that participate in the so-called unfolded protein response (UPR), or integrated stress response (ISR) (16). A number of excellent review articles have recently described the role of HIFs in adult cardiovascular physiology. Here, we describe HIF-independent mechanisms that promote tissue homeostasis within this complex organ system. Specifically, we focus on the mTORC1 and UPR pathways, their regulation of mRNA translation, and their relevance to cardiovascular function.
HYPOXIC EFFECTS ON RATES OF PROTEIN SYNTHESIS
Hypoxic Regulation of mRNA Translation via eIF4F
mRNA translation consists of three stages, namely translation initiation, elongation, and termination, each involving multiple regulatory factors (17). Initiation of eukaryotic translation is the rate-limiting step of protein synthesis. It is controlled at two levels: the assembly of the m7GpppN cap–binding eIF4F complex [consisting of the m7GpppN cap–binding protein (BP) eIF4E, the scaffolding protein eIF4G, and the ATP-dependent RNA helicase eIF4A] and of the eIF2-GTP–methionine tRNA ternary complex. O2 availability can influence the activities of both complexes, as reflected by changes in the phosphorylation status of key factors, such as 4E-BP1 and eIF2α, that regulate complex assembly (13, 18).
In most cells, hypoxia negatively impacts protein synthesis through multiple mechanisms, including the inhibition of mTOR, a kinase that in part determines the efficiency of translation initiation. mTOR exists in two distinct complexes, mTORC1 (containing raptor and GβL/mLST8) and mTORC2 (containing raptor and GβL/mLST8) (15). Whereas mTORC1 promotes ribosome biogenesis, mRNA translation, and nutrient import, mTORC2 regulates actin organization and Akt catalytic activity. Hypoxic mTORC1 inhibition causes decreased cell size and cell cycle arrest. In this section, we first introduce the mTORC1 pathway and hypoxic effects on signaling downstream of mTORC1. As shown in Figure 2a, phosphorylation of 4E-BP1 is largely controlled by mTORC1 activity. mTORC1 coordinates cell metabolism, growth (cell mass), proliferation, and survival by integrating various signals, including growth factor, nutrient, and O2 availability, to endogenous energy status (reviewed by Reference 15). mTORC1 activity is regulated by several upstream pathways, including the phosphatidylinositol 3-kinase (PI3K)/Akt, Ras/mitogen-activated protein kinase (MAPK), and AMP-activated kinase (AMPK) cascades (15, 19). All three pathways signal to mTORC1 by phosphorylating the TSC2 protein and altering the activity of the tuberous sclerosis complex (TSC). TSC, consisting of tumor suppressor proteins TSC1 and TCS2 (Figure 2a), negatively regulates mTORC1 by functioning as a GTPase-activating protein toward the small G protein Rheb (15). Whereas TSC2 phosphorylation by Akt and ERK inhibits its activity, TSC phosphorylation by AMPK enhances it activity (15, 19).
Figure 2.

Hypoxic regulation of mRNA translation and its roles in cellular tolerance to low O2 and ischemia/reperfusion. Schematic diagrams for signaling pathways activated during hypoxia that result in (a) mTORC1 inhibition or (b) activation of the unfolded protein response (UPR). (c) Ischemic preconditioning, a brief exposure to ischemia, provides a powerful temporal protection against more prolonged and deleterious ischemic/reperfusion injury. Ischemic preconditioning results in the induction of an acute protein synthesis inhibition (PSI) after ischemia/reperfusion, rather than a persistent protein synthesis inhibition in its absence (see text for more details). Other abbreviations: 4E-BP1, eIF4E-binding protein 1; AMPK, AMP-activated kinase; ATF4, activating transcription factor 4; CHOP, c/ebp-homologous protein; eIF, eukaryotic initiation factor; GADD34, DNA-damage-inducible gene 34; IRE1, inositol-requiring 1; mTORC, mammalian target of rapamycin complex; PERK, pancreatic eIF2-alpha kinase; PML, promyelocytic leukemia; PP1, protein phosphatase 1; XBP1, X box–binding protein 1.
Hypoxia, especially prolonged hypoxia, activates AMPK (20), coincident with reduced ATP levels due to lowered O2 availability for oxidative phosphorylation (see Figure 2a). Cells shift metabolism from an aerobic to an anaerobic mode, and ATP is produced primarily as a result of increased glucose uptake and glycolysis. The net increase in AMP levels in hypoxic cells triggers allosteric activation and phosphorylation of AMPK (21), promoted by growth factor withdrawal under low-O2 conditions (20). Hypoxic mTORC1 inhibition can be suppressed by TSC2-null mutations, over expression of the kinase-inactive AMPK α2 subunit, or elevated levels of the TSC2 substrate Rheb. In contrast to AMPK, neither PI3K/Akt nor Ras/MAPK pathways seem to be responsible for mTORC1 inhibition during hypoxia (22, 23). In addition to the AMPK/TSC2 pathway, hypoxia inhibits TSC2 activity in the absence of AMPK activation in mouse embryonic fibroblasts (MEFs). This regulation is partially mediated by REDD1 (24). Overexpression of Drosophila REDD1/2 orthologs is sufficient to promote larval development and survival under hypoxic conditions (9% O2) (25). As shown in Figure 2a, REDD1 also acts upstream of TSC2. Both REDD1 and TSC2 are required for a profound reduction in mTORC1 activity following exposure to moderate hypoxia (1% O2). Because REDD1 is induced during hypoxia in a HIF-1α-dependent manner (24, 25), REDD1 thus represents a feedback mechanism activated by hypoxia to regulate mTORC1.
A third mechanism involved in more long-term hypoxic regulation of mTORC1 is direct mTOR nuclear translocation mediated by the tumor suppressor protein PML. Nuclear mTOR localization is enhanced by 8–16 h of exposure to hypoxia and blocked by PML-null mutations (26). PML loss correlates with enhanced mTORC1 activity and increased tumor angiogenesis. The latter effect has been proposed to result from increased HIF-1α protein translation due to reduced hypoxic mTORC1 inhibition. Further investigation is needed to determine how rapid hypoxic mTORC1 inhibition occurs within 15–30 min, as detected in HEK293 cells and HIF-null hepatocytes and MEFs (20, 22). It is noteworthy that TSC2 is required for rapid inhibition of mTORC1 under low-O2 conditions, whereas long-term hypoxia can also inhibit mTORC1 in a TSC2-independent manner (20).
