

Abstract
The utility of quantitative molecular diagnostics for patient management depends on the ability to relate patient results to prior results or to absolute values in clinical practice guidelines. To do this, those results need to be comparable across time and methods, either by producing the same value across methods and test versions or by using reliable and stable conversions. Universally available standards and reference materials specific to quantitative molecular technologies are critical to this process but are few in number. This review describes recent history in the establishment of international standards for nucleic acid test development, organizations involved in current efforts, and future issues and initiatives.
In the 1990s, it was demonstrated that HIV viral load testing could predict and monitor the course of HIV disease.1,2 Similar studies followed for other viruses, and now quantitative molecular tests are integral to the prognosis and monitoring of several infectious diseases.3,4,5 Early assays, though, lacked adequate reproducibility and comparability, as illustrated in proficiency testing reports. This inadequacy was attributed to the abundant use of laboratory-developed protocols, inconsistent training, limited usage of standardized industry-produced assays, and the absence of adequate universal standards for test development.6,7,8
To mitigate persisting differences, current clinical practice recommendations have been developed whereby serial specimens from patients are optimally tested with the same assay each time they are submitted.9 However, the need for all tests to report consistently similar results is still critical and is driven by the increasing length of time that patients are monitored, the decreasing ability of practitioners to demand the use of specific laboratories and tests, diluted knowledge of these practice standards among the increasing number of nonspecialist treatment providers, and the fact that some medical decisions are based on absolute values instead of relative changes. The requirement for translatable quantification among different molecular tests relies on universally available, robust standard reference materials for test developers, manufacturers, and proficiency test providers.
Although molecular diagnostics have contributed to patient management for more than a decade, the establishment of standards and reference materials to support the field have lagged. The current World Health Organization International Standards address some of the needs, but they cover only six viruses and one bacterial target (National Institute for Biological Standards and Control, http://www.nibsc.ac.uk, last accessed August 20, 2009). These materials and the collective efforts to establish them have played a critical role, particularly in evaluating the sensitivity of molecular screening assays and in facilitating the relative standardization of quantitative assays. However, because of their original intended use as sensitivity standards for plasma product manufacturers' qualitative assays, they lack some of the characteristics desired for the standardization of quantitative diagnostic assays. Another possible resource, the Joint Committee on Traceability in Laboratory Medicine (JCTLM), which was established to “promote and give guidance on internationally recognized and accepted equivalence of measurements in laboratory medicine and traceability to appropriate measurement standards” maintains a database of certified reference materials and reference methods (Joint Committee on Traceability in Laboratory Medicine, http://www.bipm.org/en/committees/jc/jctlm, last accessed August 20, 2009). However, there are currently no standards for infectious disease quantitative molecular diagnostics either at the level 1 category of high-order reference materials (the level of purified chemicals) or in their level 2 category, which is designed to catalog biological products and consensus calibrated materials. Laboratory test developers and in vitro diagnostic (IVD) test manufacturers are left with very few options.
In this review we discuss the establishment of the current World Health Organization International Standards, the requirements needed specifically for quantitative molecular diagnostics, and current global efforts to address these needs. Through scientific, political, and pragmatic perspective guide opinions regarding these developments, we attempt to present them factually and to provide the clinical community with a foundation for future informed discussions.
Characterization, Establishment, and Replenishment of the World Health Organization International Standards
The World Health Organization International Standards are prepared, characterized, and established according to “The World Health Organization Guidelines for the Preparation and Establishment of Reference Standards for Biological Substances.”10 The first version of this document was written in 1978. It was revised in 1986, in 1990, and most recently in 2004 in a series of meetings with representatives from country and regional health departments, vaccine manufacturers, standards organizations, and diagnostic test manufacturers. It describes the general principles for the establishment of all World Health Organization biological reference materials, with a broad scope of intended uses: primarily vaccine preparations and immunological and biological assays. The quantification of viral targets in molecular testing is recognized in the most recent revision.
The most commonly used global standards for the calibration and characterization of quantitative molecular viral assays are the World Health Organization International Standards for hepatitis C virus (HCV), HIV, and hepatitis B virus. The World Health Organization International Standard (IS) for HCV was the first of the series and was established in 1997. It was a preparation of an HCV genotype 1a high-titered plasma unit diluted into cryosupernatant and lyophilized. The preparation was tested with two other candidate materials in a global collaborative study in 22 laboratories during 1996. The study was directed by and the data were analyzed by the World Health Organization collaborating center laboratory: the National Institute for Biological Standards and Controls (NIBSC) in South Mimms, UK. The methods examined included a variety of commercial and laboratory developed assays using single, nested, or heminested primers. Most of the data were generated by testing endpoint dilutions of the candidate standards using qualitative traditional (non-real-time) amplification assays, although quantitative data submitted by participants were included in the analysis. The first World Health Organization HCV IS (96/790) was assigned the value of 105 IU/ml (5 log10 IU/ml) lyophilized in 0.5-ml ampoules.11 The international unit is considered absolute, and, consistent with World Health Organization policies, there are no units of uncertainty associated with this or subsequent replacements. The IU for each biological preparation “has no existence other than in relation to the preparation that defines it.”10
Since 1997, the first HCV IS has been replaced by two subsequent standards: the second World Health Organization HCV IS (96/798) in 2003, which was a second lyophilization preparation of the original material12 and most recently a new material prepared and tested in 2007.13 The second HCV IS (96/798) was originally tested in the global collaborative study of 1996. Because it was the same material as the first IS and was determined in 1996 not to be statistically different from the first, there was no global collaborative study when it assumed its place as a the second IS 6 years later. Stability and comparative studies were performed in three selected laboratories before its use. The second World Health Organization HCV IS (96/798) was established with the same value as the first (96/790): 105 IU/ml (5 log10 IU/ml).
