

Abstract
Cytogenetic abnormalities play a major role in the prognosis of patients with chronic lymphocytic leukemia (CLL). Several methods have emerged to try to best identify these abnormalities. We used fluorescence in situ hybridization (FISH) to determine the frequency of cytogenetic changes in our CLL patient population. We also evaluated the effectiveness of multiplex ligation-dependent probe amplification (MLPA) in detecting these abnormalities. Sixty-two B-CLL patients and 20 healthy controls were enrolled, and FISH and MLPA analyses were performed on peripheral blood samples. Using FISH, genomic aberrations were found in 73% of patients and presented as follows: single 13q14.3 deletion (60%), trisomy 12 (7%), ATM deletion (6%), 17p13.1 deletion (2%). MLPA analyses done on 61/62 patients showed sensitivity and specificity values of 90% and 100% respectively. MLPA revealed several additional copy number changes, the most common being 19p13 (LDLR and CDKN2D). Moreover, the cost for MLPA analysis, including technical time and reagents, is 86% less than FISH. In conclusion, cytogenetic abnormalities are a common finding in CLL patients, and MLPA is a reliable approach that is more cost effective and faster than FISH. Despite MLPA limitations of sensitivity, it can be used as a first-line screen and complementary test to FISH analysis.
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the western world. It is considered an incurable disease and treatment has not changed overall survival.1,2 This diagnosis encompasses a heterogeneous group of B-cell neoplasms, in which patients demonstrate a variable clinical course. The disease progression, which is classified in the Americas according to the Rai system, ranges from stage 0 where patients display lymphocytosis in blood and marrow and survive more than 10 years, to stage IV where patients with lymphadenopathy, with or without splenomegaly and hepatomegaly, underproductive anemia, and low platelet counts have an average survival of 1.5 to 2 years.3 It is likely that patients with poor prognosis may benefit from early therapy.4 Laboratory indices such as tumor load, elevated LDH, lymphocyte doubling time, increased percentage of prolymphocytes, increased serum β2 microglobulin or soluble CD23, and diffuse bone marrow infiltration have been ineffective at predicting an impending bad prognosis in individual patients.
Two distinct disease courses have recently been identified in CLL that depend on the mutational status of immunoglobulin variable region IgVH.5,6 As B lymphocytes mature, they pass through the germinal center of the lymph node follicles where they transform into rapidly proliferating centroblasts. During this process, somatic mutations in the variable region of the immunoglobulin genes are inserted in a randomized manner. The mutational status of the immunoglobulin variable region has been shown to distinguish CLL patients with good and bad prognosis. Unfortunately, IgVH mutation testing is not practical to perform for most clinical laboratories. Investigators have therefore looked for surrogate markers to prognosticate CLL patients. The expression of ZAP-70 held initial promise but its detection by flow cytometry is fraught with technical difficulties and therefore is also problematic in many laboratories.
In many studies multiple recurring chromosomal aberrations appear to be effective prognostic markers in CLL. The most common abnormalities include 11q deletions, trisomy 12, 17p deletions and 13q deletions. In 2000, Dohner et al7 used five fluorescence in situ hybridization (FISH) probes to show that 80% of CLL cases had at least one of these genomic aberrations and that patient prognosis could be predicted even in early stages of disease. Patients with 13q deletion as the sole abnormality had the best prognosis. The worst prognosis was seen in patients with 17p deletion (or p53 mutation). Detection of chromosome abnormalities by conventional metaphase cytogenetics has been challenging due to limited proliferation of CLL cells in vitro. However, due to technical advances in culture conditions that use CpG-oligonucleotide DSP30 plus interleukin 2, conventional cytogenetics can now be used to efficiently detect complex aberrant karyotypes.8,9 Nevertheless, interphase fluorescence in situ hybridization (I-FISH) using probes specific for the most common abnormalities is generally considered the gold standard approach. Both of these technologies are expensive and time consuming.