Endoplasmic Reticulum Stress and Hypoxic Translational Inhibition
eIF2, a heterotrimeric protein complex consisting of α, β, and γ subunits, represents another important control point in mRNA translation initiation. eIF2α recruits the initiator methionine tRNA to 40S ribosomes, when it binds GTP, and eIF2α phosphorylation inhibits GDP/GTP exchange facilitated by eIF2B (18, 27). Translation of membrane-associated and extracellular proteins is performed by ribosomes residing on the cytosolic surface of the ER. Nascent polypeptide chains enter the ER, which serves as a site for protein folding, disulfide bond formation, and glycosylation before the proteins are secreted or translocated to the plasma membranes. However, ER stress occurs when cells experience dramatic variations in new protein accumulation or an overabundance of unfolded polypeptides. To alleviate ER stress, eukaryotic cells activate a series of self-defense mechanisms referred to collectively as the ER stress response (ESR), the ISR, or the UPR (27, 28). These include (a) an eIF2α-mediated reduction in protein translation to prevent the further accumulation of any unfolded proteins, (b) IRE1-XBP1-mediated transcriptional induction of ER chaperone genes to facilitate protein folding, (c) transcriptional induction of genes involved in ER-associated degradation (ERAD) of unfolded proteins, and (d) the induction of apoptosis in cells that are unable to recover from stress (27, 28). BiP is an ER chaperone protein promoting protein folding (28). When increased ER load is experienced, BiP is thought to dissociate from PERK, IRE1, and activating transcription factor (ATF)6 and sequester misfolded proteins inside the ER lumen. The dissociation of PERK-BiP complexes likely allows PERK dimerization and activation of its cytosolic kinase domain by trans-autophosphorylation. This may also result in a dramatic increase in affinity toward eIF2α (28).
Hypoxia activates both PERK and IRE1 (see Figure 2b), two of the three known UPR pathways that also include ATF6 (29). Although the precise cellular events activating PERK, an ER-resident transmembrane protein, during hypoxia are not clear, hypoxia induces eIF2α phosphorylation via PERK (14). Similar to hypoxic mTORC1 inhibition, the severity of O2 deprivation appears to affect the kinetics of eIF2α change. Severe hypoxia (0.02% O2) induces rapid eIF2α phosphorylation on Ser51 within 15 min, which plateaus after 1–2 h of low O2 (18, 29). eIF2α phosphorylation is weaker and slower under modest to moderate (0.5–1.5% O2) hypoxia, peaking after 8–20 h of low O2 exposure (14, 20). In contrast to hypoxic mTORC1 inhibition, which is enhanced by prolonged exposure, eIF2α phosphorylation is transient and decreases after reaching a plateau (14, 20, 30). The latter result is due to enhanced activity of the eIF2α phosphatase PP1 by low O2. The translation of ATF4, an important mediator of the UPR, is paradoxically enhanced by eIF2α phosphorylation (28, 31). ATF4 induces the transcription and translation of its target genes, including those encoding the proapoptotic factor CHOP and DNA-damage-inducible gene GADD34 (as also shown in Figure 2b). GADD34 is a regulatory subunit of PP1. Thus, increased GADD34 levels reverse hypoxic eIF2α phosphorylation and translation inhibition (18, 32). Moreover, hypoxia activates endonuclease activity of IRE1, resulting in the excision of a 26-nucleotide intron from XBP1 mRNA (Figure 2b), a step necessary for its activation (29). XBP1 increases the transcription of ER chaperone gene products such as BiP and CHOP (27).
Hypoxic Effects on Translation Elongation
Moderate hypoxia also affects protein synthesis by inducing elongation factor eEF2 phosphorylation and inhibiting translation elongation (20, 33). This may be a result of AMPK- and mTORC1-dependent phosphorylation of the eEF2 kinase because both AMPK and S6K (a target of mTORC1) phosphorylate this kinase (20). However, eEF2 inhibition does not seem to affect protein synthesis directly; substantial eEF2 phosphorylation is observed in serum-deprived cell after only 30 min of moderate hypoxia, but no change in the rate of protein synthesis occurs until 24 h. In contrast, hypoxic regulation of elongation seems to play a more important role in transformed breast epithelial cells. siRNA against eEF2K blocks not only hypoxic eEF2 phosphorylation but also hypoxic inhibition of protein synthesis in MCF10A cells (33).
HYPOXIC REGULATION OF TRANSLATION AND ISCHEMIC DISEASE
eIF2 regulation may increase or decrease global rates of protein synthesis, whereas eIF4F regulation may control the repertoire of mRNA being delivered to the ribosomes (34). Low levels of eIF4F activity favor the translation of more abundant mRNAs, and as eIF4F activity increases, less abundant mRNAs are recruited into the translation apparatus (34). Thus, hypoxia may regulate total protein load as well as select mRNA translation to suit cellular demands during stress. By changing the transcription and translation of specific effectors such as HIF-1α ATF4, and GADD34, both mTORC1 and ER stress are highly involved in resistance to cell death. Studies by Bi et al. (35) revealed that PERK−/− cells and MEFs with an eIF2α. Ser51Ala mutation show increased cell death during hypoxic exposure. Both PERK- an XBP1-deficient cells exhibit defective tumor growth, an effect linked to decreased hypoxia tolerance (35, 36). Some evidence suggests that pathways regulating mRNA translation activated during hypoxia may strongly depend on cell type. For example, transformed cells and nontransformed cells respond differently to low O2 (33). Nonetheless, the global translation inhibition and selective translation of subsets of regulatory proteins and enzyme in cardiac myocytes and neurons may be of additional significance because these cells are fully differentiated and no longer divide.
Clinical manifestations of hypoxia include ischemic heart disease and stroke. Cardiovascular pathologies are responsible for approximately 40% of all North American deaths; cardiac ischemia leads all other cardiovascular diseases in this regard (37). In addition, stroke, a disease state in which blood supply to areas of the brain is interrupted, can result from cardiovascular diseases such as atherosclerosis. Ischemic injuries frequently result in the loss of tissue function or tissue death. Most forms of brain ischemia, including transient global, permanent focal, and transient focal, lead to protein synthesis inhibition (38). Protein synthesis gradually recovers in the majority of regions during reperfusion, and the rate of translational recovery is affected by the duration of ischemia (39). Cell death ensues in brain regions where protein synthesis fails to recover, such as the CA1 domain of the hippocampus, following relatively short global ischemia (39). The initial translation inhibition is caused by eIF2α phosphorylation. Although eIF2α state returns to almost basal levels by 6 h of reperfusion, protein synthesis remains inhibited even 24 h after reperfusion (39). Sustained translation inhibition is not likely to be caused by 4E-BP1 phosphorylation because 4E-BP1 phosphorylation returns to control levels by 4 h of reperfusion. It is interesting that decreased eIF4F complex formation is coincident with decreases in both eIF4E and eIF4G levels in hippocampus isolated at 4 h of reperfusion (39).
Ischemic tolerance (IT) is a phenomenon in which exposure to a brief, nonlethal ischemic insult (ischemic preconditioning) produces a delayed-type protection from cell death following a second exposure to a lethal insult (Figure 2c). This could be a promising therapeutic strategy for tissues highly sensitive to ischemia, including the myocardium and central nervous system, based on the mobilization of intrinsic protective mechanisms (39). A number of molecular pathways participate in ischemic preconditioning, including pathways involving mRNA translation and ER stress. IT does not affect the acute phase of protein synthesis inhibition but enhances recovery of protein synthesis following the lethal insult and prevents extended protein synthesis inhibition (Figure 2c) (39, 40). Interestingly, although eIF2α phosphorylation is not affected by IT, IT causes a pronounced increase in GADD34 protein levels. These observations suggest that the upregulation of genes induced by IT is effective in buffering against persistent inhibition. Moreover, several reports clearly show induction of ER stress during brain and cardiac ischemia, including activation of the ATF6, IRE1, and PERK pathways; CHOP-mediated apoptosis of neurons; and the degeneration of cardiomyocytes (27). Although the role of the UPR in inducing or inhibiting cell death is likely to be affected by the duration and severity of ischemia, these studies strongly suggest that the UPR is an integral part of cellular responses and may play a critical role in maintaining cardiomyocyte and neuronal homeostasis (27).