For the recent replacement of the second by the third HCV IS, new materials were commissioned, and a full global replacement study directed by NIBSC was performed. The candidates were two lyophilized and one frozen preparation of a genotype 1a, anti-HCV-negative, HCV RNA-positive plasma unit diluted into human plasma. The candidate materials were tested with the second HCV IS (96/798) by 33 participating laboratories in 14 countries. This time most assays were quantitative, although data testing the limiting dilutions of the materials with qualitative molecular assays were also included. In addition, real-time amplification assays not present in the first study more than 10 years earlier had become common and were reflected in the submission of data. The mean quantification of the second HCV IS (96/798) was 5.10 log10 IU/ml with a 95% confidence interval of 5.02 to 5.17 log10 IU/ml, which is statistically different from the previously assigned 5.0 log10 IU/ml. The mean values of the two lyophilized candidates were 5.19 log10 IU/ml (sample 2) and 5.47 log10 IU/ml (sample 3). Sample 2 (NIBSC code 06/100) was recommended as the third HCV IS to the Expert Committee on Biological Standardization (the World Health Organization committee commissioned “to establish detailed recommendations and guidelines for the manufacturing, licensing, and control of blood products, cell regulators, vaccines and related in vitro diagnostic tests”).13
The World Health Organization HCV IS has been used as the reference to compare the quantification of HCV genotype panels provided by the NIBSC14 and regional reference preparations throughout the world.15 Although the original intended purpose for this IS was for the establishment of sensitivity criteria for molecular assays used to screen blood products in the plasma manufacturing industry, it has contributed to the standardization of blood donation screening assays as well as molecular diagnostic and monitoring assays.16,17
The same process has been applied to other World Health Organization IS preparations for nucleic acid testing, and since 1996 standards for HIV,18 hepatitis B virus,19 hepatitis A virus,20 parvovirus B19,21 human papillomavirus,22 and Plasmodium falciparum23 have been established. The primary targets have been those most needed to ensure safety of transfused blood and human blood-derived biological products (Table 1).
Table 1.
World Health Organization International Standards for Molecular Infectious Disease Testing
| Target | Year established | Assigned value* | Mean assayed value† | Characteristics‡ |
|---|---|---|---|---|
| HCV RNA First International Standard (96/790) | 1997 | 4.70 log10 IU/vial (reconstituted to 0.5 ml) 5.00 log10 IU/ml | 5.0 log10 NAT detectable units/ml | HCV-positive donation; genotype 1; diluted in cryosupernatant; freeze-dried10 |
| HBV DNA First International Standard (97/746) | 1999 | 5.70 log10 IU/vial (reconstituted to 0.5 ml) 6.00 log10 IU/ml | 6.42 log10 equivalents/ml | Single donor material, Eurohep R1; genotype A, HBsAg subtype adw2; diluted in pooled plasma; freeze-dried19 |
| HIV-1 RNA First International Standard (97/656) | 1999 | 5.00 log10 IU/vial (reconstituted to 1.0 ml) 5.00 log10 IU/ml | 4.75 log10 copies/ml in quantitative assays | HIV PCR-positive/antibody-negative plasmapheresis donation; genotype B; diluted in defibrinated plasma; freeze-dried18 |
| HCV RNA Second International Standard (96/798) | 2003 | 4.70 log10 IU/vial (reconstituted to 0.5 ml) 5.00 log10 IU/ml | 5.0 log10 NAT detectable units/ml | HCV positive donation; genotype 1; diluted in cryosupernatant; freeze dried; separate lyophilization of same material as the first HCV IS12 |
| HAV RNA First International Standard (00/560) | 2004 | 4.70 log10 IU/vial (reconstituted to 0.5 ml) 5.00 log10 IU/ml | 5.29 log10 equivalents/ml | HAV-positive donation diluted in HCV-, HAV-, HIV-, HBV and HBsAG-, anti-HCV-, anti-HIV-negative human plasma20 |
| Parvovirus B19 DNA First International Standard (99/800) | 2004 | 5.70 log10 IU/vial (reconstituted to 0.5 ml) 6.00 log10 IU/ml | 5.89 log10 copies/ml | Parvovirus B19-positive donation; diluted in pooled plasma; freeze-dried21 |
| HBV DNA Second International Standard (97/750) | 2006 | 5.70 log10 IU/vial (reconstituted to 0.5 ml) 6.00 log10 IU/ml | 6.30 log10 equivalents/ml | Single donor material, Eurohep R1; genotype A, HBsAg subtype adw2; diluted in pooled plasma; freeze-dried; second aliquot of original preparation19 |
| HIV-1 RNA Second International Standard (97/650) | 2006 | 5.56 log10 IU/vial (reconstituted to 1.0 ml) 5.56 log10 IU/ml | 5.35 log10 copies/ml in quantitative assays; original testing | Field isolate: HIV-1, genotype B env V3, gag; propagated on PBMCs, diluted in pooled human cryosupernatant18 |
| Plasmodium falciparum DNA First International Standard (04/176) | 2006 | 8.7 log10 IU/vial (reconstituted to 0.5 ml) 9.00 log10 IU/ml | 8.51 log10 equivalents/ml | Freeze-dried whole blood patient blood collected by exchange transfusion23 |
| HCV RNA Third International Standard (06/100) | 2007 | 4.89 log10 IU/vial (reconstituted to 0.5 ml) 5.19 log10 IU/ml | Not applicable | Window period anti-HCV-negative plasma, genotype 1a HCV13 |
| HPV type 16 DNA First International Standard (06/202) | 2008 | 6.70 log10 IU/vial (reconstituted to 0.5 ml) 7.00 log10 IU/ml | 1 IU/ml is traceable through calculations to 0.947 GEq/ml, determined by independent methods | Freeze-dried recombinant plasmid containing full-length HPV-16 DNA in a background of purified human genomic DNA22 |
| HPV type 18 DNA First International Standard (06/208) | 2008 | 6.70 log10 IU/vial (reconstituted to 0.5 ml) 7.00 log10 IU/ml | 1 IU/ml is traceable through calculations to 1.031 GEq/ml, determined by independent methods | Freeze-dried recombinant plasmid containing full-length HPV-18 DNA in a background of purified human genomic DNA22 |
HAV, hepatitis A virus; HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; PMBC, peripheral blood mononuclear cell; HPV, human papilloma virus; GEq, genome equivalents.
Concentration is in IU/vial. Some vials are reconstituted only to 0.5 ml.
Assayed value given when determined through global collaborative studies or when assayed by independent methods before assignment of IU.
Characteristics and instructions for use are at http://www.NIBSC.ac.uk, last accessed August 20, 2009.
Efforts to Increase the Breadth and Depth of Standards for Quantitative Molecular Infectious Disease Testing
The traditional intended purpose for international biological standards was to characterize vaccine preparations and establish the analytic sensitivity of assays used to screen blood product materials. This provides insight into the reasons that these materials today are single-point references with no specification for recoverability when diluted to prepare quantitative standardization curves. Although the establishment of the first World Health Organization HCV IS was consistent with those intended uses,16,17 the World Health Organization and NIBSC have recognized the role it and subsequent viral IS materials have had in quantitative diagnostics. A 2007 report of World Health Organization collaborating centers for biological standards and standardization describes a 5-year strategic plan “to prioritize the development of World Health Organization biological reference products for the control of blood safety-related IVD [in vitro diagnostic] tests,” reiterating the safety of the blood supply as the primary goal.24 Leaders at NIBSC acknowledge that international standards are needed to standardize quantitative and qualitative molecular infectious disease assays for patient testing and are proceeding to address this area.
