

Summary
Molecular dynamics simulation studies of protein-nucleic acid complexes are more complicated than studies of either component alone the force field has to be properly balanced, the systems tend to become very large, and a careful treatment of solvent and of electrostatic interactions is necessary. Recent investigations into several protein-DNA and protein-RNA systems have shown the feasibility of the simulation approach, yielding results of biological interest not readily accessible to experimental methods.
Introduction
In the post-genomic era interest shifts from the properties of individual components of living cells (e.g. proteins) to their many interacting complexes, of various size, composition and duration, that are at the heart of what makes the cell a living entity. Cellular components that do not communicate or interact, simply would neither benefit from, nor contribute to, the sustained organization characteristic of life. The intricate interplay between proteins and nucleic acids exhibited by all forms of life that we know of today thus forms the biological backdrop of the present review.
For complex systems molecular dynamics (MD) simulations are often the only way to obtain detailed atomic-level information, and the several program packages (some of the most commonly used are AMBER[1], CHARMM[2], GROMOS[3], GROMACS[4], and NAMD[5]) that have been developed specifically for atomistic simulations of biomolecules have over the last two decades been used in well over 10000 published studies. However, MD simulations of nucleic acids (NA) initially lagged behind protein simulations for several reasons: Nucleic acids are highly charged, usually quite non-globular, and there were not very many structures available by which to develop and validate NA simulation methodologies. Not surprising for nucleic acids it has been shown to be necessary to treat the solvent and electrostatics more carefully than what had been required for simulations of small globular proteins. Inclusion of an explicit representation of the solvent leads to a significant increase in the number of particles that have to be treated, and it is not uncommon that 80 – 90% of the computational cost comes from solvent-solvent interactions. More recently implicit solvent methods have also been used with some success for systems containing nucleic acids, but for large systems these methods are also computationally expensive. In contrast to proteins, which in many cases are quite well-behaved even in gas-phase simulations using relatively short cutoffs with group-based spherical truncation of the Coulomb interactions, nucleic acids when treated in the same way lose their structural integrity[6]. This was never a problem in CHARMM[2], which has sophisticated atom-based smoothing of electrostatic forces[6–8], but for some other programs it was not until efficient lattice sum methods became available that nucleic acids could be reliably simulated[9].
A further complication for simulations of protein-NA complexes lies in the force field, which has to be balanced in terms of pair interactions involving all combinations of solvent and the solute atoms. Much effort has gone into the development and validation of force fields, as outlined in the first section below, contributing to the success of simulation studies of protein-NA complexes.
One of the earliest studies of a protein-nucleic acid complex was of a mononucleotide bound to ribonuclease T1 [10]. Later, well-behaved simulations of duplex DNA [11] were soon accompanied by studies of transcription factors bound to DNA [12]. In the decade that followed vast increases in available computer power together with a rapidly growing number of experimental structure determinations of protein-DNA and, in particular, protein-RNA complexes stimulated an increased interest in MD simulations of these systems. In the two main sections of this review we show that it is now possible to tackle issues of relevance for molecular biology by using examples taken from some recent studies of protein-DNA and protein-RNA complexes, with the focus on all-atom simulations in explicit solvent
Force fields for nucleic acid-protein simulations
Two force fields commonly used for simulations of nucleic acid-protein complexes are AMBER[13] and CHARMM.[14,15] Both force fields treat DNA and RNA as well as modified nucleic acids. Several modifications of the AMBER nucleic acid [16–18] and protein[19–21] force fields have been published, potentially leading to complications in comparison of results from different studies. A recent extension of the CHARMM protein force field to improve treatment of the peptide backbone, termed CMAP, has been published [22] and now represents the standard for CHARMM protein and protein-NA simulations. Other force fields potentially may be used for protein-NA simulations including GROMOS, [23] OPLS, [24] MMFF [25] and CFF,[26] though the number of studies using these models is limited. Finally, an interesting development is the potential use of GB based methods for protein nucleic acid complexes.[27] While some success in the method was noted it should be emphasized that the lack of explicit solvent in the simulations, which play important roles in NA structure as well as protein-nucleic acids interactions, may lead to limitations in this approach.