In response to disrupted coronary blood flow, the heart acutely adapts to restricted O2 and nutrient supply and undergoes a dramatic switch from aerobic to anerobic metabolism. Increasing evidence shows that AMPK is quickly activated during myocardial ischemia, resulting in rapid decreases in glucose and fatty acid oxidation (37). Glycolysis, a minor source of ATP in aerobic hearts, becomes a more significant energy source (37). Although actual protein synthesis in ischemic heart has not been assessed, increased AMPK activity likely reduces protein synthesis in cardiomyocytes, a strategy enabling the heart to maintain ATP levels and ensure contractile function, ion channel activity, and overall cellular integrity. Reduced protein synthesis decreases not only protein load (which requires proper folding) but also the degradation of improperly folded proteins, which are energy-consuming processes. Data from AMPK-deficient mouse models suggest that AMPK activity likely increases heart tolerance to ischemia (41).
Several studies indicate that mTORC1 is also involved in preconditioning and confers protection against irreversible damage by short episodes of ischemia/reperfusion preceding a potentially lethal ischemia (42). Pre-treatment of mouse hearts with rapamycin protects against ischemia/reperfusion injury, suggesting that rapamycin induces a potent preconditioning-like effect against myocardial infarction (42, 43). However, other reports indicate that rapamycin blocks the late phase of preconditioning and the cardioprotective effect of insulin on reperfusion (42). Although the mechanisms for the cardioprotective effect of rapamycin await further investigation, the timing of drug administration could be critical to affecting treatment outcome, given that acute and persistent phases of protein synthesis inhibition exist. More research is needed to address this issue. Taken together, these data suggest that targeting UPR and mTORC1 is an attractive approach for treating ischemic lesions. Important questions yet to be fully elucidated include the role of hypoxic induction of ER stress on the cause of ischemia-related diseases and causes for the sustained mRNA translation inhibition.
HYPOXIC EFFECTS ON THE MAMMALIAN TARGET OF RAPAMYCIN COMPLEX 1
In this section, we discuss examples of cells, including stem cells, that respond to hypoxia by initiating processes (such as proliferation) rather than inhibiting them. We also consider tumor cells, in which hypoxic effects on mTORC1 are best characterized. Finally, we examine the mTORC1 pathway and its biological manifestations in specific cardiovascular cells in which hypoxia frequently elicits enhanced cell growth and proliferation. These differential responses to hypoxia are at least in part determined by effects on mTORC1 and its downstream targets various effects of hypoxia on this important environmental sensor, we highlight distinct responses observed in various cell types. We emphasize interesting features of hypoxic responses in cardiovascular cells that are likely due to their unique role in O2 delivery.
mTORC1 Control of Protein Synthesis Initiation
As stated above, protein synthesis intiation is regulated primarily by the formation of two critical complexes (44). The ternary complex of eIF2α, GTP, and methionine-charged tRNA is described elsewhere in this review. The eIF4F complex [composed of the eIF4G scaffolding protein, the RNA helicase eIF4A, and the m7GpppN cap–BP eIF4E (44)], once associated with the ribosome (and several other components), scans for the initiator AUG sequence and initiates translation.
eIF4E is the least abundant member of the eIF4F complex, and as such, translation efficiency is determined in large part by cellular eIF4E availability. Free eIF4E levels are regulated by a class of proteins termed the eIF4E-BPs (4E-BPs). 4E-BPs bind to the same site in eIF4E as does eIF4G and therefore prevent eIF4E binding to the scaffold and the assembly of eIF4F. 4E-BPs are regulated by phosphorylation, which determines their eIF4E binding efficiency (45). This phosphorylation occurs hierarchically, on a number of distinct sites (46). Phosphorylated 4E-BP1 binds to eIF4E poorly and is thus rendered less effective in inhibiting eIF4F complex formation. mTORC1 is at least in part responsible for 4E-BP1 phosphorylation, and so mTORC1 activity is crucial in determining translation initiation efficiency (22, 47). In addition to its role with regard to 4E-BP1, active mTORC1 phosphorylates and activates p70S6K (see Figure 3a), further enhancing protein synthesis (48). These downstream mTORC1 targets determine how effectively cells translate mRNAs, thereby dictating ultimate effects such as cell size and proliferative capacity. By activating or repressing cap-dependent translation, cells control the production of proteins essential for these processes, such as cyclin D1, c-Myc, ornithine decarboxylase, and others (49).
Figure 3.

Hypoxic effects on mTORC1. (a) In many cells, hypoxia inhibits mTORC1 activity either directly or indirectly, resulting in decreased phosphorylation of the downstream targets of mTORC1, 4E-BP1 and p70S6K. This results in an overall decrease in protein synthesis capacity and subsequent decreases in proliferation and cell growth. Many cells likely utilize this response to adapt to the hypoxic insult and await a return to homeostatic O2 levels. (b) Other cells, including cells of the cardiovascular system, respond to hypoxia with an enhancement of mTORC1 activity. mTORC1 activation increases 4E-BP1 and p70S6K phosphorylation, resulting in potentially increased protein synthesis, cell growth, and proliferation. This response may be programmed to occur upon hypoxia-induced vascular remodeling or during recovery from ischemia. Tumor cells may utilize a similar strategy through an unknown mediator or alternatively via oncogenic signaling, overcoming hypoxic inhibition.
Importantly, mTORC1 has numerous roles in environmental sensing, and in puts from various pathways converge on mTORC1 (47). In this way, mTORC1 senses the quality of the cell’s environment and determines appropriate responses. Pathways affecting mTORC1 include those that evaluate the availability of nutrients such as amino acids and glucose, growth factors, and O2 (22, 47). When these are limiting, mTORC1 activity is usually attenuated, and 4E-BP1 becomes hypophosphorylated, resulting in decreased protein synthesis (20). Conversely, nutrient and O2 sufficiency activates mTORC1, subsequently maintaining mRNA translation to generate proteins required for growth and proliferation. mTORC1 activity therefore dictates much of this decision on the basis of the cellular environment (47).
As shown in Figure 2a, hypoxia impacts mTORC1 activity through numerous mechanisms. Importantly, in many cells hypoxic exposure elicits a rapid decline in mTORC1 signaling, revealed by decreased phosphorylation of the downstream targets of mTORC1 (22). Hypoxic inhibition of mTORC1 signaling is dominant to other stimulators of mTORC1, including insulin, amino acids, and phorbol esters (22). The specific mechanism(s) of mTORC1 inhibition by hypoxia is still under investigation. It is known that hypoxia can elicit transcription of REDD1 via HIF activity, which negatively impacts mTORC1 signaling (24). However, hypoxic effects on mTORC1 targets are very rapid (less than 30 min) and occur in cells null for HIF activity, indicating that hypoxia also affects mTORC1 independently of HIF (20, 22). Despite the lack of mechanistic understanding, mTORC1 is clearly an important target of hypoxic influence.