Recommendations for the desired specifications of standards for quantitative molecular diagnostics have been communicated to World Health Organization through consultation meetings with the organization and through working groups such as Standardization of Genome Amplification Techniques (SoGAT). The group most persistent in continued discussion of performance specifications for quantitative molecular diagnostic international standards has been the Industrial Liaison Committee (ILC), an organization of molecular assay manufacturers working toward the availability of universally accepted reference standards.
The specific interest of this group has been standards to serve the development of quantitative molecular diagnostic assays used in patient testing. ILC and World Health Organization-sponsored meetings with the World Health Organization, US Food and Drug Administration, and other standards organizations were convened in 1998, 2000, and 2002 to discuss this topic. In addition, ILC has presented regularly at SoGAT meetings. Specifications discussed in these meetings included the establishment of reference materials similar to clinical samples, quantification by methods independent of the current diagnostic testing methods, preparations with concentrations adequate to evaluate the entire expected clinical range, dilution protocols and published dilution recovery expectations, and stability testing (2006).25
Studies have shown that for commercial molecular quantitative assays for which international standards have been available (HCV and HIV), variability has decreased significantly since the first generation.26,27 However, a recent survey of 28 laboratories examining the variability of Epstein-Barr virus (EBV) assays across various viral load concentrations, sample types, and assay platforms demonstrated that significant variation still exists.28 Results of the survey showed that roughly 50% of all results fell within the “acceptable” parameters of ±0.5 log10 for quantitative nucleic acid test (NAT) assays. Similar results from multicenter comparisons have also been observed.29 Furthermore, greater variation in results was observed among samples containing virally infected cellular material, suggesting that sample preparation methodology, such as DNA extraction, needs further improvements. Last, variability was significantly higher in interlaboratory comparisons versus intralaboratory comparisons, strongly suggesting a need for assay calibration to a universally accepted reference standard. This amount of variability poses significant challenges in clinical diagnosis as seen in EBV viral load testing in EBV-related tumors.29
A similar study for human cytomegalovirus (HCMV) assays in the United States, Europe, and Canada demonstrated comparable problems. Among 33 laboratories testing a panel of seven constructed and three patient-derived samples, 57.6% of quantitative results were within the range of ±0.5 log10 of expected results. Although the authors attributed the variation to several factors, they concluded that a primary measure toward resolution would be the establishment of an international reference standard.30
These observations underscore the need for continued work in providing a more diverse toolbox of international standards and the importance these materials have in patient care. Clinical molecular laboratory directors are now engaged in the discussion of needed diagnostic standards for quantitative molecular assays for infectious diseases. One challenge for IVD manufacturers will be to make available affordable reference materials so that frequent quality control protocols can be implemented in laboratories. The following presents singular and combined efforts by several groups to fill these gaps, including work on synthetic materials by the ILC and by the National Institute of Standards and Technology (NIST) (Gaithersburg, MD) and plans for a broader menu of World Health Organization international standards by SoGAT.
Evaluating the Role of Synthetic Materials
In April 2002, a World Health Organization consultation group was convened to consider the role of international reference materials in viral load monitoring by NAT and to define the characteristics of reference materials appropriate for that purpose. The specifications for an ideal international standard and what might be acceptable in light of the diverse challenges of producing and sustaining them for molecular infectious disease testing were discussed. In this meeting, HCV and the viral assays were the predominant focus as the World Health Organization HCV IS had recently been established, although many of the issues were transferrable to other infectious disease targets. Table 2, outlining desired characteristics of molecular standards and the technical and strategic challenges in producing them, is adapted from presentations and deliberations at the meeting and continued discussions into the present (Report: World Health Organization Consultation on International Standards for in Vitro Clinical Diagnostic Procedures based on Nucleic Acid Amplification, http://www.who.int/bloodproducts/publications/en/BIVD02apr22.pdf, last accessed August 25, 2009). These issues continue to confront organizations attempting to establish globally recognized standard materials.
Table 2.
Optimal Specifications and Challenges: Molecular Standards
| Characteristic | Optimal specification | Acceptable specification | Challenges |
|---|---|---|---|
| Target nucleic acid | Whole genome as found in virus; intact | Whole genome as found in virus; intact | Is there a representative consensus genome? |
| Source of target nucleic acid | Virus | Recombinant DNA (DNA viruses) or RNA synthesized in vitro (RNA viruses) | Can the virus be propagated? Is the cultured strain representative? |
| Length of target nucleic acid | Entire genome | All of the sequences recognized by current and projected future assays | Current assays may not be representative of future technologies |
| Cultured organisms, purified NA or synthetic NA may not have important secondary structure | |||
| One or several genotypes/subtypes | All relevant genotypes available as standards | One genotype for a “foundation” standard. Genotypes and subtypes calibrated from the foundation standard | One representative organism does not necessarily address technicalities of all variants |
| Resources to produce and characterize all variants and perform the global studies to value-assign them would detract from producing standards for more organisms | |||
| Which genotype/subtype? | The most globally prevalent | The most globally prevalent | The most globally prevalent or the target of most present and near future testing? |
| Sequence of target nucleic acid | Known | Known | The proposed material should be sequenced |
| Sequence of cDNA clones (from RNA viruses) or DNA clones (from DNA viruses) | Sequence of cDNA clones (from RNA viruses) or DNA clones (from DNA viruses) | ||
| Concentration of target nucleic acid | High enough to evaluate calibrators that cover levels of targets found in patients | High enough to evaluate calibrators that cover levels of targets found in patients | Some source material (patient plasma) may not have high enough titers to produce enough standard material. |
| Matrix | Natural material from which the target is predominantly assayed. (ie, blood-borne viruses: plasma) | Material that will ensure stability and intactness of target nucleic acid and that will not affect assay | Is pooled material appropriate? |
| Can the bodily fluid from one individual be representative? | |||
| Are decalcified, defibrinated materials commutable? | |||
| If the infectious target is tested from cells, should cellular standards be developed? | |||
| Temperature of storage | Ambient or refrigerated | Ambient or refrigerated | Can impact stability |
| Manufacturing process | Validated, reproducible methods within a validated quality system | Validated methods within a validated quality system | Could limit the number and type of organizations able to produce or contribute materials |
| Vial-to-vial consistency | SD ≤ 0.15 log10 copies/ml (coefficient of variation ≤ 35%) | SD in target concentration known and published | Could limit the number and type of organizations able to produce or contribute materials |
| Initial quantification of nucleic acid targets | Independent analytical method(s) | Quantitative NAT using statistically designed protocol and equal weighting of results from assays used | Absence of reference methods requires testing with relevant available methods |
| Are resources available to adequately test materials with an even representation of the available test methods? |
Because HCV cannot be propagated and measured by independent methods such as limiting dilution viral culture, participants at the 2002 meeting designated it as a model for work to explore alternative strategies. Further, considering the desired characteristics discussed, the group concluded that synthetic (in vitro synthesized) nucleic acids could be considered. The methods for producing these synthetic materials were not specified at the time; however, a well-characterized, in vitro-generated HCV RNA transcript, quantified by physicochemical assays to a primary phosphate standard was readily available for evaluation through a donation from Bayer Diagnostics (now Siemens Healthcare Diagnostics, Inc., (Deerfield, IL).31 The transcript material included target sequences for all of the technologies available.
