

Abstract
The metabotropic glutamate receptor subtype 5 (mGluR5) has been implicated in numerous neuropsychiatric disorders including addiction. We have discovered that the rigid diaryl alkyne template, derived from the potent and selective noncompetitive mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP), can serve to guide the design of novel quinoline analogues and pharmacophore optimization has resulted in potent mGluR5 noncompetitive antagonists (EC50 range 60–100 nM) in the quinoline series.
Introduction
Glutamate is the most abundant excitatory neurotransmitter in the brain. It regulates the body homeostasis through ionotropic (iGluRs) and metabotropic glutamate receptors (mGluRs). There are at least eight subtypes of the mGluRs, which have been identified and partitioned into three groups based on their localization, function, signaling pathway, and structural homology. mGluR5 belongs to group I, along with mGluR1. Accumulating evidence suggests that mGluR5 antagonists have therapeutic potential in the treatment of anxiety, depression, pain, gastro-esophageal acid reflux disease (GERD), Parkinson’s disease, epilepsy, and Fragile X Syndrome (FXS).1 To date, several mGluR5 antagonists are being tested in clinical trials for GERD, pain, and FXS.2 In addition, mGluR5s have been implicated in drug abuse.3–6
mGluR5s are located on postsynaptic glutamatergic synapses of the limbic cortex, hippocampus, amygdala, and basal ganglia (including nucleus accumbens, striatum and olfactory tubercle).1 The mGluR5 functions as a dimer, coupled to phospholipase C through Gq, and modulates the phosphatidylinositol signaling pathway. Activation of the mGluR5 increases cytosolic calcium concentrations, which initiates other signaling pathways.7
As a member of the G-protein coupled receptor (GPCR) family C, mGluR5 has a seven-transmembrane alpha-helical domain (7TM) and a large “bilobed” N-terminal domain, which contains the orthosteric binding site.8 Competitive antagonists binding to the orthosteric site have at least two disadvantages including low brain penetration and low selectivity across the different subtypes.1, 9
MPEP (2-methyl-6-(phenylethynyl)pyridine) and MTEP (3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine) are the two prototypic noncompetitive mGluR5 antagonists, which bind to the allosteric binding site located in the 7TM region.10 They are potent and selective over other mGluR subtypes.1 However, off-target actions (e.g., MPEP also acts as an inhibitor of NMDA receptor and a positive modulator of mGluR4, while MTEP is an inhibitor of cytochrome P450)2 and potential for rapid metabolic degradation (e.g., MTEP)2 have led to significant synthetic efforts to modify and improve the pharmacological and drug-like profile of these parent drugs.2, 7, 11–15 One approach has been to replace the ethynylpyridine moiety of MPEP (or ethynylthiazole moiety of MTEP) with a quinoline (or benzothiazole) structure,16 toward the discovery of new mGluR5 antagonists with a novel structural template.
Previous structure-activity relationship (SAR) studies exemplified the challenge of optimizing the mGluR5 allosteric antagonists with a parent structure that differs from the diaryl alkynes, as binding affinities to the allosteric site are sensitive to small structural changes.16–19 Typically, chemical modification of the diarylalkynyl analogues of MPEP or MTEP are better tolerated at mGluR5 than alternative templates.1, 2, 7, 19–21
In our previous SAR studies of quinoline and benzothiazole analogues, compounds 3 and 4 were discovered to bind with moderate affinity to mGluR5 by introducing the 3-cyano group into the phenyl rings at the 7-position of the quinoline or the 5-position of the benzothiazole.16 The addition of a cyano group improved the binding affinity of 3 to 110 nM from the parent compound that only displaced [3H]MPEP by 50% at 10 M.17 Milbank, et al. also published this compound in their series of quinoline analogues and showed that the addition of a 5-fluoro substitution (shown as structure 5 in Figure 1) further increased the potency ~10 fold.14 Recently, we discovered that addition of a cyano and/or fluoro group in a series of MPEP and MTEP analogues (e.g., 6 and 7 in Figure 1) also resulted in an increase in potency.19 Thus, our strategy was to use SAR derived from the MPEP and MTEP analogues to direct the design and synthesis of quinoline and benzothiazole analogues.
Figure 1.

mGluR5 antagonist structural templates.
Among our previous SAR results, we demonstrated that an additional aryl ring appended to the 4′-position of ring ‘b’ was well tolerated, shown as structure 8a–8d in Figure 1.19 Hence, using 3 and 4 as the parent structures, we incorporated additional aryl ring modifications, and designed a novel series of analogues as shown in Structure I.
Chemistry
One of the synthetic strategies toward the designed quinolines (in Structure I) is shown in Scheme 1, starting from the boronic ester 9a, which was made from commercially available 7-chloro-2-methylquinoline, under Miyaura borylation conditions.16 5-Bromo-2-hydroxybenzonitrile was coupled with 9a under modified Suzuki coupling conditions to give 10, and the free hydroxyl group of 10 was converted to the corresponding triflate 12 with trifluoromethanesulfonic anhydride in an overall 17% yield. Attempts to introduce the aryl ring by treating the triflate 12 with various substituted aryl boronic acids was undertaken using Suzuki coupling conditions, but this strategy proved to be difficult as described below. The same strategy was also applied to the benzothiazole compounds starting from 9b. This strategy was hampered by low yields in steps a to c. To improve the yield of the phenolic intermediates (10 and 11), a protection-deprotection strategy was applied as shown in Scheme 2. However, deprotection of the MOM group in compound 16, or the benzyl group in compound 17 and 18, were either low yielding or incomplete. Hence, higher yields were obtained via the direct coupling condition (step a in Scheme 1) with 5-bromo-2-hydroxybenzonitrile, on a small scale (< 2 mmol). The triflates 12 and 13, from the phenols (step b in Scheme 1) also proceeded in low yield. Triflates 12 and 13 were also easily hydrolyzed under the coupling condition of step c in Scheme 1, especially when these reactions were scaled up (>2 mmol). Utilization of a weaker base such as KF was also tried, but it did not improve the yield. Hence, only compound 14 was synthesized using this strategy.
Scheme 1a.

a Reagents and conditions: (a) Na2CO3, Pd(OAc)2, dioxane, H2O, 50 °C, overnight; (b) Trifluoromethanesulfonic anhydride, pyridine, CH2Cl2, RT, 2–5 h; (c) K3PO4, Pd(PPh3)4, Dioxane, 85 °C, overnight.
Scheme 2a.

a Reagents and conditions: (a) Na2CO3, Pd(PPh3)4, DME, H2O, 80 °C, overnight; (b) CF3COOH, CH2Cl2, RT, 3 h; (c) H2, 40 psi, Pd/C, ethanol, overnight.
Although the strategy in Scheme 2 was not successful toward the synthesis of the designed final products, several 7-substituted quinoline and 5-substituted benzothiazole analogues were obtained using this synthesis, and evaluated for mGluR5 activity.
The targeted compounds with Structure I were successfully synthesized by the strategy shown in Scheme 3. Selective bromination of 3-chlorobenzonitrile by 1,3-dibromo-5,5-dimethylhydantoin (DBH) gave 2-bromo-5-chlorobenzonitrile 19 as reported previously.22 Compound 19 was treated with various substituted aryl boronic acids under the Suzuki coupling condition to give a set of intermediates 20a-c, and final product 21. The desired compounds 22 to 26 were then synthesized by Suzuki-Miyaura coupling with quinoline boronic ester 9a or benzothiazole boron ester 9b, which were made according to our previously reported procedure.16
Scheme 3a.