Hypoxia-Induced Proliferation in Various Cell Types
Although many cells respond to hypoxia via mTORC1 inhibition and decreased growth, certain cells actually proliferate more in low O2. For example, hypoxia is present in large regions of the developing embryo and is likely a driving force for differentiation that occurs during ontogeny (50). The requirement for protein synthesis in these events (cell proliferation, growth, and differentiation) indicates that mTORC1 inhibition and thereby protein synthesis by hypoxia must not be universal. Tissues in the developing embryo demonstrate that proliferation not only is maintained in hypoxia but in many cases is enhanced.
Examples of cells that respond to hypoxia with increased proliferation are numerous. Hematopoietic stem cells proliferate more effectively in hypoxic conditions (1–1.5% O2) than in atmospheric O2 (51, 52), as do neural crest stem cells (1–5% O2) (53, 54). Additionally, cytotrophoblasts, cells derived from the placenta, multiply more readily at low O2 levels (2 %) than when grown in atmospheric O2 (21 %) (55). Finally, para-aortic splanchnopleural (P-Sp) explant cultures have been used to evaluate requirements for proper cardiovascular development in the early embryo (56). Vasculogenesis and angiogenesis in these explants require VEGF, which is induced by hypoxia in the embryo. Therefore, hypoxia plays an essential role in embryonic development, in part through increasing proliferative capacity.
In addition, hypoxia has interesting effects on embryonic stem (ES) cells. These cells also respond to low O2 with increased proliferation (B.C. Barnhat & M.C. Simon unpublished observations). Because increased proliferation presumably requires mTORC1 activity (to synthesize new proteins), our laboratory examined hypoxic effects in ES cells. Surprisingly, mTORC1 appear to be negatively regulated by hypoxia in ES cells. The mechanistic details of these observations are under investigation. These and other data demonstrate that hypoxic effects on mTORC1 differ considerably with cell type. This is likely the result of the differential requirements of some cells exposed to hypoxic conditions. It is not yet fully known, however, how these effects are regulated.
Effects of Hypoxia on Tumor Cells
Many studies have now assessed hypoxic effects on tumor cells. Solid tumors can grow quickly and develop inadequate vasculature, leading to frequent and often severe O2 deprivation (57, 58). Tumor hypoxia also selects for cells that survive the combined stresses of low O2, nutrient and growth factor deprivation, and acidosis (58). Therefore, hypoxia is an important feature of tumor development, and as such many of the underlying mechanisms of hypoxic responses are well characterized. Interestingly, neoplastic cells respond to hypoxia in different ways, depending on tumor type. For example, many tumor cells exhibit hypoxic cell cycle arrest (59–61), which is partially due to mTORC1 inhibition (B.C. Barnhart & M.C. Simon, submitted). mTORC1 target phosphorylation is blocked upon hypoxic exposure in tumor cells, although the level of inhibition varies considerably (22, 33). The mechanism behind this differential regulation is not known, but it is likely that mTORC1 activation ultimately relies on a variety of signaling pathways that are dysregulated in cancer cells. Interestingly, chemical mTORC1 inhibition (which induces cell cycle arrest) can be overcome by activation of the downstream targets of mTORC1 eIF4F and p70S6K (62, 63), indicating that mTORC1 inhibition is sufficient to block proliferation.
Some tumor cells (e.g., breast cancer cells) do not inhibit proliferation when exposed to hypoxia, and they even respond with increased proliferative capacity. This mirrors the hypoxic response of specific stem cells described above. Although the mechanisms regulating this response are unknown, one potential contributor is the PI3K/Akt pathway. Importantly, this pathway activates mTORC1 (64). Furthermore, the PTEN tumor suppressor is mutated in a wide variety of human cancers (65), and its loss leads to Akt activation, stimulating mTORC1 function. Thus, it is likely that many tumor cells are able to avoid the potentially inhibitory effects of hypoxia by altering upstream regulators of mTORC1 (see Figure 3b).
In addition to alterations upstream of mTORC1, dysregulation of its downstream targets also leads to partial liberation from hypoxic inhibition. For example, we have observed that increased eIF4F activity allows cells to partially overcome cell cycle arrest imposed by hypoxia (B.C. Barnhart & M.C. Simon, submitted). Furthermore, other mechanisms may overcome hypoxic inhibition of mTORC1. For example, cells preferentially expressing one of the HIF subunits (HIF-2α) respond to O2 deprivation with increased proliferation, a response that involves the oncogene c-Myc (61). These illustrative examples demonstrate that, although hypoxia inhibits mTORC1 signaling, blocking protein synthesis and ultimately cell growth and proliferation in many cells, other cells over ride this inhibition. In tumors, inappropriate activation of the c-Myc, HIF-2α, or PI3K/Akt pathways may lead to the ability to overcome hypoxic influences. In normal cells, however, the ability to respond to hypoxia and overcome its inhibitory effects is more likely programmed. It is important for some cells to respond to hypoxia with proliferation and growth, including cells responsible for O2 delivery. In the next subsection we discuss examples of how the mTORC1 pathway is regulated within certain cardiovascular cells, which in part dictates these cells’ hypoxic responses.
Hypoxic Effects on Cardiovascular Cells
Various cells comprising the cardiovascular system must respond to low O2 with increased growth and proliferation. Indeed, there are many examples (including smooth muscle cells, endothelial cells, and fibroblasts) of hypoxically induced proliferation, as these cells activate a program of vascular remodeling upon prolonged hypoxic exposure in tissues (for examples, see References 66–68). These mechanisms likely parallel those utilized by stem and/or malignant cells to respond to O2 starvation. In this subsection we describe known responses to hypoxia in the cardiovascular system as they relate to signaling of mTORC1 and its downstream targets. Clearly, mechanisms regulating the hypoxic response are complex; here we discuss selected work that has addressed hypoxic effects on mTORC1 in the cardiovascular system.
Pulmonary artery adventitial fibroblasts proliferate when cultured under decreased O2 tensions (e.g., Reference 67). Furthermore, pathways involved in this response have been examined; these include the MAPK and protein kinase C (PKC) pathways (67). Strikingly, rather than being inhibited, mTORC1 is activated upon hypoxic exposure in these cells (69). These data are consistent with the observation made in aortic smooth muscle cells that hypoxia activates mTORC1 (68). In pulmonary artery adventitial fibroblasts, mTORC1 stimulation is correlated with increased p70S6K activity and increased 4E-BP1 phosphorylation. These interesting results are due in part to hypoxic activation of PI3K and, ultimately, mTORC1. Additionally, protein synthesis in cardiomyocytes is unchanged when they are subjected to 0.5% O2 and is decreased only when they are exposed to 0.05% O2 (representing severe hypoxia) (70). Again, it is likely that such specialized cells utilize this response to maintain homeostasis during O2 deprivation. The lack of protein synthesis inhibition likely allows cardiomyocytes to respond to hypoxia with continued or even enhanced growth and proliferation.