At the meeting, the ILC was challenged with planning and performing a feasibility study using a synthetic RNA containing HCV sequences, and a series of studies among manufacturers were initiated. The first feasibility study was performed in 2002. Four NAT methods were included in the study: VERSANT HCV RNA 3.0 Assay (bDNA) (Siemens Healthcare Diagnostics, Inc., Deerfield, IL); COBAS AMPLICOR HCV MONITOR, version 2.0 (Roche Molecular Diagnostics, Pleasanton, CA); an internal-use quantitative HCV TMA assay (Gen-Probe, Inc., San Diego, CA); and LCx HCV RNA Quantitative Assay (Abbott Laboratories, Abbott Park, IL). The original concentration of the synthetic HCV RNA was determined to be traceable to a chemical standard (potassium phosphate) by non-NAT methods.31 Scientists at each site used the synthetic RNA transcript to quantify HCV RNA concentrations in members of a patient-derived panel. Feasibility was defined as a demonstration of panel quantification value agreement within 1.0 log10 regardless of the NAT method. Values reported by each site as synthetic RNA transcript copies per ml did not differ by more than 1.0 log10 regardless of the technology used for analysis and demonstrated the feasibility of a synthetic HCV RNA standard. Further studies in 2005 and 2006 by this group evaluated the synthetic material using automated sample preparation instruments to determine that the material could be recovered in full extraction processes. Acceptable recovery was demonstrated; however, handling the RNA transcripts to maintain their integrity required expertise that is not universally available. The results of these studies were presented at the SoGAT meeting in 2004.
The ILC continued this work in 2007 with two synthetic and two biological materials. Scientists from Roche Molecular Diagnostics, Siemens Healthcare Diagnostics, Inc. Berkeley, CA and Abbott Molecular Inc. (Des Plaines, IL) quantitatively tested dilution panels of these materials in their respective HCV assays: Roche COBAS AmpliPrep/COBAS TaqMan HCV Assay, Siemens VERSANT HCV RNA 3.0 Assay (bDNA), and Abbott RealTime HCV assay. The synthetic materials were the HCV RNA transcript supplied by Siemens Healthcare Diagnostics, Inc. (described previously) and an HCV Armored RNA Quant supplied by Asuragen, Inc. (Austin, TX). Both materials had been quantified relative to a phosphate standard by independent, non-NAT methods. The Armored RNA, an RNA transcript synthesized by in vitro transcription, is encapsulated in a bacteriophage protein coat and is stable to nucleases present in normal sample matrices. It provided a material that could be subjected to the entire assay procedure without special handling. The biological materials used were the Paul Erlich Institute regional HCV standard at 80,000 IU/ml, contributed by M. Nübling at the Paul Erlich Institute (Langen, Germany) and an unquantified high titer plasma material prediluted into negative plasma prepared by scientists at Abbott Molecular, Inc. Six to eight replicates of four to six linear points of each material were tested in each assay in the manufacturers' laboratories. The synthetic materials demonstrated full utility based on recovery, range of linearity, and variation. In all three assays, the synthetic targets could be recovered and quantified with equal or less variability than the biological materials (Figure 1). The study, findings, and suggestions of possible roles of both synthetic and biological standards were presented at the SoGAT meeting in June 2007.
Figure 1.
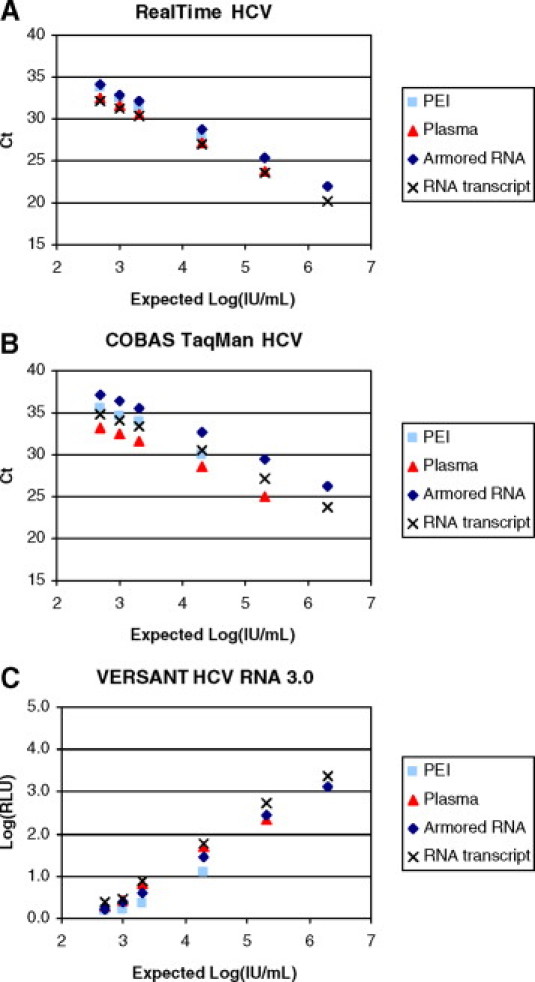
Evaluation of synthetic and biological HCV reference materials with three commercial HCV quantitative assays. Serial dilutions of the Paul Erlich Institute (PEI) regional HCV standard (80,000 IU/ml), high titer plasma, Armored RNA Quant HCV and synthetic HCV RNA transcript were tested using the RealTime HCV (A), COBAS AmpliPrep/COBAS TaqMan HCV (B), or VERSANT HCV RNA 3.0 assays (C). All test specimens were shipped and stored below −70°C. The HCV Armored RNA was prediluted, shipped, and stored in base human serum. Other specimens were formulated by each laboratory using the same base matrix immediately after thawing of the stock materials. The synthetic HCV RNA transcript was diluted in the presence of lysis reagent to inhibit potential degradation of the RNA by serum nucleases. At least six replicates of each dilution were tested in three independent runs with each assay. The synthetic materials were formulated in copies/ml of base matrix (2.5 × 103, 5 × 103, 1 × 104, 1 × 105, 1 × 106, and 1 × 107 copies/ml) and converted to IU/ml (IU/ml = copies/ml × 5). Ct, threshold cycle; RLU, relative light units.