a Reagents and conditions: (a) DBH, H2SO4, TFA;22 (b) Na2CO3, Pd(PPh3)4, DME, H2O, 70 °C, overnight; (c) K3PO4, Pd(OAc)2, ligand 1L, dioxane, H2O, 105 °C, 16–20 h
These novel quinoliness and benzothiazoles, compand 14 and 22–26, were assessed for mGluR5 activity in a binding assay and in a functional assay measuring mGluR5 activity via calcium mobilization.19 The resulting SAR led us to the synthesis of 7-pyridylquinolines, such as compound 32. The details will be discussed in the SAR section. At the same time, alkyne 28 and quinoline 3014 were synthesized for comparison.
Alkyne 28 was synthesized following the procedure in Scheme 4, in which, synthon 27 was prepared according to a literature procedure.21 Compound 27 was coupled with 5-bromonicotinonitrile under the Sonogashira coupling condition to give the target alkyne 28.
Scheme 4a.

a Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI, NEt3, RT, overnight;21 (b) Pd(PPh3)4, CuI, Et3N, DMF, TBAF, 70 °C.
The corresponding quinolines were synthesized according to Scheme 5, starting from either the commercially available 5-bromonicotinonitrile 29a or 5-bromo-2-chloronicotinonitrile 29b, which was made from a literature procedure.23 The Suzuki-Miyaura coupling reactions with boronic ester 9a gave the desired compounds 30 and 31. A Suzuki coupling reaction of 31 gave compound 32.
Scheme 5a.

a Reagents and conditions: (a) POCl3, PCl5, reflux overnight;23 (b) Na2CO3, PdCl2(dppf), dioxane, H2O, MW, 140 °C, 30 min; (c) K3PO4, Pd(OAc)2, ligand 1L, dioxane, H2O, MW 140 °C, 90 min.
All the compounds synthesized were purified by flash column chromatography, analytically characterized as the free bases, and then converted to the HBr salts for biological testing, unless otherwise described in the experimental methods.
In Vitro Pharmacology
All the compounds synthesized were assessed in a radioligand displacement binding assay for mGluR5, using [3H]MPEP as the radioligand in rat brain membranes or HEK293-T cells transfected with cloned rat mGluR5 cDNA (National Institute of Mental Health s Psychoactive Drug Screening Program).24 An assay utilizing calcium fluorescence was employed to test functional activity of compounds by measuring receptor-induced intracellular release of calcium with a kinetic imaging plate reader that makes simultaneous measurements of calcium levels in each well of a 384-well plate.19 The results of these in vitro tests for the designed quinolines, benzothiazoles and related MPEP or MTEP analogues are listed in Table 1.
Table 1.
In Vitro Data for Quinolines, Benzothiazoles and MPEP or MTEP-like Alkynyl mGluR5 Antagonistsa
 | ||||||
|---|---|---|---|---|---|---|
| Compound ID | template | Y | R | mGluR5 binding affinity Ki±SEM(nM) | mGluR5 function(Ca+2 flux) IC50±SEM(nM) | clogPb |
| 1, MPEP | A | CH | H, 3-Hc | 13±1d | 3.54±1.39d | 3.8 |
| 2, MTEP | C | N | H, 3-Hc | 16e | 13.6±2.09d | 2.1 |
| 3 | B | CH | H | 110±20f | 29±5f | 3.9 |
| 4 | D | H | 2100±580f | NT | 3.9 | |
| 6 | A | CH | H | 1.3±0.09d | 0.415±0.10d | 3.2 |
| 7 | C | CH | 3-CN-5-F | 0.9±0.2d | 0.813±0.11d | 3.2 |
| 8a | A | CH | Ph | 4.0±0.6d | 3.08±0.61d | 5.1 |
| 8b | A | CH | 4 -F-Ph | 3.0±0.5d | 7.19±1.53d | 5.3 |
| 8c | C | N | 4-Ph | 5.49±1.43d | 1.21± 1.15d | 4.2 |
| 8d | C | CH | 3-F-4-3 -Py | 11.4±3d | 3.43±0.51d | 3.0 |
| 10 | B | CH | 4-OH | >10,000 | >10,000 | 3.9 |
| 13 | D | OTf | 862±200 | 7840±1250 | 5.4 | |
| 14 | D | 4-F-Ph | 4679±1185 | >10,000 | 5.9 | |
| 16 | B | CH | 4-OCH2OCH3 | 596±109 | 294+21 | 3.6 |
| 17 | B | CH | 4-OBz | 1040±206 | >10,000 | 5.9 |
| 18 | D | OBz | 7134±2026 | >10,000 | 5.9 | |
| 21 | B | CH | 3-Hc-2-CN-4-Cl | 4720±913 | 9150±1540 | 4.6 |
| 22 | B | CH | 4-Ph | 97±20 | 1250±261 | 5.7 |
| 23 | B | CH | 4-4 -F-Ph | 64±16 | 692±64 | 5.9 |
| 24 | B | CH | 4-3 -Py | 730±190 | 1340±41 | 4.3 |
| 25 | D | Ph | 483±98 | 6890±1250 | 5.8 | |
| 26 | D | 3-Py | >10,000 | >10,000 | 4.3 | |
| 28 | A | N | H | 1.5±0.3 | 13±2.3 | 1.9 |
| 30g | B | N | H | 100±21 | 68±5.3 | 2.5 |
| 31h | B | N | 4-Cl | >10,000 | 3310±384 | 3.2 |
| 32 | B | N | 4-Ph | 97±19 | 81±8.1 | 4.7 |
Methods for binding (http://pdsp.med.unc.edu/pdspw/binding.php) and functional assays19 have been previously published;
Determined with ChemDraw Ultra 10.0;
3-H, no CN group on the 3-position of structure;
Data reported previously in literature.19;
Data reported previously in literature.25;
Data reported previously in literature.16;
Compound reported previously in literature14;
Partial antagonist; NT = not tested.
All the compounds were full antagonists, thereby inducing a complete inhibition of glutamate-induced mGluR5 activity, with the exception of 31, which exhibited partial antagonist activity reaching only 50% inhibition of the EC80 glutamate response (Figure 2)
Figure 2.

Compound 31 as a partial antagonist
All alkyne, quinoline and benzothiazole analogues were functionally inactive (<50% inhibition at 10 μM) at all the other mGluR subtypes and did not bind to NMDA receptors at a concentration of 10 μM.24
Structure-Activity Relationships
For the MPEP analogues (template A) in Table 1, the aryl ring systems introduced into the 4-position of ring b, were well tolerated (e.g., phenyl group in 8a (Ki = 4 nM) and 4-fluorophenyl group in 8b (Ki = 3 nM) as compared to their parent structure 6 (Ki = 1.3 nM).19 As predicted, the same modification on the ring ‘b’ system of the quinoline analogues (template B), 22 (Ki = 97 nM) and 23 (Ki = 64 nM) showed comparably high binding affinities to their parent structure 3 (Ki = 110 nM), which demonstrated the same SAR trend as the MPEP analogues.