Ischemia subjects cells to very low O2 (in concert with glucose, amino acid, and growth factor deprivation), frequently followed thereafter by O2 reperfusion. Changes in O2 availability that cells encounter upon ischemia and reperfusion produce interesting effects on the mTORC1 pathway. In one experimental model of ischemia/reperfusion, myocardial cells failed to respond with profound mTORC1 inhibition (71). In addition, reperfusion results in increased mTORC1 signaling (as might be expected when O2 is restored), resulting in higher levels of p70S6K and 4E-BP1 phosphorylation. These events may be involved in repairing cellular injury following ischemia. It is interesting to speculate that the attenuated mTORC1 inhibition may play some protective role for these cells, because mTORC1 activity generally protects cells from death. In addition to the role of mTORC1 in growth and proliferation, the maintenance of mTORC1 signaling during ischemia may help cells to withstand the associated stresses and aid in recovery following ischemic insult.
Hypoxia profoundly impacts gene expression patterns in all cells. As discussed above, most O2-regulated genes are induced by HIF (2). Many of these genes help cells to withstand low O2 levels; such genes include those encoding glycolytic enzymes, which increase energy production pathways less dependent on O2, as well as those involved in angiogenesis and apoptosis. The heart is especially susceptible to hypoxia from pulmonary disease, for example, or from intermittent or interrupted blood flow. One recent study examined mouse heart transcriptional activity in response to chronic or intermittent hypoxia (72). Interestingly, some of the genes transcriptionally regulated included translational mediators. Heart tissue exposed to chronic hypoxia results in eIF4E and eIF2α stimulation, which would be expected to lead to increased protein synthesis. Intermittent hypoxia, in contrast, causes a decrease in eIF4E levels. It is likely that these differential cardiac responses are activated owing to specific requirements dependent on Fan et al. (72) speculate that increased eIF4E and eIF2α levels in chronic hypoxia elicit the observed growth in heart tissue (causing cardiac hypertrophy); responses to intermittent hypoxia may involve a distinct mechanism(s). Underlying pathways promoting these responses are unknown at present.
HYPOXIA-MEDIATED PREFERENTIAL TRANSLATION OF SELECT mRNAS
The level and duration of O2 deprivation not only influence mechanisms of global translation inhibition but also are important in determining which mRNAs are selectively translated (30). Multiple research groups have examined the polysome profile of cells cultured under normoxic and various levels and the duration periods of hypoxia by microarray analysis and identified mRNA subsets that remain associated with translating polysomes (31, 73, 74). Translationally regulated transcripts show an increased association with hypoxic compared with normoxic polysomes; however, such transcripts do not exhibit a corresponding increase in gene expression at the mRNA level and therefore are more efficiently translated in response to hypoxia (31). In this portion of the review we discuss the results of these studies, the relationship of such results to O2 deprivation in cardiac tissue, possible mechanisms for the preferential translation of select mRNAs, and finally how these results are being transitioned into Phase 1 clinical trails.
Preferential Translation of mRNA Subsets under Prolonged and Severe Hypoxia
mRNA translation control is an important component of the cellular response to hypoxia and can significantly affect the expression of individual genes. Approximately 2.5% of total cellular transcripts are preferentially translated under prolonged and severe hypoxic stress (16 h O2 < 0.01 %) (31). These mRNAs, which are selectively translated despite a significant decrease in global protein synthesis, are believed to be required for cells to adapt to low O2. Microarray analysis of polysome profiles of normoxic and hypoxic HeLa cells identified a subset of mRNAs preferentially translated under prolonged and severe hypoxic stress (31). For example, the translation efficiency of ATF4 mRNA increases under these conditions. Because ATF4 is a transcription factor activating genes such as those encoding GADD153/CHOP, GADD34, and BiP, it plays a central role in the cellular adaptation to stress and the ISR. Additional translationally regulated mRNAs such as fibroblast growth factor (FGF)2 (75), VEGF (75), cyclin-dependent kinase inhibitor p57Kip2 (76), ATF3 (77), and MAPK (78) are important in the regulation of angiogenesis, apoptosis, and cell cycle progression, processes central to hypoxia-mediated cardiac development and adaptation. ATF3, VEGF, and MAP2K1 are both transcriptionally induced and more efficiently translated under hypoxia, whereas FGF2 and p57Kip2 are more efficiently translated during hypoxia, without an associated transcriptional induction (31). In addition, translation rates of an endothelium-specific receptor tyrosine kinase for angiopoietins (Tie2) increase under prolonged and severe hypoxia, as assessed by metabolic labeling of human umbilical vein endothelial cells (HUVECs) followed by immunoprecipitation with anti-Tie2 antibodies (79).
Critical differences exist in gene expression profiles between right ventricles (RVs) before either a compensated ventricular hypertrophy or heart failure occurs in response to stress (80, 81). In one study, Wistar rats received a single subcutaneous injection of pyrrolizidine alkaloid monocrotaline, and depending on the degree of pressure overload, RV hypertrophy developed into a compensated or a decompensated hypertrophy. After two weeks, although the degree of hypertrophy in the two groups was similar, significant differences in the gene expression profiles existed. The ventricles progressing to failure showed an up-regulation of multiple proapoptotic genes. In contrast, the compensated hypertrophy animals exhibited a significant upregulation of MAPK phosphatase-1, which is thought to block the apoptotic pathways via p38-MAPK. Also, ventricles ultimately developing a compensated phenotype exhibited an upregulation in VEGF levels. Because genes involved in apoptosis, angiogenesis, and MAPK pathways are translationally regulated in polysome microarray studies of HeLa cells (31), such genes may exhibit translational regulation in cardiac tissue as well and influence whether cardiac hypertrophy develops into a compensated or a decompensated phenotype.
Preferential Translation of Individual mRNAs under Acute and Severe Hypoxia
The translational efficiency of individual genes is dynamic and changes with alterations in the cellular environment (30). Analyses of polysome-associated mRNA have revealed differences in the translation efficiency of a subset of genes such as ATF4 and GADD34, depending on the duration of the hypoxic stress. In addition, although acute hypoxia (4 h O2 < 0.01%) caused a drop in the average number of ribosomes per transcript, selectively translated mRNAs such as ATF4 maintained the number of associated ribosomes and were less dependent on eIF2α availability (30). Changes in mRNA translation efficiency of select mRNAs from prostrate (DU1450) carcinoma cells cultured under acute hypoxia were assessed by microarray analysis (74). In this assay, the distribution of mRNA isolated from the translating polysome peak under normoxic and severe hypoxic conditions was compared with changes in total mRNA. After 4 h of severe hypoxia, more genes were upregulated at the translational than the transcriptional level. Both VEGF and platelet-derived growth factor (PDGF) are transcriptionally and translationally regulated under acute hypoxic stress and important in the control of angiogenesis.