Because the purpose of international standards is to provide the initial calibration point for developing assays and subsequent versions of those assays, it is critical that the standard remain reliable and unchanged. This is most challenging for biological standards in a field of evolving technologies. Each time a replacement World Health Organization International Standard is assigned a value, several years have passed, and the technologies that were used to evaluate the previous standard may not exist. Even when the former and latter are tested in unison, there may be small shifts in the valuation of the established standard, as described earlier in the World Health Organization HCV IS replacement study and documented in the HIV IS replacement studies. The impacts of these shifts are challenging to project and impossible to measure over time because of the limited amount of World Health Organization material available. For the World Health Organization global collaborative studies described in this review, four vials of candidates per method tested are available to the participants, and when the standard is established, all those requesting the material for development purposes are limited to a specific number of vials per year. This limitation is necessary so that the maximum distribution can be provided but has the unintended consequence of prohibiting statistically relevant longitudinal studies of the impact of small changes in the standards. To mitigate the resource limitation, there are several national and regional standards and secondary reference materials calibrated from the World Health Organization International Standards. In addition, diagnostic test manufacturers have procedures for developing secondary, internal use references calibrated against and traceable to the World Health Organization International Standards. Clinical scientists constructing laboratory developed tests are expected to act in a similar manner; either by generating or purchasing secondary reference materials for their internal development and calibration activities. However, with each dilution of the IS and calibration of secondary materials, there is inherent error as demonstrated in ISO 17511:2003. Because the molecular community may not be able to adequately project or quantify this impact, it is even more critical that we contribute to support, critique, and maintain the integrity of continued replacement of biological standards.
To contend with these issues and the limitations of the materials, a scenario was proposed whereby well characterized and independently tested synthetic nucleic acid material could serve as the constant in an environment in which reference methods are not available. If stable synthetic materials could be established along with the international biological standard and the relationship between the two could be quantified for relevant technologies, the synthetic material could be tested with each replacement lot of the biological standard. Test developers would continue to use the biological standards, but each replacement lot of those materials would be tested against the “unchanging” synthetic material. Because of the potential greater supply of synthetic materials, they could also be used in the characterization of secondary reference materials. This concept has not been tested for molecular diagnostics; however, current development of an HCMV nucleic acid material at the NIST and a biological standard for NIBSC provides a timely opportunity.
Development of a HCMV Standard Reference Material at NIST
Because HCMV resides intracellularly and is also present in plasma, this virus and those like it present an added level of complexity for producing standards: determining the biological matrix that most universally represents what is tested in clinical practice. Considering this, a standard that is free of biological material may be an appropriate reference for the development of several types of biological standards. The NIST has undertaken a project to develop a standard reference material for human cytomegalovirus. The material will consist of pure HCMV DNA. The goal is to develop a reference that would be traceable to the SI (International System of Units, the metric system) and suitable as a calibrant for quantitative PCR assays of HCMV viral load but perhaps more importantly as a tool to standardize various NAT assay reagents and calibrants produced by manufacturers of diagnostics or laboratory developed protocols.
Quantitative real-time PCR is rapidly becoming the method of choice for HCMV quantification over previously developed assays such as antigenemia because of its sensitivity and speed32 and the possibility of high throughput implementation. But there are issues such as DNA sequence diversity among targets used for quantification that can result in false-negative results.33,34 There is a proliferation of assays with different target sequences in the HCMV genome. In a recent survey, 10 different open reading frame targets were used, glycoprotein B being the most frequent.35
The HCMV standard reference material will consist of pure DNA from a Towne strain HCMV bacterial artificial chromosome (BAC).36 The BAC DNA insert is 10 kbp and replaces the US1 to US12 open reading frames in the Towne cytomegalovirus DNA by homologous recombination. The final construct, ΔUL147 Towne BAC, will be used to produce viral DNA.37 The Towne BAC is stable and can provide large quantities of consistent viral DNA, whereas cultured virus, especially from laboratory strains of HCMV, can produce truncated genomes.38 Genome size consistency is important for calculation of genome copy number.
The HCMV Towne BAC DNA reference material will be certified for sequence. The intent is to sequence all of the relevant regions of the genome that are used as targets for PCR assays. Two approaches will be used for quantification of the BAC DNA. Digital PCR is a primary method for counting copies of genomes or plasmids. This approach could provide traceability to the mole. Pure DNA can be accurately and precisely quantified based on phosphorus measurements using high-performance inductively coupled plasma-optical emission spectroscopy.39 Once the HCMV standard reference material has been certified, interlaboratory studies will be conducted.
SoGAT Committee for Viral Diagnostics
Originally sponsored by the World Health Organization to address the safety of transfused blood, tissue, and organs, with emphasis on viral, bacterial, and parasitic contamination, SoGAT has convened at least annually for more than 12 years. NIBSC has maintained leadership of this group with representatives from public health and other government agencies, IVD and independent control manufacturers, and diagnostic and university laboratories. The meeting provides a forum for discussion on assays, reference materials, and regulatory issues as well as for organizing collaborative studies. At the SoGAT meeting in June 2007, it was announced that a second series of meetings would be started with the focus on molecular diagnostics for clinical virology. The goals for the new group will be patterned on those for the original group, and it is expected there will be crossover interests between the two groups. SoGAT-CD (SoGAT Clinical Diagnostics) convened for the first time in June 2008.
At the first meeting of SoGAT-CD, issues specific to molecular testing for infectious diseases were discussed. The lack of quantitative reference materials for the growing number of viral molecular tests was a persistent concern. Within months of the meeting, the NIBSC issued draft protocols for comment regarding the establishment of two reference materials: one for HCMV and one for EBV quantitative assays. At the next meeting, in September 2009, participants discussed the progress and path forward of the CMV and EBV standards.
The Joint Committee for Traceability in Laboratory Medicine
The JCTLM was formed in 2002 with a Declaration of Cooperation between the International Committee of Weights and Measures, the International Federation of Clinical Chemistry and Laboratory Medicine, and the International Laboratory Accreditation Cooperation and is sponsored by the International Bureau of Weighs and Measures. The stimulus for this committee came from the IVDD, Directive 98/79/EC of the European Parliament and of the Council of October 27, 1998, on in vitro diagnostic medical devices. The work of the committee is to facilitate the implementation of traceability to higher order reference materials and measurements that are required by the IVDD. Committee participants include national measurement institutes of several countries, other government agencies (such as US Centers for Disease Control and Prevention), and industry and laboratory associations.