DNA-protein complexes
MD simulations of DNA-protein complexes have recently yielded insights into the balance between the inherent structural properties of nucleic acids and the impact of the protein partner on those properties. In many of these studies distortion of the DNA when bound to the protein was investigated, including simulations of DNA in the presence of the transcription factor p53 [28], important because of its implication in a large number of cancers. Studies of p53 have also investigated the role of Zn+2 on both recognition and stability [29] and various models of the tetrameric p53 core domain bound to DNA duplex structures as well as to Holiday junctions have been assessed based on conforomational samples generated from MD simulations. [30] An interesting study combined MD simulations with the Molecular mechanics/Generalized Born/Solvent Accessible Area (MM/GBSA) approach to investigate the cooperativity seen in the binding of the RUNT transcription factor with its DNA target sequence and with a second protein, CBFβ.[31] Results indicate that the presence of the third partner in the trimer enhances the intermolecular interactions between the partners in both the Runt-DNA and Runt-CBFβ dimers. Using a similar approach MD simulations were combined with MM/Poisson-Boltzmann-SA (MM/PBSA) free energy analysis to understand the impact of carcinogens on binding of the TATA sequence to the TATA-Box Binding Protein.[32] In general, approaches where MD simulations are used to sample the conformational space of structures with the free energies of the systems evaluated via either the MM/GBSA and MM/PBSA methods represents a powerful approach to obtain estimates of the free energies of binding for large complexes. However, it should be emphasized that adequate sampling needs to be performed to assure convergence of the free energies and care must be taken to obtain accurate estimates for the configurational entropies of the systems.
In the area of specific recognition of DNA sequences by proteins a number of interesting studies were reported. It was shown that energy calculations alone on mutant sequences modeled based on the crystal structures of DNA-protein complexes could identify consensus binding sequences for a number of complexes, including examples where indirect readout via water molecules contributes to specificity.[33] A study on Hoxc8 using a similar approach supplemented with MD simulations was also successful in identifying the cognate sequence.[34] Emphasizing the role of water in DNA-protein recognition was a simulation study of the restriction endonuclease BamHI where it was proposed that a “hydration fingerprint” associated with interfacial waters can mediate specificity.[35] Notable was the use of Grand Canonical Monte Carlo simulations to allow for proper sampling of water in regions of the DNA-protein interface not exposed to the bulk solvent. Concerning DNA binding proteins that lead to flipping of a base or bases out of the duplex and into the enzyme catalytic site, an energetic or dynamic recognition mechanism was shown to impart the specificity for GCGC sequences by the cytosine methyltransferase from HhaI (M.HhaI, Figure ).[36] Specificity is based on the ability of the enzyme to only be able to successfully flip out and stabilize the target cytosine base in the presence of the correct sequence. However, in the case of the 8-oxoguanine-glycosylase, free energy calculations in combination with crystallographic analysis indicates specificity to be related to specific recognition of 8-oxo-G versus G in the enzyme active site.[37] While yet to be experimentally determined, for the general class of “flipping proteins” it is likely that the specificity of DNA repair enzymes will rely more on unique properties associated with the chemical alterations in the DNA whereas enzymes that target specific sequences in unmodified DNA, as with the methyltransferase, may rely on an energetic or dynamic recognition mechanism.
A powerful method to overcome the inherent time limitation of MD simulations (~100 ns) is using a potential of mean force (PMF) calculation. In this approach an external or biasing potential, often referred to as an umbrella potential, is applied to overcome large energy barriers along a reaction coordinate between conformational states. The energetic contribution of the umbrella potential is then corrected for allowing the intrinsic free energy along the reaction coordinate (i.e. the PMF) to be obtained. This approach was used in base flipping studies of DNA in the presence of M.HhaI to show that the protein actively facilitates flipping of the base out of the DNA helix[38] as well as to describe the mechanism of sequence specificity discussed above.[36] PMF calculations have also been used to investigate the stereospecificity of base excision in DNA glycosylase[39] and elegant studies on conformational changes in DNA polymerase based on transition path sampling have been performed.[40] Examples of the application of biased sampling methods to large structural perturbation are the rotation of DNA around topoisomerase I[41] and the movement of PcrA Helicase along ssDNA.[42] While PMFs were not extracted in those studies, they are examples of how external potentials may be used to drive large scale structural changes in DNA-protein complexes. Again, checks must be performed to make sure that adequate sampling is performed in the MD simulations to ensure that the calculated free energy profile is adequately converged.