Recently, a polymerase chain reaction (PCR)-based approach for detecting chromosomal duplications and deletions, multiplex ligation-dependent probe amplification (MLPA), has emerged that is reported to be a fast, sensitive and cost effective method that can detect copy number changes of up to 45 nucleic acid sequences in one simple reaction, so is ideally suited to detect the deletions and trisomies associated with CLL. Each MLPA probe consists of two oligonucleotides, one consisting of a sequence specific for the forward PCR primer as well as sequence specific to a portion of the DNA region of interest. The second oligonucleotide consists of sequence specific for the reverse primer, an adjacent portion of the DNA region of interest and a stuffer sequence between the two, which is different in length for each probe. The two oligonucleotides will ligate together and amplify only if they bind side by side on the patient's DNA. All probe ligation products are amplified by PCR using only one primer pair (as they have common primer sequences flanking the gene-specific sequences). The products of individual probes are distinguishable because the length of each amplification product is unique due to variations in the length of the stuffer sequence. The resulting amplification products are size separated and quantified by capillary electrophoresis.10 Despite the apparent applicability of the MLPA technique in detecting chromosomal abnormalities in CLL, there is minimal supportive evidence. Buijs et al11 used MLPA to look for common chromosomal alterations in 54 cases of CLL and concluded that it was a powerful and economically attractive technology that detected alterations when the percentage of mutated cells was greater than 35%, which is the case for most CLL patients at diagnosis. Coll-Mulet et al came to a similar conclusion from their study of 50 CLL patients.12 Additional studies are required to validate this approach.
We have used FISH as the gold standard to prospectively determine the proportion of high and low risk patients in the Nova Scotia CLL population who might benefit from treatment tailored to their prognostic markers. Secondly, we compared MLPA and FISH as methods to detect prognostic markers in CLL, in terms of sensitivity, specificity, processing time and cost.
Materials and Methods
Case Selection and Sample Collection
Sixty three previously diagnosed (untreated and treated 6 months ago) and new untreated CLL patients were enrolled prospectively in this study. Patients with atypical CLL were excluded. All participants attended the hematology clinic at the QEII Health Sciences Centre from August 2007 to June 2008. Written informed consent was obtained from each participant as required by the Capital District Health Research Ethics Board. Peripheral blood samples were collected in EDTA and sodium heparin tubes.
Interphase FISH
FISH analysis for chromosomal abnormalities was performed on peripheral blood cells collected on sodium heparin. Leukocytes were harvested without prior culture or stimulation and preserved in a fixative solution of methanol/acetic acid at −20°C using standard methods. The cell suspension was spread on two slides for each patient in preparation for FISH. Commercial, multicolor probes provided by Vysis laboratories (New York, NY) were used that included probes for 11q22 (LSI ATM), 13q14.3 (LSI D13S319), 13q34 (LSI 13q34), 12p11.1-q11 (CEP12) and 17p13.1 (LSI P53). Probes were validated by testing 20 normal controls to establish the cut off point for lower limit of abnormal number of probe signals. We analyzed probe hybridization in 200 nuclei from each individual in the control group. The number of false positive cells for any given signal pattern were identified among these nuclei. We then used the β inverse formula in Microsoft Excel to determine the normal cut off at the upper bound of the 95th percentile. Moreover, two independent analysts scored 50 nuclei for each patient. Scoring of each sample was achieved using a fluorescence microscope. A normal control was included with every patient run.
DNA Extraction
DNA was extracted from peripheral blood collected on EDTA using a standard salting out method. It was dissolved in TE buffer at a concentration of 10–20 ng/μl and stored in the fridge at 4°C. DNA was obtained from 55 patients and 10 normal controls.
MLPA Reaction
The MLPA CLL kit (P037 and P038) from MRC-Holland (Amsterdam, The Netherlands) was used to detect chromosomal abnormalities commonly associated with CLL. The probe mix consists of 55 probes specific for chromosomal positions, 17p13, 13q14, 11q23, 10q23, 2p24, 8q24, 6q25-26, 9p21, and other regions on chromosomes 12 and 19. The manufacturer's protocol was followed. Three reaction phases included hybridization, ligation, and PCR. For hybridization, 5 μl of DNA/TE buffer solution was heated to 98°C for 5 minutes, followed by addition of the probe mix. Samples were re-heated for 1 minute at 95°C and then incubated for 16 hours at 60°C. In the ligation phase, the annealed oligonucleotide probes were incubated with ligase at 54°C. In the third phase, 35 cycles of PCR were performed using PCR master mix and SALSA polymerase that was added to the ligated products at 60°C. PCR reactions were performed in an MJ thermal cycler with a heated lid. Finally, a gene scan ABI3130XL capillary electrophoresis (Burlington, Canada) was used to size separate and analyze PCR products.
MLPA Data Analysis
Genemarker software (Soft Genetics, State College, PA) performed the MLPA analysis by dividing relative peak area of each probe by the sum of all probe peak areas. The ratio was then normalized in relation to a normal control. Threshold of detection was set at 0.85–1.2. The presence of more than 2 outliers was required to call a positive result for any chromosomal abnormality identified by FISH.