In general, mGluR5 binding affinity (Ki) values were comparable to functional potency (IC50) values in the calcium fluorescence assay. However, in the case of these two quinolines, 22 and 23, their functional potencies were lower than expected (e.g., 22 (IC50 = 1250 nM) and 23 (IC50 = 692 nM) v. 3 (IC50 = 29 nM)). Considering this and their high cLogP values (e.g., 22, cLogP =5.7 and 23, cLogP = 5.9), we reasoned that addition of the third aromatic ring system may be problematic for future in vivo investigation. Hence, we introduced the heteroatom “N” into the ring systems of our target compound, including the 3rd additional aryl ring and ring ‘b’, to give compounds 24, 30–32. This modification lowered the cLogP values below 5. The corresponding alkyne was also synthesized for comparison (e.g., 28 v. 6).
The cLogP value of alkyne 28 was decreased to 1.9; high binding affinity (Ki = 1.5 nM) and functional activity (IC50 = 13 nM) were retained. The same trend was observed in quinoline 30 (Ki = 100 nM, IC50 = 68 nM) compared to its parent compound 3 (Ki = 110 nM, IC50 = 29 nM). Importantly, the additional heteroaryl ring was well tolerated (32 v. 30) in both binding affinity (32 (Ki = 97 nM) v. 30 (Ki = 100 nM)), and functional activity (32 (IC50 = 81 nM) v. 30 (IC50 = 68 nM)).
On the other hand, the relatively low binding affinity of 24 implied that the “N” introduced to the third ring system was detrimental to the binding affinity (Ki = 730 nM) and the functional activity (IC50 = 1340 nM), although its cLogP value is comparable to compound 32 (4.3 v. 4.7). This suggested the 4–3 -pyridyl substituent was not favored for the template B structure.
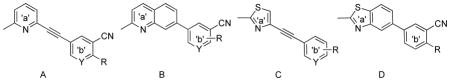
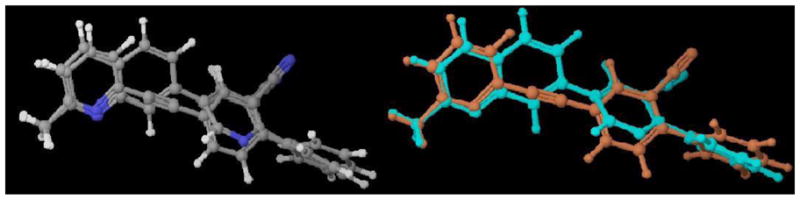
3D-superimposition comparison of 8a and 32, shown in Figure 3,26 demonstrated that with the additional phenyl ring, alkyne 8a and quinoline 32 aligned quite well. Conversely, 3D-superimposition of 8d and 26, showed that the benzothiazole 26 did not align well with alkyne 8d (Figure 4),26 which may explain why the benzothiazoles (template D) did not have comparable binding affinities as seen in the quinoline series. However, although they only showed moderate potencies, improved tolerability for the additional ring system was observed for compound 25 (Ki = 483 nM), which showed ~4-fold increase in binding affinity compared to its parent structure 4 (Ki = 2100 nM). This corresponds to the trend observed in the alkyne series (template C) 8c (Ki = 5.49 nM) v. 2, MTEP (Ki = 16 nM).19 Other aryl-substituted benzothiazoles, such as 14, only showed low binding affinity (Ki = 4679 nM) and compound 26 was completely inactive at mGluR5.
Figure 3. 3D-superimposition of 8a (alkyne) and 32 (quinoline).26.
Note: Compound 32 is shown in turquoise blue and 8a in orange on right side for clarity.
Figure 4. 3D-superimposition of 8d (alkyne) and 26 (benzothiazole).26.
Note: Compound 8d is shown in turquoise blue and 26 in orange on right side for clarity.
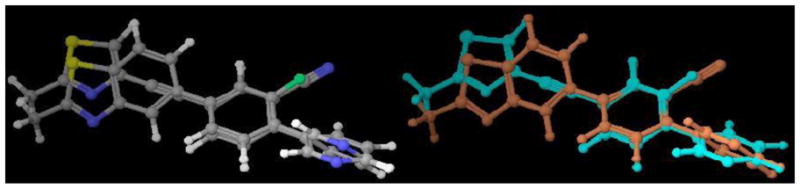
The SAR results of other 7-aryl-substituted quinoline analogues with structural template B also provided additional SAR for mGluR5. In Figure 5,26 the small substitutions on the 4-position of ring b in template B (e.g., OH in compound 10 and Cl in compound 31) were not favored for binding or functional potency. This also corresponded to our previously reported results.16 Compound 31 was observed to be a weak partial antagonist. Similar conversions of functional efficacy from mGluR5 antagonists to partial antagonists, or even to the positive allosteric modulators has been reported recently.2, 27, 28 Hence, we provide additional evidence suggesting that mGluR5 intrinsic activities are very sensitive to small structural modifications.
Figure 5.
3D-superimposition of quinoline analogues.26
The flexibility of this 4-position on ring ‘b’ in template B is also limited to the size of the substitution. For example, compound 17 extends the phenyl ring away from the quinoline, which decreased binding affinity (Ki =1040 nM) ~10 fold compared to compound 22 (Ki = 97 nM). A similar 15-fold decrease in potency was also observed in the 6-aryl substituted benzothiazole (18 (Ki = 7134 nM) v. 25 (Ki = 483nM)) in template D.
In addition to these in vitro pharmacological results, computational toxicology analyses29 predicted that these quinoline compounds would not show toxicity, adding promise to the development of quinoline compounds as mGluR5 antagonist.
Summary
A series of quinoline and benzothiazole analogues of the parent compounds 3 and 4 were synthesized as selective mGluR5 antagonists using the SAR results we obtained for MPEP and MTEP analogues.19 As hypothesized, the mGluR5 binding affinity and functional results of these analogues demonstrated overlapping SAR between the quinolines and their corresponding MPEP analogues. The additional ring modifications on ring ‘b’ were tolerated in both alkyne and quinoline structures, with the alkynes showing higher potencies, uniformly, than the quinolines. This suggests that the SAR results of MPEP analogues can be used to direct the further investigation of novel quinoline analogues. In vivo evaluation of the lead compounds herein will provide valuable direction toward future drug design.
Experimental Methods
Reaction conditions and yields were not optimized, and spectroscopic data refer to the free base unless otherwise described in each compound. Microwave reactions were performed using a CEM Corp. (Matthews, NC) Discover LabMate system using the standard 10 mL reaction vessel. Flash chromatography was performed using silica gel (EMD Chemicals, Inc.; 230–400 mesh, 60 Å). 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer. Chemical shifts are reported in parts-per-million (ppm) and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26; (CD3)2SO, 2.50), 13C spectra (CDCl3, 77.2; (CD3)2SO, 39.5), 19F spectra (CFCl3, 0). Infrared spectra were recorded as a KBr pellet using a Perkin-Elmer Spectrum RZ I FT-IR spectrometer or recorded as powder using an Avatar 370 FT-IR thermo Nicolet spectrometer. Gas chromatography-mass spectrometry (GC/MS) data were acquired using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μM film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and agrees within 0.5% of calculated values. Melting point determination was conducted using a Thomas-Hoover melting point apparatus and are uncorrected. Anhydrous solvents were purchased from Aldrich or J. T. Baker and were used without further purification, except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Aldrich Chemical Co., Combi-Blocks, TCI, America., Matrix Scientific, Lancaster Synthesis, Inc. (Alfa Aesar) and AK Scientific, Inc. The final products were converted into HBr salts, typically by treating the free base with methanolic HBr followed by precipitation from a combination of organic solvents. On the basis of NMR, GC-MS, and combustion data, all final compounds are >95% pure. Methods for binding affinity and functional assay for mGluR5 have been previously reported.19
General Procedure A: Coupling of heteroaryl bromides with boronic ester or acid
To a suspension of boronic ester (0.6 equiv.) or boronic acid (1.2 equiv.), heteroaryl bromide (1 equiv.), and Na2CO3 or KF (3 equiv.) in a mixture of solvents DME/H2O (3/1, 4 mL for 1 mmol scale reaction) was added Pd(PPh3)4 (5 mol%) under Argon. The mixture was warmed to 70–80 °C for 3 h to overnight with TLC monitoring. The solvent was then removed under reduced pressure, and the residue was extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated to crude product, which was then purified by flash column chromatography to give the pure product.