RNA-protein complexes
Simulation studies have been performed on a number of RNA-protein complexes in the cellular machinery involved in processing genetic information, from transcription and splicing to translation of the mature mRNA. MD simulation of a model of the RNA Polymerase II (PolII) elongation complex with both RNA and DNA, including the unwound DNA bubble, in the pre-translocation state indicated that several loops in the protein are involved in keeping the DNA bubble open, and that fork loop 1 of PolII is implicated in the unidirectionality of PolII motion along the DNA.[43] Special hardware was used for simulation of the large 0.5 million particle system, though only 2 ns of simulation time was reported, so that robust conclusions from the simulation are limited. A 50 ns simulation of a complete virus[44] and a shorter targeted MD simulation of the entire ribosome[45] are additional noteworthy efforts that establish the stability of large scale simulations on heterogeneous systems.
The spliceosome, which in eukaryotes carries out removal of introns and splicing of the coding regions to form mature mRNA, is a dynamic ribonucleoprotein complex consisting of five small nuclear RNAs and associated proteins. It has attracted considerable attention from the simulation community, and several interesting studies of the U1A protein-RNA complex have been reported. The U1A protein is one of the best characterized proteins containing an RNA binding domain (RBD, also known as an RNA-recognition motif (RRM)). The first studies[46,47] focused on elucidating the molecular basis for the high affinity of the protein for the RNA hairpin and internal loop (Figure 2f), and on possible mechanistic effects of salt concentration on the stability of the complex [48]; their main importance was in establishing the feasibility of simulating RNA-protein complexes using either proper spherical cutoff[47] or lattice sum[46,48] methods. A recent extension[49] of the study by Tang and Nilsson[47], showed the key role of a set of positively charged amino acid residues for proper recognition of the RNA, and a simulation analysis[50] of the structural consequences of experimental point mutations involving aromatic acids in U1A provides a foundation for further attempts to modify the stability of RBD-RNA complexes. Other studies of U1A-RNA have shown the importance of correlated motions for the identification of cooperative networks involved in the binding[51–53], and that such collective motions are absent in a weakly binding mutant.[54] A simulation of another RBD-RNA complex highlighted the flexibility of residues, as well as the presence of several quite rapidly exchanging water molecules at the protein-RNA interface.[55]
Figure 2.

Basic amino acid residues in the U1A protein (cartoon) contacting the RNA hairpin structure (spacefilling representation with A/U bases in yellow and G/C bases in blue). The two lysines (K22 and K50, green spacefilling) contact the RNA backbone, whereas the arginine (R52, cyan spacefilling) contacts the major groove edge of G16. Image of the U1A-RNA complex[66] (PDB id 1URN) was created with the program RasMol[67].
Most studies of translation deal with the ribosome, but in at least one study[56] the MM/PBSA approach was used to show that the affinity of tRNAGln for its synthetase depends on entropic contributions from the internal mobility in the tRNA core. Several studies have used part of the ribosome, typically around the decoding center in the small subunit. Replica exchange MD simulations indicate that the energy barrier for the conformational change of the key nucleotides (A1492 and A1493 in the small ribosomal subunit) upon tRNA binding is low enough to allow binding of ligands to influence the equilibrium.[57] Specificity of codon-anticodon interactions in the ribosomal decoding center has been analyzed in structural terms using standard MD simulations [58] and also in energetic terms using the linear interaction energy method.[59] Free energy perturbation calculations, together with an empirical valence bond approach indicate an entropic origin for the rate enhancement of the peptide bond synthesis reaction at the other end of the tRNA.[60] Interactions between ribosomal RNA and individual ribosomal proteins have also been investigated in terms of conformational[61] and hydration[62] properties.
Conclusions
From the above it is clear that at this time, December 2007, MD simulations have advanced to a point that allows for a wide range of DNA-protein and RNA-protein studies to be performed with confidence that the obtained results are representative of the experimental regimen. Accordingly, it is anticipated that future studies will more closely link MD studies with experimental results, with such collaborative efforts taking advantage of free-energy methods beyond free energy perturbations calculations, including studies based on PMF calculations and using MM/GBSA and MM/PBSA approaches. Explicit solvent MD simulations will also allow for a more detailed understanding of the role of hydration, including interfacial waters, in protein-NA interactions. Finally, the ever increasing availability of computational power will allow for increased sampling, in the form of longer simulations as well as multiple simulations[63] as required to obtain more precise free energy estimates and perform statistical analysis of simulation results, not to mention studies on extremely large macromolecular complexes.