Global standardization can lead to the ultimate goal of achieving correct and equivalent measurements on patients' samples anywhere, at any time, and independent of specific assay. Different assays should be able to produce comparable results with consistent uncertainties and intervals. As an important step in that direction, there needs to be higher-order reference materials and reference measurements. The term “higher-order” was not defined in the IVDD; but the requirements are described in ISO 15193 and ISO 15194.40,41 The JCTLM Working Group 1 evaluates submitted nominations for reference materials and methods by determining compliance with these ISO standards. Based on this, two lists are published in the JCTLM database. List 1 describes certified reference materials and measurement methods that are traceable to the SI. The kinds of materials include small molecules, such as electrolytes, metabolites, and some proteins. List 2 includes reference materials, not traceable to the SI, but traceable to an internationally agreed on protocol. Examples here are coagulation factors or blood typing. Working Group 1 is divided into review teams for nine categories of analytes, including nucleic acids. Announcements for new nominations to be considered for inclusion on the lists are made periodically, and listed materials are removed when they are no longer available. The JCTLM database is hosted on the International Bureau of Weighs and Measures website, where documentation to provide transparency to the decision-making process is available (Joint Committee for Traceability in Laboratory Medicine, http://www.bipm.org/en/committees/jc/jctlm, last accessed August 26, 2009). Armbruster and Miller42 have written a comprehensive review article on JCTLM and its role in global standardization.
The JCTLM Working Group 1 Nucleic Acid Review Team has experts from Japan, Europe, and the United States representing bioindustry, molecular diagnostics manufacturers, reference material producers, and public health organizations. The group is preparing the criteria that will be applied to assess the “quality” and “higher-order traceability” of JCTLM database-nominated nucleic acids reference materials, which only have stated “nominal qualities,” most usually sequence. There is limited ISO standard guidance on the criteria that need to be fulfilled to consider a reference material's nominal properties as those of higher-order or high-quality (although issues of identity, stability, homogeneity, and commutability need to be considered). Developing clear guidance on internationally accepted criteria against which the traceability of nucleic acid reference materials can be reviewed will hopefully encourage the nomination of more nucleic acids reference materials to the JCTLM database.
Association for Molecular Pathology
The Association for Molecular Pathology is a not-for-profit scientific society dedicated to the advancement, practice, and science of clinical molecular laboratory medicine and translational research based on the applications of genomics and proteomics (Association for Molecular Pathology, http://www.amp.org, last accessed August 27, 2009). The Association for Molecular Pathology Clinical Practice Committee Working Group on Infectious Disease has focused on the need for standardization of quantitative molecular viral load testing. Quantitative PCR procedures, in particular real-time PCR, are popular ways to monitor viral load because of the reliability and broad linear range of quantification. Many different assays have been developed, often in individual laboratories. The need for reference standards for normalization of laboratory measurements is continually recognized and emphasized by the Association for Molecular Pathology in meetings, workshops, discussion threads, and collaborative studies.28,29,32 The organization is working with the NIST to produce an HCMV Standard Reference Material. Future projects include assessing the need for reference standards for quantitative BK virus and EBV viral load testing.
Perspective and Future Issues
The benefit of having internationally recognized reference materials for quantitative molecular tests is now well recognized by all of the constituencies involved in development and testing: clinical laboratories, independent control and reference material manufacturers, IVD manufacturers, and regulatory and policy makers. Challenges now involve communication of the complex issues across these diverse stakeholders and exploring different approaches to strategic, technical, and applications issues. Strategic issues include agreement on target priorities and design and management of studies. For technical issues, linearity of the materials, the relationship of synthetics to biological products, biological matrices, possible standard reference methods, and commutability are topics that are surfacing.
Until recently, the impact of commutability on standardization of laboratory results has not been adequately stressed. A review in 2006 of the Joint Committee for Traceability in Laboratory Medicine's list of approved reference materials revealed very few that were validated for commutability against native clinical samples.43 One of the most important considerations in the development of effective standards and reference materials is that the material be commutable, ie, behave as closely as possible to the test samples in the full range of measurement procedures expected to make use of the material. Commutability is described as equivalence of the mathematical relationships, or ratios, between the results of different measurement procedures for a reference material and for test samples of normal and pathological origin.44
Commutability does not apply solely to synthetic reference materials. Biological standards are often assumed to be commutable because of their native origin but may not have been specifically validated as such. In 2005 a committee was formed by the Clinical and Laboratory Standards Institute for the development of guidelines to establish consistent assessment of commutability of reference materials.45 Application of commutability to standards and secondary reference materials is expected to be a seminal topic for diagnostics in the future and molecular diagnostics will necessarily be part of that discussion.
Other important technical issues include understanding the relationship between the different approaches to standards that technological advancements may provide in the near future. Plans for the establishment of both synthetic and biological standard materials for HCMV have already been discussed in this review. Experiments to understand the relationship of each of these materials to each other and the role each will play in standardization of HCMV assays will include discussions of traceability, commutability, and strategy.
The recent interest from all levels of stakeholders in the effort to establish international standards for quantitative molecular diagnostics will lead to further inclusivity, transparency, and critical evaluation. It will be important to maintain avenues of communication that have opened up through professional organizations such as Association for Molecular Pathology, European Society for Clinical Virology, and Pan American Society for Clinical Virology. The ILC has reaffirmed its primary objective to work in the area of these technical and strategic issues regarding the establishment and replenishment of international standards and is encouraged by the significant attention being given to these critical issues by others. Discussion, critique, and constructive efforts specific to the issues in quantitative molecular diagnostic standards are really just beginning. Continued and active involvement of all stakeholders will be necessary to achieve the common goal of reliable and commutable patient results in molecular infectious disease testing.
Acknowledgements
We acknowledge Jan Turczyn and the scientists at Siemens Healthcare Diagnostics (Berkeley, CA) for their contribution of HCV standard materials and their participation in the synthetic materials feasibility experiments. We also acknowledge Micha Nübling and the Paul Erlich Institute for the contribution of the HCV Paul Erlich Institute biological standard reference used in the synthetic materials feasibility experiments. Finally, we acknowledge the framework and support of the Industrial Liaison Committee and the chair, Jim Reid. The need for this review was recognized, formed, and supported through the organization's discussions, workshops, and experimentation. Particular thanks are given to Giuseppe Colucci for editorial input.
Footnotes
Certain commercial equipment and materials are identified to specify the experimental procedure. This does not imply endorsement by the National Institute of Standards and Technology nor does it imply that the material or equipment is the best available for the purpose.