Cost-Effectiveness Analysis
The overall testing time and cost for analysis of each sample, including patients and normal controls were estimated taking into account hands-on technician time, technologist salary per hour, and the cost of materials and supplies. Calculations assumed that samples were processed in batches of 10. The aforementioned analysis applied for both FISH and MLPA.
Statistical Analysis
A Wilcoxon rank sum test and a Kruskal-Wallis nonparametric test was performed to compare variables between subjects with normal results and subjects with cytogenetic abnormalities.
Results
Demographic and Clinical Data
The demographic data for 62 patients are summarized in Table 1.
Table 1.
Demographic and Clinical Data
| Age (years) | Frequency |
|---|---|
| <50 | 6 |
| 50–70 | 35 |
| >70 | 21 |
| Sex | Frequency |
| Male | 46 |
| Female | 16 |
| Median white blood cell count /L | 25 × 109 |
| White blood cell count range | 2–202 |
| Median absolute lymphocyte count | 14 × 109 |
| Absolute lymphocyte count range | 1–110 |
| Median smear cells (%) | 44 |
| Rai stage, no (%) | |
| Stage 0 | 46 (74%) |
| Stage 1 | 14 (22%) |
| Stage 2 | 0 (0%) |
| Stage 3 | 1 (2%) |
| Stage 4 | 1 (2%) |
FISH
Interphase FISH was used to detect chromosome abnormalities commonly associated with CLL. Validation procedures established lower limits for positivity of 3%, 5%, 2%, 5%, and 1% for 13q14.3, 13q34, 11q22, and 17p13.1 deletions and trisomy 12, respectively. The frequency of each defect, in the patients tested, is shown in Table 2. Forty-seven of 62 patients (75%) had an abnormal karyotype. While 42 of these had a single abnormality detected, five had two abnormalities. The most common defect was 13q14.3, which was present in 39 patients. The deletion was heterozygous in the majority of these, but 4 cases were homozygous. Trisomy 12 was observed as an isolated abnormality in 5% of patients. Deletions of 17P13.1, 11q22 or 13q34 were not detected as the sole aberration in any of our patients. 13q14.3 deletion accompanied p53 (17p13.1) deletion in one patient and ATM (11q22) deletion in three patients. Table 3 shows the proportion of abnormal cells detected with each probe.
Table 2.
Frequency of Recurring Cytogenetic Abnormalities Detected by FISH in CLL Patients
| Frequency | Percent | |
|---|---|---|
| Normal | 15 | 25 |
| Abnormal | 47 | 75 |
| Isolated abnormalities | ||
| 13q14.3 deletion | ||
| Monoallelic | 27 | 44 |
| Biallelic | 4 | 6 |
| Monoallelic and biallelic | 8 | 13 |
| Trisomy 12 | 3 | 5 |
| 11q22 deletion | 0 | 0 |
| 17p13.1 deletion | 0 | 0 |
| Combined abnormalities | ||
| 11q22 deletion and 13q14.3 deletion | 3 | 5 |
| Trisomy 12 and 13q14.3 deletion | 1 | 2 |
| 17p13.1 deletion and 13q14.3 deletion | 1 | 2 |
Table 3.
Median and Range of Abnormal Cells Detected by FISH
| Chromosomal aberrations | Median of abnormal cells (range %) |
|---|---|
| 13q14.3 deletion | 60 (10–97) |
| Trisomy 12 | 68 (58–82) |
| 11q22 deletion | 66 (18–96) |
| 17p13.1 deletion | Only one patient with 19% abnormal cells |
MLPA
MLPA analysis for chromosomal abnormalities was performed on 61 of 62 patients to determine its validity as an alternate to FISH for clinical diagnostics. DNA was not recovered from the peripheral blood sample of one patient for unknown reasons. MLPA results were in concordance with FISH results in 54 cases. All 15 cases that were normal by FISH were also normal for those particular abnormalities by MLPA. Discordant results were observed in seven cases. In five of these, 13q14.3 deletions were detected by FISH but not MLPA. MLPA findings partially agreed with FISH results in the remaining two cases with dual abnormalities. FISH detected 11q22/13q14.3 and 17p13.1/13q14.3 deletions, while MLPA only detected the 13q14.3 deletion. However, the 11q22 and 17p13.1 abnormalities were present in only a small proportion of cells. These latter two cases were excluded from the reliability analysis.