Figure 1.
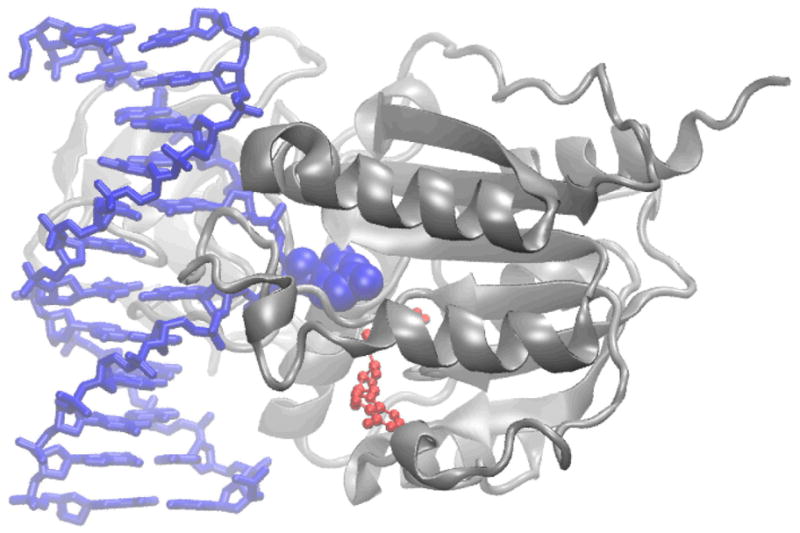
Cytosine-5-methyltransferase from HhaI showing the protein in gray ribbon, the DNA in blue with the flipped cytosine base in space filling representation and the cofactor S-homocysteine in red CPK (PDB id 5MHT[64], Image created with the program VMD[65]).
Acknowledgments
Financial support is acknowledged from the NIH (GM051501, ADM Jr.) and the Swedish Research Council (LN).
Contributor Information
Alexander D. MacKerell, Jr., Email: alex@outerbanks.umaryland.edu, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore MD 21201, USA
Lennart Nilsson, Email: Lennart.Nilsson@biosci.ki.se, Department of Biosciences and Nutrition, Karolinska Institutet, SE-141 57 HUDDINGE, Sweden.
References
- 1.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The AMBER biomolecular simulation programs. Journal of Computational Chemistry. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 3.Christen M, Hunenberger PH, Bakowies D, Baron R, Burgi R, Geerke DP, Heinz TN, Kastenholz MA, Krautler V, Oostenbrink C, et al. The GROMOS software for biomolecular simulation: GROMOS05. Journal of Computational Chemistry. 2005;26:1719–1751. doi: 10.1002/jcc.20303. [DOI] [PubMed] [Google Scholar]
- 4.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 5.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norberg J, Nilsson L. On the Truncation of Long-Range Electrostatic Interactions in DNA. Biophys J. 2000;79:1537–1553. doi: 10.1016/S0006-3495(00)76405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks CL, Pettitt BM, Karplus M. Structural and Energetic Effects of Truncating Long Ranged Interactions in Ionic and Polar Fluids. J Chem Phys. 1985;83:5897–5908. [Google Scholar]
- 8.Steinbach PJ, Brooks BR. New Spherical-Cutoff Methods For Long-Range Forces in Macromolecular Simulation. J Comp Chem. 1994;15:667–683. [Google Scholar]
- 9.Cheatham TE, III, Miller JL, Fox T, Darden TA, Kollman PA. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J Am Chem Soc. 1995;117:4193–4194. [Google Scholar]
- 10.MacKerell AD, Jr, Rigler R, Nilsson L, Hahn U, Saenger W. Protein Dynamics: A time-resolved fluorescence, energetic and molecular dynamics study of ribonuclease T1. Biophysical Chemistry. 1987;26:247–261. doi: 10.1016/0301-4622(87)80027-3. [DOI] [PubMed] [Google Scholar]
- 11.Norberg J, Nilsson L. High-Pressure Molecular Dynamics of a Nucleic Acid Fragment. Chem Phys Lett. 1994;224:219–224. [Google Scholar]
- 12.Eriksson MAL, Hard T, Nilsson L. Molecular Dynamics Simulations of the Glucocorticoid Receptor DNA-Binding Domain in Complex With DNA and Free in Solution. Biophys J. 1995;68:402–426. doi: 10.1016/S0006-3495(95)80203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society. 1995;117:5179–5197. [Google Scholar]
- 14.Foloppe N, MacKerell AD., Jr All-atom empirical force field for nucleic acids: 1) Parameter optimization based on small molecule and condensed phase macromolecular target data. J Comp Chem. 2000;21:86–104. [Google Scholar]
- 15.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 16.Cheatham TE, III, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J Biomol Struct Dyn. 1999;16:845–861. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 17.Ryjacek F, Kubar T, Hobza P. New Parameterization of the Cornell et al. Empirical Force Field Covering Amino Group Nonplanarity in Nucleic Acid Bases. J Comp Chem. 2003;24:1891–1901. doi: 10.1002/jcc.10352. [DOI] [PubMed] [Google Scholar]