References
- 1.Mellors JW, Muñoz A, Giorgi JV, Margolick JB, Tassoni CJ, Gupta P, Kingsley LA, Todd JA, Saah AJ, Detels R, Phair JP, Rinaldo CR. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann Intern Med. 1997;126:946–954. doi: 10.7326/0003-4819-126-12-199706150-00003. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien WA, Hartigan PM, Daar ES, Simberkoff MS, Hamilton JD. Changes in plasma HIV RNA levels and CD4+ counts predict both response to antiretroviral therapy and therapeutic failure: VA cooperative study group on AIDS. Ann Intern Med. 1997;126:983–985. doi: 10.7326/0003-4819-126-12-199706150-00002. [DOI] [PubMed] [Google Scholar]
- 3.Asian Pacific Association for the Study of the Liver (APASL) Hepatitis C Working Party Asian Pacific Association for the Study of the Liver consensus statements on the diagnosis, management and treatment of hepatitis C virus infection. J Gastroenterol Hepatol. 2007;22:615–633. doi: 10.1111/j.1440-1746.2007.04883.x. [DOI] [PubMed] [Google Scholar]
- 4.Hammer SM, Saag MS, Schechter M, Montaner J, Schooley RT, Jacobsen DM, Thompson MA, Carpenter CCJ, Fischel MA, Gazzard BG, Gatell JM, Hirsch MS, Katzenstein DA, Richman DD, Vella S, Yeni PG, Volberding PA. Treatment for adult HIV infection. 2006 recommendations of the AIDS society—USA panel. JAMA. 2006;296:827–843. doi: 10.1001/jama.296.7.827. [DOI] [PubMed] [Google Scholar]
- 5.Keeffe EB, Dieterich DT, Han SH, Jacobson IM, Martin P, Schiff ER, Tobias H, Wright TL. A treatment algorithm for the management of chronic Hepatitis B virus infection in the United States: an update. Clin Gastroenterol Hepatol. 2006;4:936–962. doi: 10.1016/j.cgh.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Best SJ, Gust AP, Johnson EIM, McGavin CH, Dax E. Quality of human immunodeficiency virus viral load testing in Australia. J Clin Microbiol. 2000;38:4015–4020. doi: 10.1128/jcm.38.11.4015-4020.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schirm J, van Loon AM, Valetnine-Thon E, Klapper P, Reid J, Cleator G. External quality assessment program for qualitative and quantitative detection of hepatitis C virus RNA in diagnostic virology. J Clin Microbiol. 2002;40:2973–2980. doi: 10.1128/JCM.40.8.2973-2980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaaijer HL, Cuypers HT, Reesink HW, Winkel IN, Gerken G, Lelie PN. Reliability of polymerase chain reaction for detection of hepatitis C virus. Lancet. 1993;341:722–724. doi: 10.1016/0140-6736(93)90488-3. [DOI] [PubMed] [Google Scholar]
- 9.Clinical and Laboratory Standards Institute . Quantitative Molecular Methods for Infectious Diseases; Approved Guideline. CLSI Document MM6-A. Clinical and Laboratory Standards Institute; Wayne, PA: 2003. p. 30. [Google Scholar]
- 10.World Health Organization . Annex Ii Recommendations for the Preparation, Characterization and Establishment of International and Other Biological Reference Standards (revised 2004). WHO Technical Report Series, No. 932. World Health Organization; Geneva: 2006. p. 80. [Google Scholar]
- 11.Saldanha J, Heath A, Lelie N, WHO Collaborative Study Group Establishment of the first international standard for nucleic acid amplification technology assays for HCV RNA. Vox Sang. 1999;76:149–158. doi: 10.1159/000031040. [DOI] [PubMed] [Google Scholar]
- 12.Saldanha J, Heath A, Aberham C, Albrecht J, Gentili G, Gessner M, Pisani G. World Health Organization collaborative study to establish a replacement WHO international standard for hepatitis C virus RNA nucleic acid amplification technology assays. Vox Sang. 2005;88:202–204. doi: 10.1111/j.1423-0410.2005.00606.x. [DOI] [PubMed] [Google Scholar]
- 13.Baylis SA, Heath AB, Collaborative Study Group . WHO collaborative study to establish a replacement WHO international standard for hepatitis C virus RNA nucleic acid amplification technology (NAT)-based assays, BS07.2055. World Health Organization; Geneva: 2007. [Google Scholar]
- 14.Saldanha J, Heath A, Lelie N, Collaborative Study Group Collaborative study to calibrate hepatitis C virus genotypes 2–6 against the HCV international standard 96/790 (genotype 1) Vox Sang. 2003;84:20–27. doi: 10.1046/j.1423-0410.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 15.Saldanha J, Heath A, Lelie N, Pisani G, Nubling M, Yu R, Collaborative Study Group Calibration of HCV working reagents for NAT assays against the HCV international standard. Vox Sang. 2000;78:217–224. doi: 10.1159/000031184. [DOI] [PubMed] [Google Scholar]
- 16.Saldanha J. Standardization: a progress report. Biologicals. 1999;27:285–289. doi: 10.1006/biol.1999.0222. [DOI] [PubMed] [Google Scholar]
- 17.Saldanha J. Validation and standardization of nucleic acid amplification technology (NAT) assays for the detection of viral contamination of blood and blood products. J Clin Virol. 2001;20:7–13. doi: 10.1016/s1386-6532(00)00149-9. [DOI] [PubMed] [Google Scholar]
- 18.Holmes H, Davis C, Heath A, Hewlett I, Lelie N. An international collaborative study to establish the 1st international standard for HIV-1 RNA for use in nucleic acid-based techniques. J Virol Methods. 2001;92:141–150. doi: 10.1016/s0166-0934(00)00283-4. [DOI] [PubMed] [Google Scholar]
- 19.Saldanha J, Gerlach W, Lelie N, Dawson P, Heerman K, Health A, WHO Collaborative Study Group An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang. 2001;80:63–71. doi: 10.1046/j.1423-0410.2001.00003.x. [DOI] [PubMed] [Google Scholar]
- 20.Saldanha J, Heath A, Lelie N, Pisani G, Yu MY, Collaborative Study Group A World Health Organization International Standard for hepatitis A virus RNA nucleic acid amplification technology assays. Vox Sang. 2005;89:52–58. doi: 10.1111/j.1423-0410.2005.00633.x. [DOI] [PubMed] [Google Scholar]
- 21.Saldanha J, Lelie N, Yu MW, B19 Collaborative Study Group Establishment of the first World Health Organization International Standard for human parvovirus B19 DNA nucleic acid amplification techniques. Vox Sang. 2002;82:24–31. doi: 10.1046/j.1423-0410.2002.00132.x. [DOI] [PubMed] [Google Scholar]
- 22.Wilkinson DE, Heath AB, Ferguson M, World Health Organization Human Papillomavirus DNA International Collaborative Study Group WHO collaborative study to establish WHO international standards for human papillomavirus type 16 DNA and type 18 DNA nucleic acid amplification technology (NAT)-based assays. Int J Cancer. 2009 [Epub PMID: 19904756] [Google Scholar]