General Procedure B: Coupling of heteroaryl chlorides with boronic ester or acid
To a suspension of boronic ester (1 equiv.) or boronic acid (1.2 equiv.), heteroaryl chloride (1 equiv.), biphenyl phosphine ligand (1L, 4 mol %) and K3PO4 (3 equiv.) in a solvent mixture of dioxane/H2O (10/1, 4 mL for 1 mmol scale reaction) was added Pd(OAc)2 (2 mol %) under Argon. The mixture was warmed to 105 °C and kept stirring overnight. Solvents were then removed under reduced pressure, and the residue was extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated to dryness. The crude product was purified by flash column chromatography to give the pure product.
2-Hydroxy-5-(2-methylquinolin-7-yl)benzonitrile (10)
A suspension of 5-bromo-2-hydroxybenzonitrile (0.24 g, 1.2 mmol), 9a (0.3 g, 1.1 mmol), Na2CO3 (0.32 g, 3 mmol), and Pd(OAc)2 (0.011 g, 5%) in a mixture of dioxane (4 mL) and water (0.4 mL) was degassed and heated at 50 °C overnight, under Argon protection. Solvent was removed under vacuum. The residue was dissolved in methanol and filtered. The filtrate was concentrated and purified by a flash chromatography eluting with EtOAc to provide the solid product (0.19 g) in 60% yield; 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H), 8.10 (s, 1H), 7.98 (m, 2H), 7.84 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 2.65 (s, 3H); 13C NMR (100MHz, DMSO-d6) δ 160.6, 160.2, 151.8, 136.7, 134.1, 132.2, 129.2, 126.0, 125.3, 125.3, 122.8, 117.6, 110.0, 100.4, 25.3; HBr salt precipitated from MeOH; mp 225–226 °C; IR (powder) 3463, 2226 cm−1; Anal. (C17H12N2O·HBr·1/2 H2O) C, H, N.
2-Hydroxy-5-(2-methylbenzo[d]thiazol-5-yl)benzonitrile (11)
A suspension of 5-bromo-2-hydroxybenzonitrile (0.24 g, 1.2 mmol), 9b (0.3 g, 1 mmol), Na2CO3 (0.32 g, 3 mmol), and Pd(OAc)2 (0.011 g, 5%) in a mixture of dioxane (4 mL) and water (0.4 mL) was degassed and heated at 50 °C overnight, under Argon protection. Solvent was removed under vacuum. The residue was dissolved in methanol and filtered. The filtrate was concentrated and purified using a flash chromatography eluting with EtOAc to provide the solid product (0.11 g) in 40% yield; mp 195–197 °C (dec); 1H NMR (400 MHz, DMSO-d6) δ 11.2 (br, 1H), 8.15 (s, 1H), 8.06 (d, J = 8.4 Hz, 1H), 8.01 (s, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 2.80 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.7, 160.4, 154.4, 137.2, 134.8, 134.0, 132.0, 131.9, 123.9, 123.1, 120.0, 117.6, 117.5, 100.2, 20.5; IR (powder) 3096, 2232 cm−1; Anal. (C15H10N2OS·5/8 H2O) C, H, N.
2-Cyano-4-(2-methylquinolin-7-yl)phenyl triflate (12)
To a suspension of 10 (0.6 g, 1.7 mmol) in 8 mL CH2Cl2 on an ice bath, pyridine (0.6 mL, 7.4 mmol) and trifloromethanesulfonic anhydride (0.6 mL, 3.4 mmol) were added successively. The reaction mixture was stirred at RT for 2 h. The reaction mixture was extracted with CH2Cl2. The organic layer was concentrated and purified by flash chromatography eluting with hexane/EtOAc (4:1) to give the product as syrup (0.2 g) in 30% yield. 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 8.10 (d, J = 8.8 Hz, 1H), 8.10 (s, 1H), 8.05 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 8.8Hz, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 2.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.1, 159.5, 149.2, 145.5, 134.8, 133.5, 129.5, 129.2, 126.8, 126.5, 123.8, 123.6, 123.5, 109.8, 29.5; GC-MS (EI) m/z 392 (M+)
2-Cyano-4-(2-methylbenzo[d]thiazol-5-yl)phenyl triflate (13)
To a solution of 11 (0.4 g, 1.5 mmol) in 20 mL CH2Cl2 on an ice bath, pyridine (0.3 mL, 3.7 mmol) and trifloromethanesulfonic anhydride (0.3 mL, 1.7 mmol) were added successively. The reaction mixture was stirred at RT for 5 h and then extracted with CH2Cl2. The organic layer was concentrated and purified by flash chromatography eluting with hexane/EtOAc (4:1) to give the product as a solid (0.1 g) in 18% yield; mp 87–89 °C (dec); 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 1.6 Hz, 1H), 8.00 (d, J = 2.0 Hz, 1H), 7.96 (s, 1H), 7.94 (s, 1H); 7.59 (d, J = 8.4 Hz, 1H), 7.53 (dd, J = 8.4, 2.0 Hz, 1H), 2.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.2, 163.8, 159.1, 149.8, 143.8, 142.3, 138.4, 135.5, 133.5, 133.0, 123.9, 123.4, 122.6, 121.1, 20.28; 19F NMR (376 MHz, CDCl3) δ -72.9; IR (powder) 2232 cm−1; GC-MS (EI) m/z 398 (M+); Anal. (C16H9F3N2O3S2) C, H, N.
2-(4-Fluorophenyl)-4-(2-methylbenzo[d]thiazol-5-yl)benzonitrile (14)
A suspension of 13 (0.4 g, 0.75 mmol), 4-fluorophenylboronic acid (0.135 g, 1 mmol), K3PO4 (0.64 g, 3 mmol), and Pd(PPh3)4 (0.06 g, 0.05 mmol) in dioxane (5 mL) was degassed and heated at 85 °C overnight, under Argon protection. Solvent was removed under vacuum. The residue was extracted with CH2Cl2 and washed with brine. The organic layer was dried over magnesium sulfate and concentrated to give a crude product. It was purified by flash chromatography eluting with hexane/EtOAc (6:1, 4:1) to give the product (50 mg) in 25% yield; mp 101–103 °C; 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.04 (s, 1H), 7.93 (dd, J = 8.4, 8.8 Hz, 2H), 7.60 (m, 4H), 7.22 (dd, J = 8.4, 8.8 Hz, 2H), 2.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.7, 154.4, 143.4, 140.8, 136.9, 136.1, 132.6, 131.9, 130.9, 130.8, 130.8, 123.9, 122.4, 120.9, 118.8, 116.2, 116.0, 112.1, 20.5; 19F NMR (376 MHz, CDCl3) δ -113.3; IR (powder) 2232 cm−1; GC-MS (EI) m/z 44 (M+); Anal. (C21H13FN2S) C, H, N. HBr salt precipitated from MeOH. Anal.(C21H13FN2S·HBr) C, H, N.