- 18.Perez A, Marchan I, Svozil D, Sponer J, Cheatham ITE, Laughton CA, Orozco M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of {alpha}/{gamma} Conformers. Biophy J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comp Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 20.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Structure, Function, and Bioinformatics. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang ZX, Zhang W, Wu C, Lei HX, Cieplak P, Duan Y. Strike a balance: Optimization of backbone torsion parameters of AMBER polarizable force field for simulations of proteins and peptides. Journal of Computational Chemistry. 2006;27:781–790. doi: 10.1002/jcc.20386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKerell AD, Jr, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comp Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 23.Soares TA, Hunenberger PH, Kastenholz MA, Kraeutler V, Lenz T, Lins R, Oostenbrink C, van Gunsteren W. An improved nucleic acid parameter set for the GROMOS force field. J Comp Chem. 2005;26:725–737. doi: 10.1002/jcc.20193. [DOI] [PubMed] [Google Scholar]
- 24.Kaminski G, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 25.Halgren TA. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. Journal of Computational Chemistry. 1996;17:490–519. [Google Scholar]
- 26.Ewig CS, Berry R, Dinur U, Hill J-R, Hwang M-J, Li H, Liang C, Maple J, Peng Z, Stockfisch TP, et al. Derivation of Class II Force Fields. VIII. Derivation of a General Quantum Mechanical Force Field for Organic Compounds. J Comp Chem. 2001;22:1782–1800. doi: 10.1002/jcc.1131. [DOI] [PubMed] [Google Scholar]
- 27.Chocholousova J, Feig M. Implicit solvent simulations of DNA and DNA-protein complexes: agreement with explicit solvent vs experiment. J Phys Chem B. 2006;110:17240–17251. doi: 10.1021/jp0627675. [DOI] [PubMed] [Google Scholar]
- 28.Pan YP, Nussinov R. Structural basis for p53 binding-induced DNA bending. Journal of Biological Chemistry. 2007;282:691–699. doi: 10.1074/jbc.M605908200. [DOI] [PubMed] [Google Scholar]
- 29.Duan JX, Nilsson L. Effect of Zn2+ on DNA recognition and stability of the p53 DNA-binding domain. Biochemistry. 2006;45:7483–7492. doi: 10.1021/bi0603165. [DOI] [PubMed] [Google Scholar]
- 30.Ma B, Levine AJ. Probing potential binding modes of the p53 tetramer to DNA based on the symmetries encoded in p53 response elements. Nucl Acids Res. 2007 doi: 10.1093/nar/gkm1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31**.Habtemariam B, Anisimov VM, MacKerell AD., Jr Cooperative Binding of DNA and CBFβ to the Runt Domain of the CBFα Studied via MD simulations. Nucl Acid Res. 2005;33:4212–4222. doi: 10.1093/nar/gki724. Application MM/GBSA to characterize the free energy of binding of DNA-protein complexes, including mechanism of binding cooperativity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Zhang Q, Schlick T. Stereochemistry and position-dependent effects of carcinogens on TATA/TBP binding. Biophys J. 2006;90:1865–1877. doi: 10.1529/biophysj.105.074344. Application of MM/PBSA to characterize the free energy of binding of DNA-protein complexes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paillard G, Lavery R. Analyzing protein-DNA recognition mechanisms. Structure. 2004;12:113–122. doi: 10.1016/j.str.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 34.Roy S, Sen S. Exploring the potential of complex formation between a mutant DNA and the wild type protein counterpart: a MM and MD simulation approach. J Mol Graph Model. 2006;25:158–168. doi: 10.1016/j.jmgm.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 35**.Fuxreiter M, Mezei M, Simon I, Osman R. Interfacial water as a "hydration fingerprint" in the noncognate complex of BamHI. Biophys J. 2005;89:903–911. doi: 10.1529/biophysj.105.063263. Application Grand Canonical MC simulations to rigorously solvate DNA-protein complexes, including regions not exposed to bulk solvent. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Huang N, MacKerell AD., Jr Specificity in protein-DNA interactions: energetic recognition by the (cytosine-C5)-methyltransferase from HhaI. J Mol Biol. 2005;345:265–274. doi: 10.1016/j.jmb.2004.10.042. Application of PMF approaches to study base flipping and identification of energetic or dynamic recognition as the mode of sequence specificity. [DOI] [PubMed] [Google Scholar]