- 23.Padley D, Heath A, Sutherland A, Chiodini PL, Baylis S, Collaborative Study Group Establishment of the 1st World Health Organization International Standard for Plasmodium falciparum DNA for nucleic acid amplification technique (NAT)-based assays. Malar J. 2008;7:139–147. doi: 10.1186/1475-2875-7-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.World Health Organization . Development of WHO biological reference preparations for blood safety related in vitro diagnostic tests: report of a meeting with the WHO Collaborating Centres for Biological Standards and Standardization. World Health Organization; Geneva: 2007. [Google Scholar]
- 25.Hendricks DA. WHO Consultation on International Standards for in Vitro Clinical Diagnostics Procedures Based on Nucleic Acid Techniques (NAT) World Health Organization; Geneva: 2002. Ideal design characteristics of international standards for quantification of viruses; pp. 4–5. [Google Scholar]
- 26.Halfon P, Bourliere M, Penaranda G, Khiri H, Ouzan D. Real-time PCR assays for hepatitis C virus (HCV) RNA quantification are adequate for clinical management of patients with chronic HCV infection. J Clin Microbiol. 2006;44:2507–2511. doi: 10.1128/JCM.00163-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller Z, Stelzl E, Bozic M, Haas J, March E, Kessler HH. Evaluation of automated sample preparation and quantitative PCR LCx assay for determination of human immunodeficiency virus type 1 RNA. J Clin Microbiol. 2004;42:1439–1443. doi: 10.1128/JCM.42.4.1439-1443.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preiksaitis JK, Pang XL, Fox JD, Fenton JM, Caliendo AM, Miller GG. Interlaboratory comparison of Epstein-Barr virus viral load assays. Am J Transplant. 2009;9:269–279. doi: 10.1111/j.1600-6143.2008.02514.x. [DOI] [PubMed] [Google Scholar]
- 29.Hayden RT, Hokanson KM, Pounds SB, Bankowski MJ, Belzer SW, Carr J, Diorio D, Forman MS, Joshi Y, Hillyard D, Hodinka RL, Nikiforova MN, Romain CA, Stevenson J, Valsamakis A, Balfour HH., Jr Multicenter comparison of different real-time PCR assays for quantitative detection of Epstein-Barr virus. J Clin Microbiol. 2008;46:157–163. doi: 10.1128/JCM.01252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pang XL, Fox JD, Fenton JM, Miller GG, Caliendo AM, Preiksaitis JK. Interlaboratory Comparison of Cytomegalovirus Viral Load Assays. Am J Transplant. 2009;9:258–268. doi: 10.1111/j.1600-6143.2008.02513.x. [DOI] [PubMed] [Google Scholar]
- 31.Collins ML, Zayati C, Detmer J, Daly B, Kolberg JA, Cha T-A, Irving BD, Tucker J, Urdea MS. Preparation and characterization of RNA standards for use in quantitative branched DNA hybridization assays. Anal Biochem. 1995;226:120–129. doi: 10.1006/abio.1995.1199. [DOI] [PubMed] [Google Scholar]
- 32.Caliendo AM, St. George K, Kao S-Y, Allega J, Tan B-H, LaFontaine R, Bui L, Rinaldo CR. Comparison of quantitative cytomegalovirus (CMV) PCR in plasma and CMV antigenemia assay: clinical utility of the prototype Amplicor CMV monitor test in transplant recipients. J Clin Microbiol. 2000;3:2122–2127. doi: 10.1128/jcm.38.6.2122-2127.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nye MB, Leman AR, Meyer ME, Menegus MA, Rothberg PG. Sequence diversity in the glycoprotein B gene complicates real-time PCR assays for detection and quantification of cytomegalovirus. J Clin Microbiol. 2005;43:4968–4971. doi: 10.1128/JCM.43.10.4968-4971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lengerova M, Racil Z, Volfova P, Lochmanova J, Berkovcova J, Dvorakava D, Vorlicek J, Mayer J. Real-time PCR diagnostics failure caused by nucleotide variability within exon 4 of the human cytomegalovirus major immediate-early gene. J Clin Microbiol. 2007;45:1042–1044. doi: 10.1128/JCM.01109-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JDC, Wengenack NL, Rosenblatt JE, Cockerill FR, Smith TF. Real-time PCR in clinical microbiology: applications for a routine laboratory testing. Clin Microbiol Rev. 2006;19:165–200. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchini A, Liu H, Zhu H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001;75:1870–1878. doi: 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, Patterson CE, Yang S, Zhu H. Coupling generation of cytomegalovirus deletion mutants and amplification of viral BAC clones. J Virol Methods. 2004;121:137–143. doi: 10.1016/j.jviromet.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Stinski MF, Mocarski ES, Thomsen DR. DNA of human cytomegalovirus size heterogeneity and defectiveness resulting from serial undiluted passage. J Virol. 1979;31:231–239. doi: 10.1128/jvi.31.1.231-239.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holden MJ, Rabb SA, Tewari YB, Winchester MR. Traceable phosphorus measurements by ICP-OES and HPLC for the quantification of DNA. Anal Chem. 2007;79:1536–1541. doi: 10.1021/ac061463b. [DOI] [PubMed] [Google Scholar]
- 40.International Standards Organization . In vitro diagnostic medical devices—measurement of quantities in samples of biological origin. Presentation of reference measurement procedures. ISO Document 15193. International Standards Organization; Geneva: 2002. pp. 9–16. [Google Scholar]
- 41.International Standards Organization . In vitro diagnostic medical devices—;Measurement of quantities in samples of biological origin. Description of reference materials. ISO Document 15194. International Standards Organization; Geneva: 2002. [Google Scholar]
- 42.Armbruster D, Miller RR. The Joint Committee for Traceability in Laboratory Medicine (JCTLM): a global approach to promote the standardization of clinical laboratory test results. Clin Biochem Rev. 2007;28:105–113. [PMC free article] [PubMed] [Google Scholar]
- 43.Miller WG, Myers GL, Rej R. Why commutability matters. Clin Chem. 2006;52:553–554. doi: 10.1373/clinchem.2005.063511. [DOI] [PubMed] [Google Scholar]
- 44.Vesper HW, Miller WG, Myers GL. Reference materials and commutability. Clin Biochem Rev. 2007;28:139–147. [PMC free article] [PubMed] [Google Scholar]
- 45.Clinical and Laboratory Standards Institute . Characterization and quantification of commutable reference materials for laboratory medicine, proposed guideline. CLSI Document C53-P. Clinical and Laboratory Standards Institute; Wayne, PA: 2008. [Google Scholar]