Whereas changes in transcription can take hours to achieve, translational regulation is rapid and reversible (82). The ability to respond quickly to changes in O2 levels to induce a cardioprotective pattern in gene expression may be quite important in cardiac tissue in which O2 tension can change rapidly. Acute responses may increase cardiac resistance to injury (preconditioning) and guard against secondary injury after an acute ischemic insult (see Figure 2c). Microarray analysis has also been used to study changes in gene expression patterns caused by brief ischemia in a rat acute ischemic heart model (75). Total RNA was isolated from rats that underwent 20 min of coronary occlusion followed by 4 h of reperfusion. This total RNA was compared with total RNA isolated from sham-operated control rats. Increased mRNA abundance was observed for those mRNAs encoding heat shock proteins, growth factors (VEGF), and antiapoptotic factors (ATF3 and the GADD 45α). Because VEGF is both transcriptionally and translationally regulated in DU1450 and HeLa cells, it seems likely that the same is true in endothelial cells. The ability to rapidly upregulate VEGF levels under ischemic injury increases the vascularization of the ischemic tissue and is likely important for the adaptation of endothelial cells to O2 deprivation.
PREFERENTIAL TRANSLATION OF PROANGIOGENIC FACTORS DURING THE INTEGRATED STRESS RESPONSE
As described above, PERK+/+ tumors facilitate endothelial survival and functional vessel formation, whereas PERK−/− tumors form irregularly structured vessels (35). PERK promotes a tumor microenvironment favoring the formation of vessels by promoting selective translation of proangiogenic transcripts. Microarray analyses of polysomes from K-Ras-transformed PERK+/+ and PERK−/− MEFs demonstrate that PERK affects the translation of multiple proangiogenic genes during hypoxic stress. Validated candidates include adhesion molecules; VEGF and type 1 collagen inducible protein (VCIP), which favors the formation of functional microvessels; and matrix metalloproteinase (MMP) 13, which is essential for endothelial cell activation, migration, and capillary formation (73). It appears that PERK plays a role in tumor and, presumably, normal, cellular adaptation to hypoxic stress by regulating the translation of proangiogenic factors necessary for the development of functional microvessels. The ISR is an important mediator of hypoxic adaptation, and it seems likely that it is activated by both acute cardiac ischemia and reperfusion and that this response promotes recovery from O2 deprivation by enhancing the selective translation of proangiogenic factors (se Figure 4).
Figure 4.
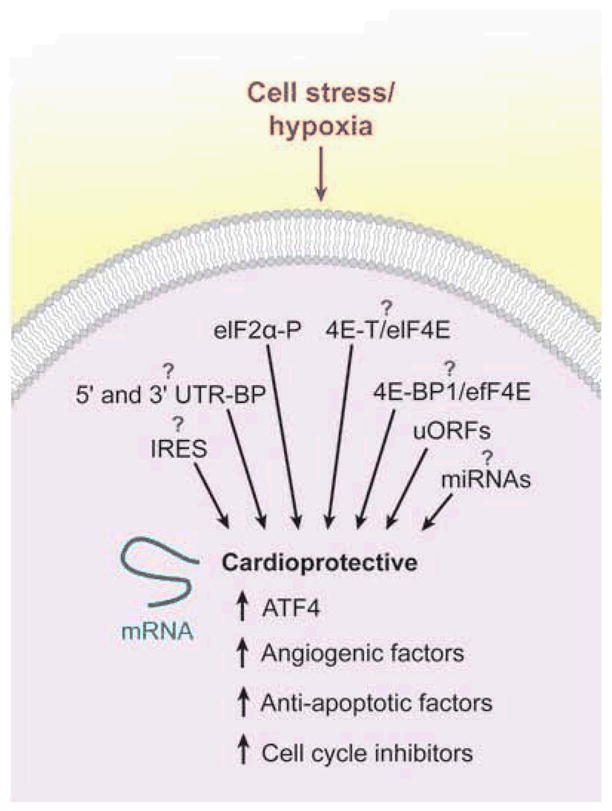
Theoretical model and potential mechanisms for the selective translation of individual mRNAs whose protein products may contribute to a cardioprotective phenotype. Cellular stresses such as hypoxia can lead to the selective translation of mRNAs involved in the integrated stress response, angiogenesis, antiapoptotic factors, and cell cycle inhibitors. These factors may be involved in the adaptation of cardiac tissue to hypoxic stress. The mechanism(s) for the hypoxia-mediated preferential translation of individual mRNAs is largely unknown, but contributing regulatory pathways are shown. Of note, both 4E-BP1 and 4E-T can inhibit eIF4E activity in O2-starved cells. Abbreviations: 4E-BP1, eIF4E-binding protein 1; ATF4, activating transcription factor 4; eIF, eukaryotic initiation factor; IRES, internal ribosome entry site; miRNA, microRNA; uORF, upstream open reading frame; UTR-BP, untranslated region-binding protein.
POSSIBLE MECHANISMS FOR HYPOXIA-MEDIATED TRANSLATION OF SELECT mRNAS
Preferential translation of mRNA subsets is thought to be an important component of the cellular response to O2 deprivation. However, it is not clear how individual genes are selectively translated, despite decreases in global protein synthesis. As we discuss above, the regulation of global protein synthesis occurs primarily through the modification of translation initiation and elongation factors. In contrast, the regulation of specific mRNAs is thought to be mediated by protein complexes that recognize elements located in the 5′ and 3′ untranslated regions (UTRs) of target mRNAs (83), as shown in Figure 4. ATF4 contains three evolutionarily conserved upstream open reading frames (uORFs) in its 5′ UTR, and like yeast GCN4, these may play an important regulatory role in basal repression and translational activation (84). Interestingly, Tie2 mRNA contains five uORFs in the 5′ UTR, which are inhibitory for translation (85). Furthermore, both CHOP and GADD34 contain uORFs in their 5′ UTRs and are translationally regulated as part of the ISR (18). It is unclear at present whether uORFs are a general mechanism for stimulating the translation of select mRNAs under hypoxia. Multiple stress-regulated mRNAs, such as HIF-1α, VEGF, and Tie2, have been reported to contain internal ribosome entry sites (IRESs), and these elements may allow mRNAs to continue to be efficiently translated under hypoxia when cap-dependent translation is diminished (79, 86, 87). However, monocistronic transcripts can be generated from cryptic promoters or cryptic splice sites within the 5′ UTR in this assay, and these transcripts can give the false appearance of IRES activity (88). Transgenic mice expressing reporter genes under the control of the VEGF or FGF2 IRES were developed (89). Interestingly, although VEGF IRES activity is low in embryonic and adult tissue, both the VEGF and FGF2 IRESs exhibit increased translational activity under severe hind limb ischemia. The existence of cellular IRESs and their biological role in the cell’s adaptation to stress remain a controversial field (90). Recently, microRNAs that change in response to hypoxia (Figure 4) have been identified, and these may provide an additional mechanism for the preferential translation of individual mRNAs (91, 92). Much work remains to elucidate how individual mRNAs that facilitate the cells’ adaptation to O2 deprivation are preferentially translated under hypoxic stress.