- 37.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 38.Huang N, Banavali NK, MacKerell AD., Jr Protein-facilitated base flipping in DNA by cytosine-5-methyltransferase. Proc Natl Acad Sci U S A. 2003;100:68–73. doi: 10.1073/pnas.0135427100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nat Struct Biol. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- 40**.Wang Y, Schlick T. Distinct energetics and closing pathways for DNA polymerase beta with 8-oxoG template and different incoming nucleotides. BMC Struct Biol. 2007;7:7. doi: 10.1186/1472-6807-7-7. Detailed investigation using restraint methods of energetics associated with conformational changes in DNA Polymerase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sari L, Andricioaei I. Rotation of DNA around intact strand in human topoisomerase I implies distinct mechanisms for positive and negative supercoil relaxation. Nucleic Acids Res. 2005;33:6621–6634. doi: 10.1093/nar/gki935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu J, Ha T, Schulten K. Structure-based model of the stepping motor of PcrA helicase. Biophys J. 2006;91:2097–2114. doi: 10.1529/biophysj.106.088203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suenaga A, Okimoto N, Futatsugi N, Hirano Y, Narumi T, Ohno Y, Yanai R, Hirokawa T, Ebisuzaki T, Konagaya A, et al. Structure and dynamics of RNA polymerase II elongation complex. Biochemical and Biophysical Research Communications. 2006;343:90–98. doi: 10.1016/j.bbrc.2006.02.124. [DOI] [PubMed] [Google Scholar]
- 44*.Freddolino PL, Arkhipov AS, Larson SB, McPherson A, Schulten K. Molecular Dynamics Simulations of the Complete Satellite Tobacco Mosaic Virus. Structure. 2006;14:437–449. doi: 10.1016/j.str.2005.11.014. An impressive simulation of a very large system over a significant length of time. [DOI] [PubMed] [Google Scholar]
- 45*.Sanbonmatsu KY, Joseph S, Tung C-S. Simulating movement of tRNA into the ribosome during decoding. Proceedings of the National Academy of Sciences. 2005;102:15854–15859. doi: 10.1073/pnas.0503456102. Information rich simulations of a crucial process for ensuring the correctness of protein synthesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reyes CM, Kollman PA. Molecular dynamics studies of U1A-RNA complexes. RNA. 1999;5:235–244. doi: 10.1017/s1355838299981657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang Y, Nilsson L. Molecular dynamics simulations of the complex between human U1A protein and hairpin II of U1 small nuclear RNA and of free RNA in solution. Biophys J. 1999;77:1284–1305. doi: 10.1016/S0006-3495(99)76979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hermann T, Westhof E. Simulations of the dynamics at an RNA protein interface. Nat Struct Biol. 1999;6:540–544. doi: 10.1038/9310. [DOI] [PubMed] [Google Scholar]
- 49.Law MJ, Linde ME, Chambers EJ, Oubridge C, Katsamba PS, Nilsson L, Haworth IS, Laird-Offringa IA. The role of positively charged amino acids and electrostatic interactions in the complex of U1A protein and U1 hairpin II RNA. Nucl Acids Res. 2006;34:275–285. doi: 10.1093/nar/gkj436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Kormos BL, Beveridge DL, Baranger AM. Molecular dynamics simulation studies of a protein-RNA complex with a selectively modified binding interface. Biopolymers. 2006;81:256–269. doi: 10.1002/bip.20408. [DOI] [PubMed] [Google Scholar]