When abnormal cells were present at a low level, they were undetectable by the MLPA technique. The 13q14.3 deletions were detected at a minimum concentration of 26%. The sensitivity of MLPA in detecting 13q14.3 deletions was assessed as described below. Due to a small sample size, the lower limits for detection of the remaining abnormalities could not be determined, but 11q22 and 17p13.1 deletions were not detectable at an abnormal cell concentration of 18 and 19%, respectively.
Because the MLPA kit has the capacity to detect abnormalities that are not included in the FISH analysis, additional abnormalities were detected in some patients using this approach. Since this was not the focus of our study, these findings were not confirmed by FISH. These copy number changes included gains in 8q24 MYC (2/61), 2p24 MYCN (4/61), 17p13 (2/61), 19p13 LDLR and CDKN2D (14/61) and loss of 2p24 MYCN (4/61) and 6q25 ESR1 (1/61). No abnormalities with 9p21 CDKN2A and CDKN2B or 10q23.31 PTEN were detected in this population.
Representative MLPA analyses are demonstrated in Figure 1, A–C. Analyses of all MLPA data show a sensitivity of 0.9, specificity of 1, and positive and negative predictive values of 1 and 0.75, respectively.
Figure 1.
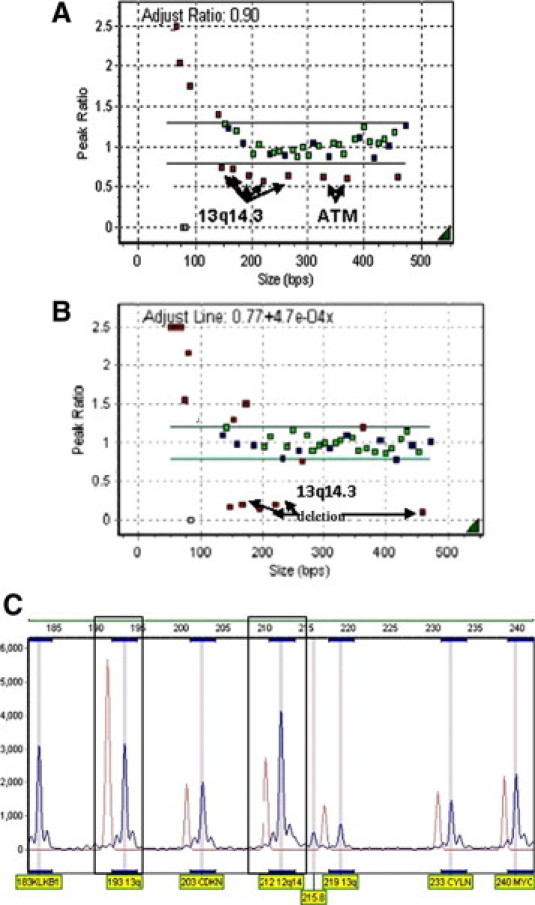
MLPA analysis of three CLL patients. A: Height ratio plot from a patient with heterozygous 13q14.1 and ATM deletions, normalized to that of a normal control. Copy number changes are seen as small red squares that lie outside the threshold detection (green line), which was set at 0.8–1.2. Small green and blue squares represent normal copy number of genes detected by a probe mix specific for CLL chromosomal sequences (green squares) and internal control probes (blue squares) targeting other chromosomes. MLPA probe amplification products below 130 nt represent DNA quantity control fragments. B: A CLL patient with homozygous 13q14.1 deletion. C: A peak profile method of viewing MLPA analysis. Each peak represents a specific probe amplification product. We show a CLL patient (blue) with heterozygous 13q14.3 deletion and trisomy 12 compared with a normal control group (red).
Sensitivity of MLPA Analysis
The sensitivity of MLPA in detecting copy number variation in decreasing numbers of abnormal cells in leukemic peripheral blood was determined and the results are shown in Table 4. DNA from a patient shown by FISH to have 97% CLL cells heterozygous for the 13q14.3 deletion was diluted serially with normal DNA. MLPA reliably detected the 13q14.3 deletion only when it was present in greater than 36.4% of cells.
Table 4.
Sensitivity of MLPA for Detecting a 13q14.3 Deletion in Decreasing Number of CLL Cells
| Proportion of CLL/normal DNA (%) | FISH | MLPA* |
|---|---|---|
| 1. 97.0 | 98 | + |
| 2. 48.5 | 49 | + |
| 3. 36.4 | 23 | + |
| 4. 24.3 | 9 | +† |
| 5. 12.1 | 18 | − |
| 6. 0.0 | 3 | − |
A positive signal indicates that more than two probes for this deletion had a RCN outside the normal range of 0.85–1.2. The patient was heterozygous for this deletion.