CLINICAL APPLICATIONS
Angiogenic factors such as VEGF and FGF2 are protective in situations in which cell injury occurs (93). Upregulation of VEGF and FGF2 gene expression synergistically stimulates blood vessel density and results in increased vascularization of ischemic tissue. In addition, VEGF also affects blood vessel maturation and functionality. For these reasons, VEGF and FGF2 have become the targets of intensive experimental and clinical gene therapy studies. Because of the central role that VEGF plays in angiogenesis, the VEGF-encoding sequence was the first to be transferred to patients in Phase 1 clinical trails (75). In these studies, VEGF was incorporated into plasmid DNA or an adenovirus vector and administered by direct injection into ischemic myocardium. These early uncontrolled studies showed promise for the treatment of myocardial perfusion and decreased angina frequency and nitroglycerin use. However, results from subsequent larger controlled clinical trails have not been conclusive. In addition, Phase 1 clinical trials have been performed with FGF2, and the results have also been inconclusive. The implantation of erythroid colony-forming cells supplies angiogenic cytokines (VEGF and FGF2) and may be a novel strategy for stimulating therapeutic angiogenesis (94).
SUMMARY AND FUTURE DIRECTIONS
Low O2 levels cause profound adaptive effects on cellular metabolism and gene expression, with implications for development, physiology, and disease. Cells within solid tumors frequently encounter hypoxia, given the poor vascular function of tumor blood vessels. Tumor hypoxia and responses to O2 deprivation have been established as integral components of neoplastic progression. However, the O2- and nutrient-delivering cardiovascular system is also modulated by O2 availability to meet the needs of both developing and mature organisms. Ongoing studies in the field continue to address questions of central importance, such as the following: How does O2 availability fine-tune cardiovascular function? What are the molecular mechanisms by which hypoxia regulates the mTORCl and UPR/ISR pathways? How are select mRNAs (e.g., VEGF and Tie2) Preferentially translated in O2-starved cells? How do cardiovascular cells sense O2? How can hypoxic adaptations be exploited to promote tissue health during cardiovascular disease? The ultimate goal is to better define hypoxic responses to develop novel therapies that combat deleterious results from cells encountering low O2during tissue ischemia, myocardial infraction, and stroke.
- VEGF
vascular endothelial growth factor
- eIF2 control
the formation of the 40S ribosome–binding eIF2α-GTP-methione tRNA ternary complex, which determines the rate of global protein translation initiation
- eIF4F control
the formation of the eIF4E-eIF4G-eIF4A complex that binds to the m7GpppN cap, which controls the translation of cap-containing mRNAs
- mTORC1
mammalian target of rapamycin complex 1
- Unfolded protein response (UPR)
the series of molecular signals generated as a consequence of the presence of unfolded proteins in the endoplasmic reticulum (ER) or other ER-related stress; results in changes in the regulation of transcription and translation
- ISR
integrated stress response
- BP
binding protein
- 4E-BP
eIF4E-binding protein
- MAPK
mitogen-activated protein kinase
- ATF
activating transcription factor
- GADD
DNA-damage-inducible gene
- Ischemic tolerance (IT)
a brief, nonlethal ischemic insult that protects cells from death against a second exposure to a lethal insult
- FGF
fibroblast growth factor
- Tie2
endothelium-specific receptor tyrosine kinase for angiopoietins
- RV
right ventricle
- VCIP
VEGF and type 1 collagen inducible protein
- MMP
matrix metalloproteinase
- UTRs
untranslated regions
- uORF
upstream open reading frame
- IRESs
internal ribosome entry sites
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any biases that might be perceived as affecting this review.
LITERATURE CITED
- 1.Ramirez-Bergeron DL, Simon MC. Hypoxia-inducible factor and the development of stem cells of the cardiovascular system. Stem Cells. 2001;19:279–86. doi: 10.1634/stemcells.19-4-279. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–78. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 3.Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004;18:2183–94. doi: 10.1101/gad.1243304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 5.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–7. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 6.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan HE, Lo J, Johnson RS. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest. 1999;103:691–96. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, et al. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–27. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, et al. Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–10. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 11.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, et al. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–95. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 12.Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA. 1996;93:9493–98. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–97. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 14.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol Cell Biol. 2002;22:7405–16. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 17.Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593–612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 18.Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4:423–36. doi: 10.1158/1541-7786.MCR-06-0150. [DOI] [PubMed] [Google Scholar]
- 19.Averous J, Proud CG. When translation meets transformation: the mTOR story. Oncogene. 2006;25:6423–35. doi: 10.1038/sj.onc.1209887. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–31. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999;277:E1–10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 22.Arsham AM, Howell JJ, Simon MC. A novel HIF-independent hypoxic response regulating mTOR and its targets. J Biol Chem. 2003;278:29655–60. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 23.Brugarolas J, Kaelin WG., Jr Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell. 2004;6:7–10. doi: 10.1016/j.ccr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 24.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–92. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, et al. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–85. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–58. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 28.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–49. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 29.Magagnin MG, Koritzinsky M, Wouters BG. Patterns of tumor oxygenation and their influence on the cellular hypoxic response and hypoxia-directed therapies. Drug Resist Updat. 2006;9:185–97. doi: 10.1016/j.drup.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25:1114–25. doi: 10.1038/sj.emboj.7600998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blais JD, Filipenko V, Bi M, Harding HP, Ron D, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469–82. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wouters BG, van den Beucken T, Magagnin MG, Koritzinsky M, Fels D, Koumenis C. Control of the hypoxic response through regulation of mRNA translation. Semin Cell Dev Biol. 2005;16:487–501. doi: 10.1016/j.semcdb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006;26:3955–65. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 35.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–81. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–47. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 37.Sambandam N, Lopaschuk GD. AMP-activated protein kinase (AMPK) control of fatty acid and glucose metabolism in the ischemic heart. Prog Lipid Res. 2003;42:238–56. doi: 10.1016/s0163-7827(02)00065-6. [DOI] [PubMed] [Google Scholar]
- 38.Hossmann KA. Disturbances of cerebral protein synthesis and ischemic cell death. Prog Brain Res. 1993;96:161–77. doi: 10.1016/s0079-6123(08)63265-3. [DOI] [PubMed] [Google Scholar]
- 39.DeGracia DJ. Acute and persistent protein synthesis inhibition following cerebral reperfusion. J Neurosci Res. 2004;77:771–76. doi: 10.1002/jnr.20225. [DOI] [PubMed] [Google Scholar]