2-Benzoxy-5-bromobenzonitrile (15a)
A suspension of 5-bromo-2-hydroxybenzonitrile (10 g, 50 mmol), benzyl bromide (6 mL, 50 mmol), and K2CO3 (7.6 g, 55 mmol) in acetone (100 mL) was refluxed for 5 h. The reaction mixture was filtered. The filtrate was concentrated and solidified to give crude product. It was washed with hexane to give the pure product (12 g) in 83% yield; mp 74–76 °C. 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.57 (d, J = 9.2 Hz, 1H), 7.44–7.33 (m, 5H), 6.88 (d, J = 9.2 Hz, 1H) 5.21 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 159.6, 137.3, 136.2, 135.3, 129.3, 29.1, 128.7, 128.1, 127.2, 115.2, 114.9, 112.9, 104.5, 71.2,; IR (powder) 2232, 1135 cm−1; GC-MS (EI) m/z 287, 289 (M+).
5-Bromo-2-methoxymethoxylbenzonitrile (15b)
To a suspension of 5-bromo-2-hydroxybenzonitrile (1 g, 5 mmol) and K2CO3 (0.76 g, 5.5 mmol) in DMF (10 mL) was added methoxymethyl chloride (4.18 mL, 5.5 mmol). The reaction mixture was kept at RT overnight and extracted with ether. The organic layer was concentrated to give the product (0.93 g) in 77% yield; mp 65–67 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (m, 1H), 7.65 (m, 1H), 7.16 (m, 1H), 5.29 (s, 2 H), 3.53 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.5, 137.5, 135.9, 116.9, 115.1, 113.9, 104.9, 95.2, 57.0; IR (powder) 2236, 1129 cm−1; GC-MS (EI) m/z 241, 243 (M+).
2-Methoxymethoxy-5-(2-methylquinolin-7-yl)benzonitrile (16)
Prepared by following the general procedure A using 9a (0.27 g, 1 mmol) and 15b (0.24 g, 1 mmol), eluting with hexane/EtOAc (4:1) to give the product (0.18 g) in 59% yield; mp 97–99 °C; 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 2.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 2.4 Hz, 1H), 7.91 (dd, J = 8.8, 2.4 Hz, 1H), 7.88 (d, J = 8.8 Hz, 1H), 7.67 (dd, J = 8.4, 2.0 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 5.35 (s, 2H), 3.57 (s, 3H), 2.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.2, 158.9, 148.3, 139.6, 136.1, 134.8, 133.4, 132.4, 128.6, 126.3, 126.0, 124.7, 122.6, 115.7, 103.7, 95.1, 57.0, 25.6; IR (powder) 2232, 1258 cm−1; GC-MS (EI) m/z 304 (M+); Anal. (C19H16N2O2·1/4 H2O) C, H, N.
2-Benzoxy-5-(2-methylquinolin-7-yl)benzonitrile (17)
Prepared by following the general procedure A using 9a (0.27 g, 1 mmol) and 15a (0.35 g, 1.1 mmol), eluting with hexane/EtOAc (4:1) to give the solid product (0.14 g) in 40% yield. HBr salt precipitated from MeOH/Acetone. mp 190–191 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.86 (m, 1H), 8.31 (m, 3H), 8.17 (dd, J = 7.2, 9.2 Hz, 2H), 7.82 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 9.2 Hz, 1H), 7.52 (m, 2H), 7.43 (dd, J = 8.0, 7.2 Hz, 2H), 7.37 (d, J = 6.4 Hz, 1H), 5.37 (s, 2H), 2.84 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 160.6, 159.5, 136.6, 134.6, 133.3, 132.0, 130.2, 129.3, 129.0, 128.3, 127.7, 126.5, 124.1, 116.7, 115.2, 102.6, 71.2, 22.8; IR (powder) 2349, 1135 cm−1; GC-MS (EI) m/z 350 (M+); Anal. (C24H18N2O·HBr·2/3 H2O) C, H, N.
2-Benzoxy-5-(2-methylbenzo[d]thiazol-5-yl)benzonitrile (18)
Prepared by following the general procedure A using 9b (0.48 g, 1.8 mmol) and 15a (0.65 g, 2.7 mmol), eluting with hexane/EtOAc (5:2) to give the solid product (0.6 g) in 94% yield; mp 102–103 °C; 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.89 (s, 1H), 7.86 (d, J = 9.2 Hz, 1H), 7.76 (d, J = 7.2 Hz, 1H), 7.5–7.4 (m, 5H), 7.36 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 9.2 Hz, 1H), 5.29 (s, 2 H), 2.87 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 168.5, 159.9, 154.3, 137.2, 135.8, 135.3, 134.4, 133.3, 132.6, 129.0, 128.5, 127.2, 123.8, 122.2, 120.5, 116.6, 113.7, 103.2, 71.1, 20.5; IR (powder) 2232, 1170 cm−1; GC-MS (EI) m/z 356 (M+); Anal. (C22H16N2OS) C, H, N.
2-Bromo-5-chloro-benzonitrile (19)22
Yield: 4.45 g (76%). 1H NMR (400 MHz, CDCl3) δ7.64 (s, 1H), 7.63 (d, J = 10.8 Hz, 1H), 7.44 (d, J = 10.8 Hz, 1H); GC-MS (EI) m/z 217 (M+).
5-Chloro-2-phenylbenzonitrile (20a)
Prepared by following the general procedure A using 19 (1 g, 5 mmol) and phenyl boronic acid (0.67 g, 5.5 mmol), eluting with hexane/EtOAc (6:1) to give the product (0.52 g) in 50% yield; mp 87–89 °C; 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.48 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 144.2, 137.2, 133.9, 133.4, 131.6, 129.3, 129.1, 128.9, 117.7, 112.9; IR (powder) 2226 cm−1; GC-MS (EI) m/z 213 (M+).
5-Chloro-2-(4-fluorophenyl)benzonitrile (20b)
Prepared by following the general procedure A using 19 (0.54 g, 2.5 mmol) and 4-fluorophenyl boronic acid (0.27 g, 2.7 mmol). The crude product was solidified by adding MeOH and filtered to give the pure product (0.22 g) in 40% yield; mp 101–102 °C; 1H NMR (400 MHz, CDCl3) δ 7.74 (s, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.51 (dd, J = 8.8, 8.8 Hz, 2H), 7.43 (d, J = 8.8 Hz, 1H), 7.19 (dd, J = 8.4, 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 158.3, 139.2, 130.6, 130.5, 130.2, 129.5, 127.6, 126.9, 126.8, 124.4, 119.7, 112.4, 112.2; 19F NMR (376 MHz, CDCl3) δ -112.8; IR (powder) 2226 cm−1; GC-MS (EI) m/z 231 (M+).