- 51.Kormos BL, Baranger AM, Beveridge DL. Do collective atomic fluctuations account for cooperative effects? Molecular dynamics studies of the U1A-RNA complex. Journal of the American Chemical Society. 2006;128:8992–8993. doi: 10.1021/ja0606071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kormos BL, Baranger AM, Beveridge DL. A study of collective atomic fluctuations and cooperativity in the U1A-RNA complex based on molecular dynamics simulations. Journal of Structural Biology. 2007;157:500–513. doi: 10.1016/j.jsb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Showalter SA, Hall KB. Correlated Motions in the U1 snRNA Stem/Loop 2:U1A RBD1 Complex. Biophys J. 2005;89:2046–2058. doi: 10.1529/biophysj.104.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Showalter SA, Hall KB. A Functional Role for Correlated Motion in the N-terminal RNA-binding Domain of Human U1A Protein. J Mol Biol. 2002;322:533–542. doi: 10.1016/s0022-2836(02)00804-5. [DOI] [PubMed] [Google Scholar]
- 55.Castrignano T, Chillemi G, Varani G, Desideri A. Molecular Dynamics Simulation of the RNA Complex of a Double-Stranded RNA-Binding Domain Reveals Dynamic Features of the Intermolecular Interface and Its Hydration. Biophys J. 2002;83:3542–3552. doi: 10.1016/S0006-3495(02)75354-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamasaki S, Nakamura S, Terada T, Shimizu K. Mechanism of the Difference in the Binding Affinity of E. coli tRNAGln to Glutaminyl-tRNA Synthetase Caused by Noninterface Nucleotides in Variable Loop. Biophys J. 2007;92:192–200. doi: 10.1529/biophysj.106.093351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Sanbonmatsu KY. Energy landscape of the ribosomal decoding center. Biochimie. 2006;88:1053–1059. doi: 10.1016/j.biochi.2006.06.012. Information rich simulations of a crucial process for ensuring the correctness of protein synthesis. [DOI] [PubMed] [Google Scholar]
- 58.Sanbonmatsu KY, Joseph S. Understanding Discrimination by the Ribosome: Stability Testing and Groove Measurement of Codon-Anticodon Pairs. J Mol Biol. 2003;328:33–47. doi: 10.1016/s0022-2836(03)00236-5. [DOI] [PubMed] [Google Scholar]
- 59*.Almlof M, Ander M, Aqvist J. Energetics of Codon-Anticodon Recognition on the Small Ribosomal Subunit. Biochemistry. 2007;46:200–209. doi: 10.1021/bi061713i. Information rich simulations of a crucial process for ensuring the correctness of protein synthesis. [DOI] [PubMed] [Google Scholar]
- 60**.Trobro S, Aqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc Natl Acad Sci USA. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. This focused study of an important part of the ribosomal machinery provides new insights into its catalytic properties. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li W, Sengupta J, Rath BK, Frank J. Functional conformations of the L11-ribosomal RNA complex revealed by correlative analysis of cryo-EM and molecular dynamics simulations. RNA-a Publication of the RNA Society. 2006;12:1240–1253. doi: 10.1261/rna.2294806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reblova K, Spackova Na, Koca J, Leontis NB, Sponer J. Long-Residency Hydration, Cation Binding, and Dynamics of Loop E/Helix IV rRNA-L25 Protein Complex. Biophys J. 2004;87:3397–3412. doi: 10.1529/biophysj.104.047126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elofsson A, Nilsson L. How Consistent Are Molecular-Dynamics Simulations - Comparing Structure and Dynamics in Reduced and Oxidized Escherichia-Coli Thioredoxin. Journal of Molecular Biology. 1993;233:766–780. doi: 10.1006/jmbi.1993.1551. [DOI] [PubMed] [Google Scholar]
- 64.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI Methyltransferase Flips Its Target Base Out of the DNA Helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 65.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Molec Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 66.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 67.Sayle R, Milner-White EJ. RasMol: Biomolecular graphics for all. Trends Biochem Sci. 1995;20:374–376. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]