The abnormality was only detected in one of three assays at this dilution. All other results were consistent between three assays.
Cost and Time-Effectiveness for FISH Versus MLPA
The time and cost for sample processing and analysis of CLL cases by both FISH and MLPA are shown in Table 5. These values are based on the assumption that samples are batched and analyzed in groups of 10 and include appropriate controls. Analysis by FISH includes scoring of slides by two independent readers. Turnaround time for FISH includes sample processing, overnight hybridization of probes and scoring. For MLPA, it includes DNA extraction, overnight hybridization of probes, PCR, and analysis.
Table 5.
Cost and Time of Analysis for Recurring Abnormalities in CLL Patients by FISH Versus MLPA
| FISH | MLPA | |
|---|---|---|
| Material and supplies | $234 | $40 |
| Technical time for processing | $94 (1.8 hours/sample × 2 readers) | $52 (0.2 hours/sample) |
| Total cost | $328 | $45 |
| Turnaround time | 48–72 hours | 48–72 hours |
Discussion
CLL has generally been considered a disease of the elderly with male predominance. According to the Surveillance Epidemiology and End Results (SEER) cancer statistics review (http://seer.cancer.gov/statfacts/html/clyl.html?statfacts_page=clyl.html&x=19&y=19 (accessed July 8, 2008), the median age of CLL patients at the time of diagnosis is 72 years.13 Approximately 11% are diagnosed below the age of 55 years, with 9% between age 45 and 54. Similarly, 16% of our patients were diagnosed below age 55 with 3% below age 45. We also report a predominance of males with a ratio of 2.9:1. While this male predominance is somewhat higher than the 2:1 ratio recorded in SEER, Omoti's demographic data from a single Nigerian Centre contradicts the North American data showing a female preponderance with a male to female ratio of 1:3.14
In accord with Dohner et al7 we have shown that 97% of our patient population, including those with poor prognostic cytogenetic markers such as P53 and ATM deletions, presented at an early stage of disease. The fact that some early stage asymptomatic CLL cases present with poor prognostic cytogenetic markers has induced investigators to start a therapy trial in such patients in the hope that it prolongs survival.15 The current clinical staging system does not take into account cytogenetic markers and is not able to predict patients' clinical course and their treatment requirement, especially at the time of diagnosis.
The first objective of this study was to prospectively determine the proportion of Nova Scotia CLL patients who are at risk of disease progression, based on the presence of well-established cytogenetic prognostic markers,16,17,18,19 with a view to individualized therapy. FISH analysis is reported to consistently detect abnormalities in 64% of CLL patients4,20 and was used here as the gold standard to identify these markers. Despite the small sample size, our observations showed a frequency similar to previously published results. The most common aberration was the 13q14.3 deletion (44%), while 11q and 17p deletions and trisomy 12 were less frequent (5%, 2%, and 7% respectively). See Table 6 for a summary of other reports.
Table 6.
Published Studies on the Frequency of Cytogenetic Aberrations in CLL by FISH
| Published study reference number |
|||||||
|---|---|---|---|---|---|---|---|
| 7 | 4 | 5 | 6 | 7 | 18 | 19 | |
| Number of pts | 325 | 113 | 153 | 509 | 100 | 132 | 500 |
| Normal karyotype | 18 | 108 | 36 | ||||
| 13q deletion | 178 | 56 | 81 | 160 | 44 | 76 | 287 |
| 11q deletion | 58 | 13 | 14 | 17 | 23 | 21 | 60 |
| 12q trisomy | 53 | 21 | 26 | 71 | 11 | 17 | 68 |
| 17p deletion | 23 | 7 | 16 | 17 | 12 | 7 | 35 |
| 6q deletion | 21 | 9 | 23 | ||||
The number of samples per column can be greater than the number of patients if multiple abnormalities are present.