5-Chloro-2-(pyridin-3-yl)benzonitrile (20c)
Prepared by following the general procedure A using 19 (0.44 g, 2 mmol) and 3-pyridylboronic acid (0.27 g, 2.2 mmol), eluting with hexane/EtOAc (1:1) to give the product (0.35 g) in 75% yield; mp 137–139 °C. 1H NMR (400 MHz, CDCl3) δ 8.76(s, 1H), 8.73 (d, J = 7.2 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.80 (s, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.48 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 150.4, 149.4, 140.5, 136.2, 135.1, 133.8, 133.7, 133.1, 131.5, 123.7, 117.1, 113.3; IR (powder) 2232 cm−1; GC-MS (EI) m/z 214 (M+).
5-Chloro-2-(2-methylquinolin-7-yl)benzonitrile (21)
Prepared by following the general procedure A using 9a (0.19 g, 0.7 mmol) and 19 (0.2 g, 0.7 mmol), eluting with hexane/EtOAc (4:1) to provide the product (0.1 g) in 36% yield; mp 98–100 °C; 1H NMR (400 MHz, CDCl3 δ 8.15(s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.79 (s, 1H), 7.69 (dd, J = 8.4, 8.8 Hz, 2H), 7.59 (d, J = 8.8 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 2.78 (s, 3H); 13C NMR (100MHz, CDCl3) δ 160.4, 152.7, 147.8, 143.6, 138.2, 136.3, 133.6, 133.7, 131.8, 129.1, 128.4, 126.0, 123.3, 117.5, 113.2, 25.6; IR (powder) 2232 cm−1; GC-MS (EI) m/z 278 (M+); HBr salt precipitated from MeOH/Acetone/ether. Anal. (C17H11ClN2·HBr·1/3H2O) C, H, N.
5-(2-Methylquinolin-7-yl)-2-phenylbenzonitrile (22)
Prepared by following the general procedure B using 9a (0.4 g, 1.5 mmol) and 20a (0.26 g, 1.2 mmol), eluting with hexane/EtOAc (4:1) to provide the product (0.16 g) in 42% yield. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.14 (s, 1H), 8.11 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.67- 7.46 (m, 6H), 7.35 (d, J = 8.8 Hz, 1H), 2.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.05, 148.06, 144.49, 140.06, 135.90, 132.45, 131.64, 128.81, 126.72, 124.54, 122.58, 118.66, 111.94, 24.87. GC-MS (EI) m/z 320 (M+). HBr salt precipitated from MeOH/Acetone. mp 273–274 °C (dec); IR (powder) 2220 cm−1; Anal. (C23H16N2·HBr·5/4 H2O) C, H, N.
5-(2-Methylquinolin-7-yl)-2-(4-fluorophenyl)benzonitrile (23)
Prepared by following the general procedure B using 9a (0.28 g, 1.1 mmol) and 20b (0.27 g, 1 mmol), eluting with hexane/EtOAc (4:1, 2.5:1) to give the product (0.16 g) in 42% yield. HBr salt precipitated from MeOH/Acetone; mp 192–193 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.95 (d, J = 6.4 Hz, 1H), 8.48 (s, 1H), 8.40 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 8.27 (m, 2H), 7.88 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 9.2 Hz, 1H), 7.72 (m, 2H), 7.42 (m, 2H), 2.79 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 165.2, 162.0, 159.8, 144.3, 138.8, 133.3, 132.9, 131.9, 131.8, 131.7, 130.4, 127.0, 124.5, 118.9, 116.7, 116.4, 112.1, 22.8; 19F NMR (376 MHz, DMSO-d6) δ -113.0; IR (powder) 2226 cm−1; GC-MS (EI) m/z 338 (M+); Anal. (C23H15FN2·HBr·7/4 H2O) C, H, N.
5-(2-Methylquinolin-7-yl)-2-(pyridin-3-yl)benzonitrile (24)
Prepared by following the general procedure B using 9a (0.36 g, 1.33 mmol) and 20c (0.28 g, 1.3 mmol), eluting with EtOAc to provide the product (0.14 g) in 33% yield. 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 8.74 (d, J = 6.0 Hz, 1H), 8.30 (s, 1H), 8.18 (s, 1H), 8.11 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 6.0 Hz, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.48 (m, 1H), 7.36 (d, J = 8.4 Hz, 1H), 2.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.18, 149.98, 149.31, 141.06, 138.90, 136.08, 132.65, 131.93, 130.71, 128.64, 126.9, 126.28, 124.45, 123.43, 122.73, 112.26, 25.48; GC-MS (EI) m/z 321 (M+); Anal. (C22H15N3·1/2 H2O) C, H, N. HBr salt precipitated from acetone. mp 171–172 °C; IR (powder) 2220 cm−1; Anal. (C22H15N3·2HBr·3/2 H2O) C, H, N.
2-Phenyl-4-(2-methylbenzo[d]thiazol-5-yl)benzonitrile (25)
Prepared by following the general procedure B using 9b (0.38 g, 1.38 mmol) and 20a (0.26 g, 1.26 mmol), eluting with hexane/EtOAc (4:1) to give the solid product (0.1 g) in 24% yield. 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 8.05 (s, 1H), 7.94 (dd, J = 7.2, 7.2 Hz, 2H), 7.65–7.47 (m, 7H), 2.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.4, 154.2, 140.4, 137.7, 136.8, 132.4, 131.6, 130.7, 128.7, 123.7, 122.1, 120.7, 118.7, 111.9, 20.31; GC-MS (EI) m/z 326 (M+). HBr salt precipitated from MeOH/Acetone; mp 234–235 °C (dec); IR (powder) 2226 cm−1; Anal. (C21H14N2S·HBr) C, H, N.
2-(Pyridin-3-yl)-4-(2-methylbenzo[d]thiazol-5-yl)benzonitrile (26)
Prepared by following the general procedure B using 9b (0.3 g, 1.1 mmol) and 20c (0.21 g, 1 mmol), eluting with EtOAc and MeOH/NH4OH (trace amount) to give the product (0.18 g) in 55% yield. 1H NMR (400 MHz, CDCl3) δ 8.84 (s, 1H), 8.73 (d, J = 6.4 Hz, 1H), 8.19 (d, J = 2.0 Hz, 1H), 8.09 (d, J = 2.0 Hz, 1H), 8.00 (m, 1H), 7.97 (s, 1H), 7.95 (s, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.61 (m, 1H), 7.49 (dd, J = 8.0, 4.8 Hz, 1H), 2.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.5, 154.2, 150.0, 149.3, 141.4, 140.5, 136.1, 133.6, 132.6, 131.9, 130.7, 123.4, 122.2, 120.7, 118.2, 112.2, 20.3; GC-MS (EI) m/z 327 (M+). HBr salt precipitated from MeOH/Acetone. mp 264–266 °C(dec); IR (powder) 2220 cm−1; Anal. (C20H13N3S·2HBr·H2O) C, H, N.
2-Methyl-6-((trimethylsilanyl)ethynyl)pyridine (27)21
Yield: 2.35 g (71%). 1H NMR (400 MHz, CDCl3) δ 7.54 (m, 1H), 7.30 (m, 1H), 7.10 (m, 1H), 2.55 (s, 3H), 0.26 (s, 9H). GC-MS (EI) m/z 189 (M+).