- 40.Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–96. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- 41.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100:474–88. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 42.Hedhli N, Pelat M, Depre C. Protein turnover in cardiac cell growth and survival. Cardiovasc Res. 2005;68:186–96. doi: 10.1016/j.cardiores.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 43.Force T, Kuida K, Namchuk M, Parang K, Kyriakis JM. Inhibitors of protein kinase signaling pathways: emerging therapies for cardiovascular disease. Circulation. 2004;109:1196–205. doi: 10.1161/01.CIR.0000118538.21306.A9. [DOI] [PubMed] [Google Scholar]
- 44.Kapp LD, Lorsch JR. The molecular mechanics of eukaryotic translation. Annu Rev Biochem. 2004;73:657–704. doi: 10.1146/annurev.biochem.73.030403.080419. [DOI] [PubMed] [Google Scholar]
- 45.Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- 46.Mothe-Satney I, Yang D, Fadden P, Haystead TA, Lawrence JC., Jr Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol. 2000;20:3558–67. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–71. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 48.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 49.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 50.Lee YM, Jeong CH, Koo SY, Son MJ, Song HS, et al. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. 2001;220:175–86. doi: 10.1002/1097-0177(20010201)220:2<175::AID-DVDY1101>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 51.Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–37. [PubMed] [Google Scholar]
- 52.Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. F Clin Invest. 2003;112:126–35. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenie sympathoadrenal differentiation by isolated neural crest stem cells. F Neurosci. 2000;20:7370–76. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377–83. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–72. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 56.Ramirez-Bergeron DL, Runge A, Adelman DM, Gohil M, Simon MC. HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev Cell. 2006;11:81–92. doi: 10.1016/j.devcel.2006.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 58.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–76. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 59.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–56. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1α is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–69. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BPl/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBPl/eIF4E. Genes Dev. 2002;16:1472–87. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 65.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 66.Cogo A, Napolitano G, Michoud MC, Barbon DR, Ward M, Martin JG. Effects of hypoxia on rat airway smooth muscle cell proliferation. J Appl Physiol. 2003;94:1403–9. doi: 10.1152/japplphysiol.00363.2002. [DOI] [PubMed] [Google Scholar]
- 67.Das M, Bouchey DM, Moore MJ, Hopkins DC, Nemenoff RA, Stenmark KR. Hypoxia-induced proliferative response of vascular adventitial fibroblasts is dependent on G protein-mediated activation of mitogen-activated protein kinases. J Biol Chem. 2001;276:15631–40. doi: 10.1074/jbc.M010690200. [DOI] [PubMed] [Google Scholar]
- 68.Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16:771–80. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 69.Gerasimovskaya EV, Tucker DA, Stenmark KR. Activation of phosphatidylinositol 3-kinase, Akt, and mammalian target of rapamycin is necessary for hypoxia-induced pulmonary artery adventitial fibroblast proliferation. J Appl Physiol. 2005;98:722–31. doi: 10.1152/japplphysiol.00715.2004. [DOI] [PubMed] [Google Scholar]
- 70.Casey TM, Pakay JL, Guppy M, Arthur PG. Hypoxia causes downregulation and RNA synthesis in noncontracting mammalian cardiomyocytes. Circ Res. 2002;90:777–83. doi: 10.1161/01.res.0000015592.95986.03. [DOI] [PubMed] [Google Scholar]
- 71.Crozier SJ, Zhang X, Wang J, Cheung J, Kimball SR, Jefferson LS. signaling pathways and regulatory mechanisms of mRNA translation following myocardial ischemia-reperfusion. J Appl Physiol. 2006;101:576–82. doi: 10.1152/japplphysiol.01122.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan C, Iacobas DA, Zhou D, Chen Q, Lai JK, et al. Gene expression and phenotypic characterization of mouse heart after chronic constant or intermittent hypoxia. Physiol Genomics. 2005;22:292–307. doi: 10.1152/physiolgenomics.00217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blais JD, Addison CL, Edge R, Falls T, Zhao H, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. 2006;26:9517–32. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koritzinsky M, Seigneuric R, Magagnin MG, van den Beucken T, Lambin P, Wouters BG. The hypoxic proteome is influenced by gene-specific changes in mRNA translational. Radiother Oncol. 2005;76:177–86. doi: 10.1016/j.radonc.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 75.Nordlie MA, Wold LE, Simkhovich BZ, Sesti C, Kloner RA. Molecular aspects of ischemic heart disease: ischemia/reperfusion-induced genetic changes and potential applications of gene and RNA interference therapy. J Cardiovasc Pharmacol Ther. 2006;11:17–30. doi: 10.1177/107424840601100102. [DOI] [PubMed] [Google Scholar]
- 76.Chen H, Shi S, Acosta L, Li W, Lu J, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–31. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nobori K, Ito H, Tamamori-Adachi M, Adachi S, Ono Y, et al. ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: a novel cardioprotective role of ATF3. J Mol Cell Cardiol. 2002;34:1387–97. doi: 10.1006/jmcc.2002.2091. [DOI] [PubMed] [Google Scholar]
- 78.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 79.Park EH, Lee JM, Blais JD, Bell JC, Pelletier J. Internal translation initiation mediated by the angiogenic factor Tie2. J Biol Chem. 2005;280:20945–53. doi: 10.1074/jbc.M412744200. [DOI] [PubMed] [Google Scholar]
- 80.Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, et al. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol Genomics. 2005;21:314–23. doi: 10.1152/physiolgenomics.00185.2004. [DOI] [PubMed] [Google Scholar]
- 81.Bull TM, Coldren CD, Geraci MW, Voelkel NF. Gene expression profiling in pulmonary hypertension. Proc Am Thorac Soc. 2007;4:117–20. doi: 10.1513/pats.200605-128JG. [DOI] [PubMed] [Google Scholar]
- 82.Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–35. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van den Beucken T, Koritzinsky M, Wouters BG. Translational control of gene expression during hypoxia. Cancer Biol Ther. 2006;5:749–55. doi: 10.4161/cbt.5.7.2972. [DOI] [PubMed] [Google Scholar]
- 84.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, et al. Regulated translational initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 85.Park EH, Lee JM, Pelletier J. The Tie2 5′ untranslated region is inhibitory to 5′ end-mediated translation initiation. FEBS Lett. 2006;580:1309–19. doi: 10.1016/j.febslet.2006.01.049. [DOI] [PubMed] [Google Scholar]
- 86.Lang KJ, Kappel A, Goodall GJ. Hypoxia-inducible factor-1α mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell. 2002;13:1792–801. doi: 10.1091/mbc.02-02-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huez I, Bornes S, Bresson D, Creancier L, Prats H. New vascular endothelial growth factor isoform generated by internal ribosome entry site-driven CUG translation initiation. Mol Endocrinol. 2001;15:2197–210. doi: 10.1210/mend.15.12.0738. [DOI] [PubMed] [Google Scholar]
- 88.Bert AG, Grepin R, Vadas MA, Goodall GJ. Assessing IRES activity in the HIF-1α and other cellular 5′ UTRs. RNA. 2006;12:1074–83. doi: 10.1261/rna.2320506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bornes S, Prado-Lourenco L, Bastide A, Zanibellato C, Iacovoni JS, et al. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ Res. 2007;100:305–8. doi: 10.1161/01.RES.0000258873.08041.c9. [DOI] [PubMed] [Google Scholar]
- 90.Kozak M. A second look at cellular mRNA sequences said to function as internal ribosome entry sites. Nucleic Acids Res. 2005;33:6593–602. doi: 10.1093/nar/gki958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, et al. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859–67. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Donker RB, Mouillet JF, Nelson DM, Sadovsky Y. The expression of Argonaute2 and related microRNA biogenesis proteins in normal and hypoxic trophoblasts. Mol Hum Reprod. 2007;13:273–79. doi: 10.1093/molehr/gam006. [DOI] [PubMed] [Google Scholar]
- 93.Das DK, Maulik N, Moraru II. Gene expression in acute myocardial stress. Induction by hypoxia, ischemia, reperfusion, hyperthermia and oxidative stress. J Mol Cell Cardiol. 1995;27:181–93. doi: 10.1016/s0022-2828(08)80017-x. [DOI] [PubMed] [Google Scholar]
- 94.Hampton T. Search is on for cells that repair heart. JAMA. 2006;296:2541. doi: 10.1001/jama.296.21.2541. [DOI] [PubMed] [Google Scholar]