FISH has been validated as a reliable diagnostic approach. However, it is expensive and time consuming. For these reasons our second objective was to compare MLPA to FISH as an alternate approach for detecting deletions and duplications associated with CLL. The results were highly concordant. Compared with FISH, MLPA was highly specific, with no false positive results. MLPA also had a good detection sensitivity of 90%, as has been previously suggested.11,21,22 False negative results were seen in seven patients (five completely discordant and two partially discordant cases with dual abnormalities). In all cases these negative results can be attributed to a low proportion of mutant cells, ranging from 10% to 23%. These results are consistent with the MLPA kit manufacturer's claim of probe mixes P037 and P038 being able to detect abnormalities in cases with greater than 30% to 60% abnormal cells. Indeed, our data showed that we were able to reliably detect the 13q14.3 abnormality in DNA samples containing approximately 36% leukemia cells, by using a reliable method of adjusting the detection threshold and using multiple normal controls. We recommend a separate control for each MLPA assay to decrease the rate for false positive results. Our detection rate is superior to that of Buijs11 who reported a threshold for detection of 40% for RB1 deletion, 54% for ATM deletion, and 54% for trisomy 12. Coll-Mulet et al12 reported a threshold for detection of abnormalities of 25%. They may have achieved a higher level of sensitivity from their analyses because they extracted DNA for MLPA from purified B cells in all cases with fewer than 80% lymphocytes in whole blood, while we extracted DNA directly from whole blood in all cases. These studies show that MLPA has a very high level of specificity and, although less sensitive than FISH, has an adequate level of sensitivity for detecting cytogenetic abnormalities in CLL, especially in the setting of symptomatic disease progression, which usually presents with an increase in lymphocyte count.
MLPA was found to be superior to FISH according to other criteria. Because of the multiplex nature of this approach it has the ability to detect a large number of genetic alterations that are not present in the standard FISH analysis for CLL. For Example, the current CLL MLPA kits contain probes targeting several areas on 13q14 including MIR15-16, which belong to the microRNA class of genes.23 miR15a and miR16-1 down-regulate BCL-2 and thus induce apoptosis. They are reported to be deleted in 15–65% of patients with CLL.23,24 Likewise, we have found that 52% of the study patients showed reduced copy number of those genes. miR15a and miR16-1 are also reported to be associated with high expression of ZAP-70 and disease progression.25 Therefore, the ability to identify the microRNA signature in CLL patients is an additional advantage of the MLPA test.
Several additional defects were detected in our patient population, although their presence was not confirmed by FISH due to the unavailability of FISH probes for these chromosomal regions in our laboratory. These additional markers were detected as the sole defect in only one case. This suggests that they are typically secondary to the defects more commonly associated with CLL. In two of our patients who displayed a 13q14.3 deletion, a gain of MYC on 8q24, which is a proto-oncogene that drives cell proliferation, was also found. In contrast, to this increase, Korz et al26 reported that 55% of the B-CLL patients showed down-regulated expression of MYC proto-oncogene. Hence, the role of MYC is still unknown in these patients. A second oncogene gain, 2p24 MYCN was noted in four cases. MYCN is found in CLL patients with mutated and unmutated IgVH.,27 which predict favorable and unfavorable prognosis respectively, therefore, the implication of MYCN as an isolated abnormality is not yet clear. We also observed a gain of 19p13 LDLR and CDKN2D in 14 patients. Previous reports demonstrated a strong association between trisomy 19 and trisomy 12, but of the four patients reported here with trisomy 12, only one showed a copy number gain of 19p13. A strong association between trisomy 12, trisomy 19 and IgVH hypermutation27 has also been demonstrated, whereas an isolated trisomy 12 is associated with unmutated IgVH and progressive disease.9 MLPA is therefore a helpful tool to stratify patients with trisomy 12 abnormalities that are characterized by heterogeneity. Four other patients showed a loss of 2p24 MYCN, which is as an uncommon finding in CLL.27 One final case showed deleted 6q25 ESR1, which was seen in 6% of CLL patients.28 Deletions of CDKN2A or CDKN2B on 9p21 were not detected. The lack of 9p21 deletions in our patients compared with those studied by Buijs, may reflect the fact that his patients had advanced stage disease, while the majority of our patients were at an early stage.
We compared the time required from sample processing to reporting of results, using FISH versus MLPA. Results were obtained in approximately the same time with either technique. However, only 1/12 of the hands-on technical time was required for MLPA. This, together with the lower cost of reagents would reduce the cost for detecting cytogenetic markers in CLL by 86%.
In summary, compared with FISH as the current gold standard testing method, the MLPA technique was less costly; it was reproducible and allowed simultaneous analyses for large numbers of samples in a shorter time. On the other hand, it was less sensitive than FISH, being able to detect only those abnormalities that were present in a significant proportion of cells. Since FISH analysis is limited by the number of probes used, it underestimates the complexity of prognostic cytogenetic markers in CLL.8,9 Similar findings have been reported in the detection of chromosomal abnormalities associated with other diseases.21,29,30 We conclude, therefore, that MLPA is an ideal screening technique, and could be used as a complementary method to FISH.
Acknowledgements
We thank the division of Anatomical Pathology for allowing us to share their equipment, all technicians in the molecular biology division, especially Marissa Gaudet for technical assistance and Kara Thompson for help with statistical analyses.