5-((6-Methylpyridin-2-yl)ethynyl)nicotinonitrile (28)
To a suspension of 5-bromonicotinonitrile (0.5 g, 2.5 mmol), 27 (0.5 g, 2.6 mmol), CuI (0.05 g, 0.25 mmol), Et3N (1.47 mL) and Pd(PPh)4) (0.15 g) in degassed DMF (8 mL) was added tributylamonium fluoride (TBAF, 2.64 mL, 2.6 mmol) in THF (1.0 M) dropwise at 55°C for 30 min. The reaction mixture was kept at 70°C overnight, then extracted with ether and washed with brine. The organic layer was dried over magnesium sulfate and concentrated to give the crude product. It was purified by flash chromatography eluting with hexane/EtOAc (1:1) to provide the pure product (0.3 g) in 52% yield; mp 162–164 °C; 1H NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 8.83 (s, 1H), 8.13 (d, J = 2.0 Hz, 1H), 7.64 (m, 1H), 7.41 (d, J = 7.6 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 2.61 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.6, 151.3, 149.0, 144.4, 141.7, 136.9, 133.4, 126.7, 125.1, 124.0, 114.7, 20.59; IR (powder) 2226 cm−1; GC-MS (EI) m/z 219 (M+); Anal. (C14H9N3) C, H, N.
5-Bromo-2-chloronicotinonitrile (29b)23
A solution of 5-bromo-2-hydroxynicotinonitrile (0.2 g, 1 mmol) and PCl5 (0.21 g, 1 mmol) in POCl3 (3 mL) was refluxed overnight. Aqueous NaOH solution (1 M) was added slowly to quench the reaction with an ice bath. Product was extracted with ether. The solvent was removed to give crude product, which was used in the followed reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.12 (s, 1H); GC-MS (EI) m/z 216, 218 (M+).
5-(2-Methylquinolin-7-yl)nicotinonitrile (30)14
A suspension of 5-bromonicotinonitrile (0.09 g, 0.5 mmol), 9a (0.2 g, 0.5 mmol), Na2CO3 (0.16 g, 1.5 mmol), and PdCl2(dppf) (0.02 g) in DME (4.5 mL) and H2O (0.75 mL) was degassed and reacted under microwave condition at 140 °C for 30 min. Solvent was removed under vacuum. The residue was extracted with EtOAc and washed with brine. The organic layer was dried over magnesium sulfate and evaporated to give the crude product. It was purified by flash chromatography eluting with hexane/EtOAc (1:1) to give the product (0.1 g) in 80% yield; mp 195–196 °C; 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H), 8.91 (s, 1H), 8.28 (d, J = 2.0 Hz, 1H), 8.26 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.38 (dd, J = 8.4, 2.0 Hz, 1H), 2.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.1, 160.5, 151.8, 151.2, 137.5, 136.3, 129.1, 127.9, 124.5, 123.5, 110.2, 100.5, 25.9; GC-MS (EI) m/z 245 (M+); Anal. (C16H11N3) C, H, N. HBr salt precipitated from MeOH/Acetone. Anal. (C16H11N3·2HBr·3/4 H2O) C, H, N.
2-Chloro-5-(2-methylquinolin-7-yl)nicotinonitrile (31)
Prepared by following the general procedure A using 9a (0.11 g, 0.4 mmol) and 29 (0.1 g, 0.4 mmol), eluting with hexane/EtOAc (4:1, 1:1) to provide the product (70 mg) in 65% yield; mp 187–189 °C (dec); 1H NMR (400 MHz, CDCl3) δ 8.96 (s, 1H), 8.32 (s, 1H), 8.24 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 2.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 165.3. 152.8, 141.5, 140.0, 139.9, 134.3, 131.7, 129.8, 128.5, 120.5, 115.4, 30.3; IR (powder) 2232 cm−1; GC-MS (EI) m/z 279 (M+); Anal. (C16H10ClN3·1/8 H2O) C, H, N. HBr salt precipitated from MeOH/Acetone. Anal. (C16H10ClN3·HBr·5/4 H2O) C, H, N.
5-(2-Methylquinolin-7-yl)-2-phenylnicotinonitrile (32)
To a microwave reaction vessel was added 31 (0.28g, 1 mmol), phenylboronic acid (0.13 g, 1.05 mmol), Pd(OAc)2 (0.045g, 0.20 mmol, 3mol%), ligand 1L (0.17g, 0.40 mmol, 6 mol%), K3PO4 ( 0.64 g, 3 mmol), dioxane (5mL). The reaction mixture was degassed, protected under Argon, and reacted under microwave condition at 140°C for 90 min, followed the same work-up procedure in general procedure B, eluting with hexane/EtOAc (4:1, 2.5:1) to provide the product (70 mg) in 30% yield; mp 105–107 °C (dec); 1H NMR (400 MHz, CDCl3) δ 9.26 (t, J = 2.8 Hz, 1H), 8.40 (t, J = 2.8 Hz, 1H), 8.32 (s, 1H), 8.12 (dd, J = 8.0, 2.8 Hz, 1H), 8.02 (m, 2H), 7.96 (dd, J = 8.0, 2.8 Hz, 1H), 7.55 (dt, J = 8.4, 2.0 Hz, 1H), 7.56 (m, 3H), 7.38 (dd, J = 6.8, 2.8 Hz, 1H), 2.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.7, 159.9, 151.5, 148.2, 140.2, 137.0, 136.2, 134.4, 130.6, 129.7, 129.2, 129.1, 129.0, 127.3, 126.7, 124.4, 123.3, 117.9, 107.8, 25.7; IR (powder) 2226 cm−1; GC-MS (EI) m/z 321 (M+); Anal. (C22H15N3·1/8 H2O) C, H, N. HBr salt precipitated from MeOH/Acetone. Anal. (C22H15N3·2HBr·H2O) C, H, N.
Supplementary Material
Acknowledgments
This work was funded by the NIDA-Intramural Research Program and an NRSA Fellowship F32 NS049865 awarded by NINDS to ALR. mGluR5 binding data, and a functional screens across all mGluRs were provided by the National Institute of Mental Health s Psychoactive Drug Screening Program, Contract no. NO1MH32004 (NIMH PDSP). The NIDA Addiction Treatment Discovery Program provided the computational toxicology data, thanks to Dr. Rik Kline.