Footnotes
Supported by grants from the Capital District Health Research Services and Hematology Tumor Site Group.
References
- 1.Richard Lee G, Foerster J. Chronic lymphocytic leukemia. In: Maxwell MW, Foerster J, Lukens J, Paraskevas F, John PG, Rodgers GM, editors. Wintrobe's Clinical Hematology. ed 10. Lippincott Williams & Wilkins; 1999. p. 2415. [Google Scholar]
- 2.Dighiero G. CLL biology and prognosis. Hematology Am Soc Hematol Educ Program. 2005:278–284. doi: 10.1182/asheducation-2005.1.278. [DOI] [PubMed] [Google Scholar]
- 3.Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood. 1975;46:219–234. doi: 10.1182/blood-2016-08-737650. [DOI] [PubMed] [Google Scholar]
- 4.Boyd K, Dearden CE. Alemtuzumab in the treatment of chronic lymphocytic lymphoma. Expert Rev Anticancer Ther. 2008;8:525–533. doi: 10.1586/14737140.8.4.525. [DOI] [PubMed] [Google Scholar]
- 5.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–1854. [PubMed] [Google Scholar]
- 6.Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, Lichtman SM, Schulman P, Vinciguerra VP, Rai KR, Ferrarini M, Chiorazzi N. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–1847. [PubMed] [Google Scholar]
- 7.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, Döhner K, Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 8.Dicker F, Schnittger S, Haferlach T, Kern W, Schoch C. Immunostimulatory oligonucleotide-induced metaphase cytogenetics detect chromosomal aberrations in 80% of CLL patients: a study of 132 CLL cases with correlation to FISH, IgVH status, and CD38 expression. Blood. 2006;108:3152–3160. doi: 10.1182/blood-2006-02-005322. [DOI] [PubMed] [Google Scholar]
- 9.Haferlach C, Dicker F, Schnittger S, Kern W, Haferlach T. Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia. 2007;21:2442–2451. doi: 10.1038/sj.leu.2404935. [DOI] [PubMed] [Google Scholar]
- 10.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buijs A, Krijtenburg PJ, Meijer E. Detection of risk-identifying chromosomal abnormalities and genomic profiling by multiplex ligation-dependent probe amplification in chronic lymphocytic leukemia. Haematologica. 2006;91:1434–1435. [PubMed] [Google Scholar]
- 12.Coll-Mulet L, Santidrian AF, Cosialls AM, Iglesias-Serret D, de Frias M, Grau J, Menoyo A, Gonzalez-Barca E, Pons G, Domingo A, Gil J. Multiplex ligation-dependent probe amplification for detection of genomic alterations in chronic lymphocytic leukaemia. Br J Haematol. 2008;142:793–801. doi: 10.1111/j.1365-2141.2008.07268.x. [DOI] [PubMed] [Google Scholar]
- 13.National Cancer Institute . Surveillance Epidemiology and End Results (SEER) Cancer Statistics Review 1975–2001. US National Institutes of Health; 2001. [Google Scholar]
- 14.Omoti CE, Awodu OA, Bazuaye GN. Chronic lymphoid leukaemia: clinico-haematological correlation and outcome in a single institution in Niger Delta region of Nigeria. Int J Lab Hematol. 2007;29:426–432. doi: 10.1111/j.1751-553X.2007.00888.x. [DOI] [PubMed] [Google Scholar]
- 15.Dighiero G, Maloum K, Desablens B, Cazin B, Navarro M, Leblay R, Leporrier M, Jaubert J, Lepeu G, Dreyfus B, Binet JL, Travade P. Chlorambucil in indolent chronic lymphocytic leukemia: French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med. 1998;338:1506–1514. doi: 10.1056/NEJM199805213382104. [DOI] [PubMed] [Google Scholar]
- 16.Dewald GW, Brockman SR, Paternoster SF, Bone ND, O'Fallon JR, Allmer C, James CD, Jelinek DF, Tschumper RC, Hanson CA, Pruthi RK, Witzig TE, Call TG, Kay NE. Chromosome anomalies detected by interphase fluorescence in situ hybridization: correlation with significant biological features of B-cell chronic lymphocytic leukaemia. Br J Haematol. 2003;121:287–295. doi: 10.1046/j.1365-2141.2003.04265.x. [DOI] [PubMed] [Google Scholar]