Abbreviations
- mGluR
metabotropic glutamate receptor
- iGluR
ionotropic glutamate receptor
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- MTEP
3-((2-Methyl-4-thiazolyl)ethynyl)pyridine
Footnotes
Supporting Information Available
Elemental analysis results are available free of charge via the Internet at http://pubs.acs.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaescheke GWJG, Nordquist RE, Spooren W. mGlu5 receptor antagonist and their therapeutic potentail. Expert Opin Ther Patents. 2008;18(2):123–142. [Google Scholar]
- 2.Lindsley CW, Emmitte KA. Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr Opin Drug Discov Devel. 2009;12(4):446–457. [PubMed] [Google Scholar]
- 3.Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4(9):873–4. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 4.Kenny PJ, Markou A. The ups and downs of addiction: role of metabotropic glutamate receptors. Trends Pharmacol Sci. 2004;25(5):265–72. doi: 10.1016/j.tips.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Lee B, Platt DM, Rowlett JK, Adewale AS, Spealman RD. Attenuation of behavioral effects of cocaine by the Metabotropic Glutamate Receptor 5 Antagonist 2-Methyl-6-(phenylethynyl)-pyridine in squirrel monkeys: comparison with dizocilpine. J Pharmacol Exp Ther. 2005;312(3):1232–40. doi: 10.1124/jpet.104.078733. [DOI] [PubMed] [Google Scholar]
- 6.Platt DM, Rowlett JK, Spealman RD. Attenuation of cocaine self-administration in squirrel monkeys following repeated administration of the mGluR5 antagonist MPEP: comparison with dizocilpine. Psychopharmacology (Berl) 2008;200(2):167–76. doi: 10.1007/s00213-008-1191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll FI. Antagonists at metabotropic glutamate receptor subtype 5: structure activity relationships and therapeutic potential for addiction. Ann N Y Acad Sci. 2008;1141:221–32. doi: 10.1196/annals.1441.015. [DOI] [PubMed] [Google Scholar]
- 8.Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets. 2007;8(1):169–84. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- 9.Bridges TM, Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3(9):530–41. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 10.Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezeau L, Carroll F, Pin JP, Cambria A, Vranesic I, Flor PJ, Gasparini F, Kuhn R. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem. 2000;275(43):33750–8. doi: 10.1074/jbc.M006230200. [DOI] [PubMed] [Google Scholar]
- 11.Bach P, Nilsson K, Wallberg A, Bauer U, Hammerland LG, Peterson A, Svensson T, Osterlund K, Karis D, Boije M, Wensbo D. A new series of pyridinyl-alkynes as antagonists of the metabotropic glutamate receptor 5 (mGluR5) Bioorg Med Chem Lett. 2006;16(18):4792–5. doi: 10.1016/j.bmcl.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 12.Bach P, Nilsson K, Svensson T, Bauer U, Hammerland LG, Peterson A, Wallberg A, Osterlund K, Karis D, Boije M, Wensbo D. Structure-activity relationships for the linker in a series of pyridinyl-alkynes that are antagonists of the metabotropic glutamate receptor 5 (mGluR5) Bioorg Med Chem Lett. 2006;16(18):4788–91. doi: 10.1016/j.bmcl.2006.06.078. [DOI] [PubMed] [Google Scholar]
- 13.Wendt JA, Deeter SD, Bove SE, Knauer CS, Brooker RM, Augelli-Szafran CE, Schwarz RD, Kinsora JJ, Kilgore KS. Synthesis and SAR of 2-aryl pyrido[2,3-d]pyrimidines as potent mGlu5 receptor antagonists. Bioorg Med Chem Lett. 2007;17(19):5396–9. doi: 10.1016/j.bmcl.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 14.Milbank JB, Knauer CS, Augelli-Szafran CE, Sakkab-Tan AT, Lin KK, Yamagata K, Hoffman JK, Zhuang N, Thomas J, Galatsis P, Wendt JA, Mickelson JW, Schwarz RD, Kinsora JJ, Lotarski SM, Stakich K, Gillespie KK, Lam WW, Mutlib AE. Rational design of 7-arylquinolines as non-competitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg Med Chem Lett. 2007;17(16):4415–8. doi: 10.1016/j.bmcl.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 15.Galatsis P, Yamagata K, Wendt JA, Connolly CJ, Mickelson JW, Milbank JB, Bove SE, Knauer CS, Brooker RM, Augelli-Szafran CE, Schwarz RD, Kinsora JJ, Kilgore KS. Synthesis and SAR comparison of regioisomeric aryl naphthyridines as potent mGlu5 receptor antagonists. Bioorg Med Chem Lett. 2007;17(23):6525–8. doi: 10.1016/j.bmcl.2007.09.083. [DOI] [PubMed] [Google Scholar]
- 16.Kulkarni SS, Newman AH. Discovery of heterobicyclic templates for novel metabotropic glutamate receptor subtype 5 antagonists. Bioorg Med Chem Lett. 2007;17(11):2987–91. doi: 10.1016/j.bmcl.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulkarni SS, Newman AH. Design and synthesis of novel heterobiaryl amides as metabotropic glutamate receptor subtype 5 antagonists. Bioorg Med Chem Lett. 2007;17(7):2074–9. doi: 10.1016/j.bmcl.2006.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulkarni SS, Nightingale B, Dersch CM, Rothman RB, Newman AH. Design and synthesis of noncompetitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg Med Chem Lett. 2006;16(13):3371–5. doi: 10.1016/j.bmcl.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 19.Kulkarni SS, Zou MF, Cao J, Deschamps JR, Rodriguez AL, Conn PJ, Newman AH. Structure-activity relationships comparing N-(6-methylpyridin-yl)-substituted aryl amides to 2-methyl-6-(substituted-arylethynyl)pyridines or 2-methyl-4-(substituted-arylethynyl)thiazoles as novel metabotropic glutamate receptor subtype 5 antagonists. J Med Chem. 2009;52(11):3563–3575. doi: 10.1021/jm900172f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alagille D, Baldwin RM, Roth BL, Wroblewski JT, Grajkowska E, Tamagnan GD. Functionalization at position 3 of the phenyl ring of the potent mGluR5 noncompetitive antagonists MPEP. Bioorg Med Chem Lett. 2005;15(4):945–9. doi: 10.1016/j.bmcl.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 21.Alagille D, Baldwin RM, Roth BL, Wroblewski JT, Grajkowska E, Tamagnan GD. Synthesis and receptor assay of aromatic-ethynyl-aromatic derivatives with potent mGluR5 antagonist activity. Bioorg Med Chem. 2005;13(1):197–209. doi: 10.1016/j.bmc.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 22.Boice GN, Savarin CG, Murry JA, Conrad K, Matty L, Corley EG, Smitrovich JH, Hughes D. An efficient synthesis of a highly functionalized 4-arylpiperidine. Tetrahedron. 2004;60:11367–11374. [Google Scholar]
- 23.Witherington J, Bordas V, Garland SL, Hickey DM, Ife RJ, Liddle J, Saunders M, Smith DG, Ward RW. 5-aryl-pyrazolo[3,4-b]pyridines: potent inhibitors of glycogen synthase kinase-3 (GSK-3) Bioorg Med Chem Lett. 2003;13(9):1577–80. doi: 10.1016/s0960-894x(03)00134-3. [DOI] [PubMed] [Google Scholar]
- 24.PDSP. NIMH Psychoactive Drug Screening Program. 2009 http://pdsp.med.unc.edu/indexR.html.
- 25.Busse CS, Brodkin J, Tattersall D, Anderson JJ, Warren N, Tehrani L, Bristow LJ, Varney MA, Cosford ND. The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology. 2004;29(11):1971–9. doi: 10.1038/sj.npp.1300540. [DOI] [PubMed] [Google Scholar]
- 26.MacroModel. Maestro using the default setting of commond "flexible ligand alignment". 9.6; 8.5. Schrödinger, LLC; New York, NY: 2008. [Google Scholar]
- 27.Sharma S, Kedrowski J, Rook JM, Smith RL, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. Discovery of molecular switches that modulate modes of metabotropic glutamate receptor subtype 5 (mGlu5) pharmacology in vitro and in vivo within a series of functionalized, regioisomeric 2- and 5-(phenylethynyl)pyrimidines. J Med Chem. 2009;52(14):4103–4106. doi: 10.1021/jm900654c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg Med Chem Lett. 2008;18(14):4098–101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ATDP. The Addiction Treatment Discovery Program. Division of Pharmacotherapies and Medical Consequences of Drug Abuse; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.