- 17.Bacher U, Kern W, Schoch C, Hiddemann W, Haferlach T. Discrimination of chronic lymphocytic leukemia (CLL) and CLL/PL by cytomorphology can clearly be correlated to specific genetic markers as investigated by interphase fluorescence in situ hybridization (FISH) Ann Hematol. 2004;83:349–355. doi: 10.1007/s00277-004-0869-4. [DOI] [PubMed] [Google Scholar]
- 18.Reddy KS. Chronic lymphocytic leukaemia profiled for prognosis using a fluorescence in situ hybridisation panel. Br J Haematol. 2006;132:705–722. doi: 10.1111/j.1365-2141.2005.05919.x. [DOI] [PubMed] [Google Scholar]
- 19.Glassman AB, Hayes KJ. The value of fluorescence in situ hybridization in the diagnosis and prognosis of chronic lymphocytic leukemia. Cancer Genet Cytogenet. 2005;158:88–91. doi: 10.1016/j.cancergencyto.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Goorha S, Glenn MJ, Drozd-Borysiuk E, Chen Z. A set of commercially available fluorescent in-situ hybridization probes efficiently detects cytogenetic abnormalities in patients with chronic lymphocytic leukemia. Genet Med. 2004;6:48–53. doi: 10.1097/01.gim.0000105741.57923.08. [DOI] [PubMed] [Google Scholar]
- 21.Vorstman JA, Jalali GR, Rappaport EF, Hacker AM, Scott C, Emanuel BS. MLPA: a rapid, reliable, and sensitive method for detection and analysis of abnormalities of 22q. Hum Mutat. 2006;27:814–821. doi: 10.1002/humu.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moerland E, van Hezik RL, van der Aa TC, van Beek MW, van den Brule AJ. Detection of HER2 amplification in breast carcinomas: comparison of multiplex ligation-dependent probe amplification (MLPA) and fluorescence in situ hybridization (FISH) combined with automated spot counting. Cell Oncol. 2006;28:151–159. doi: 10.1155/2006/741586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouillette P, Erba H, Kujawski L, Kaminski M, Shedden K, Malek SN. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res. 2008;68:1012–1021. doi: 10.1158/0008-5472.CAN-07-3105. [DOI] [PubMed] [Google Scholar]
- 25.Calin GA, Croce CM. Genomics of chronic lymphocytic leukemia microRNAs as new players with clinical significance. Semin Oncol. 2006;33:167–173. doi: 10.1053/j.seminoncol.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 26.Korz C, Pscherer A, Benner A, Mertens D, Schaffner C, Leupolt E, Döhner H, Stilgenbauer S, Lichter P. Evidence for distinct pathomechanisms in B-cell chronic lymphocytic leukemia and mantle cell lymphoma by quantitative expression analysis of cell cycle and apoptosis-associated genes. Blood. 2002;99:4554–4561. doi: 10.1182/blood.v99.12.4554. [DOI] [PubMed] [Google Scholar]
- 27.Schwaenen C, Nessling M, Wessendorf S, Salvi T, Wrobel G, Radlwimmer B, Kestler HA, Haslinger C, Stilgenbauer S, Döhner H, Bentz M, Lichter P. Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations. Proc Natl Acad Sci USA. 2004;101:1039–1044. doi: 10.1073/pnas.0304717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juliusson G, Gahrton G, Oscier, Fitchett M, Ross F, Brito-Babapulle V, Catovsky D, Knuutila S, Elonen E, Lechleitner M, Tanzer J, Schoenwald M, Castoldi GL, Cuneo A, Nowell P, Peterson L, Kay N. Cytogenetic findings and survival in B-cell chronic lymphocytic leukemia: Second IWCCLL compilation of data on 662 patients. Leukemia Lymphoma. 1991;5(Suppl 1):21–25. doi: 10.3109/10428199109103374. [DOI] [PubMed] [Google Scholar]
- 29.Takata M, Suzuki T, Ansai S, Kimura T, Shirasaki F, Hatta N, Saida T. Genome profiling of melanocytic tumors using multiplex ligation-dependent probe amplification (MLPA): its usefulness as an adjunctive diagnostic tool for melanocytic tumors. J Dermatol Sci. 2005;40:51–57. doi: 10.1016/j.jdermsci.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Slater H, Bruno D, Ren H, La P, Burgess T, Hills L, Nouri S, Schouten J, Choo KH. Improved testing for CMT1A and HNPP using multiplex ligation-dependent probe amplification (MLPA) with rapid DNA preparations: comparison with the interphase FISH method. Hum Mutat. 2004;24:164–171. doi: 10.1002/humu.20072. [DOI] [PubMed] [Google Scholar]