

Abstract
In this part of a series on cardiogenetic founder mutations in the Netherlands, we review the Dutch founder mutations in hypertrophic cardiomyopathy (HCM) patients.
HCM is a common autosomal dominant genetic disease affecting at least one in 500 persons in the general population. Worldwide, most mutations in HCM patients are identified in genes encoding sarcomeric proteins, mainly in the myosin-binding protein C gene (MYBPC3, OMIM #600958) and the beta myosin heavy chain gene (MYH7, OMIM #160760). In the Netherlands, the great majority of mutations occur in the MYBPC3, involving mainly three Dutch founder mutations in the MYBPC3 gene, the c.2373_2374insG, the c.2864_2865delCT and the c.2827C>T mutation. In this review, we describe the genetics of HCM, the genotype-phenotype relation of Dutch founder MYBPC3 gene mutations, the prevalence and the geographic distribution of the Dutch founder mutations, and the consequences for genetic counselling and testing. (Neth Heart J 2010;18:248–54.)
Keywords: Cardiomyopathy, Founder Effect, Mutation, Myosin-binding Protein C
Hypertrophic cardiomyopathy (HCM) is a common genetic disease affecting at least one in 500 persons in the general population.1,2 In 1958 Teare gave a description of HCM when he reported a series of eight young patients who died suddenly from a disorder of the heart muscle.3,4 He was the first to describe the asymmetrical appearance of hypertrophy and its familial nature. He also described a disordered arrangement of muscle fibres at microscopic examination of the hearts of his cases, now known as myocyte disarray.
Nowadays, the diagnosis of HCM is most frequently made at two-dimensional echocardiography. The clinical diagnosis rests on the presence of a hypertrophied, non-dilated left ventricle on echocardiography (left ventricle wall thickness ≥15 mm or ≥13 mm in a patient’s relative) in the absence of other cardiac or systemic diseases that may cause cardiac hypertrophy, such as aortic valve stenosis and Hypertension.5-8
HCM has long been regarded as a disease that mainly affects young people. It was thought that penetrance, i.e. the presence of left ventricular hypertrophy, was complete at approximately 20 to 30 years of age.9 It is more and more recognised that not only symptoms but also hypertrophy can develop at any age and that the clinical course of the disease varies from person to person. Patients may remain asymptomatic throughout life, but the disease can also give rise to heart failure and other adverse events such as sudden cardiac death (SCD) and embolic Stroke. Annual mortality rates from overt HCM in non-selected populations nowadays are 1 to 2% (SCD and end-stage heart failure).10-16
The treatment of patients with HCM is complex and requires understanding of the pathophysiology in each individual patient. Basically, management of the disease is based on relief of symptoms and on risk stratification to prevent SCD. Consensus documents are available to guide the treatment in HCM patients.17-19
Genetics of HCM
HCM is inherited as an autosomal dominant trait. Currently, in more than half of the HCM patients a disease-causing mutation can be identified.20-27 Mutations may occur in a large number of different genes, but are usually found in the genes encoding sarcomeric proteins (table 1). Most HCM patients carry one heterozygous mutation, but in 3 to 5% of cases, patients carry two mutations in the same gene: on both copies of the gene (compound heterozygote or homozygote) or in different genes (digenic). This is generally associated with a more severe phenotype with a younger age of onset (often <10 years) and more adverse events, such as sudden cardiac death (SCD).26,28-31
The two most frequently mutated genes worldwide are the MYBPC3 gene (OMIM #600958) and the MYH7 gene (OMIM #160760). These encode the sarcomeric proteins cardiac myosin-binding protein C and beta myosin heavy chain, respectively. The majority of the mutations worldwide, around 13 to 32%, are identified in the MYBPC3 gene. Around 4 to 25% of the mutations are found in the MYH7 gene.9,23-26,32 Missense mutations (a point mutation in which a single nucleotide is changed, resulting in a codon that codes for a different amino acid) are the most frequent type of mutations in HCM patients. Missense mutations create a mutant protein that interferes with normal function and has a dominant negative effect on function (in which mutant protein adversely affects the normal, wild-type gene product). However, in the MYBPC3 gene most mutations are nonsense or frameshift mutations that are presumed to result in truncated proteins,33,34 suggesting haploinsufficiency (in which the total level of a particular protein produced by the cell is reduced and therefore not sufficient to permit the cell to function normally).
Since the discovery of the first genes for HCM, much has been speculated on specific genotype-phenotype correlations. At first specific mutations, predominantly in the MYH7 gene, were described and were associated with a ‘malignant’ phenotype (more SCD).35-37 Whereas ‘benign’ mutations were reported in families with normal longevity as well.35,36,38-43 The supposed ‘malignant’ and ‘benign’ effects of these mutations, however, have been contradicted in many subsequent studies. Nowadays it is believed that in general there are no clear genotype-phenotype relations with respect to magnitude of left ventricular hypertrophy and incidence of SCD.27,35,39,41,44-46 These studies on genotype-phenotype correlations have also revealed that not all mutation carriers in a family have the same phenotype or are affected. This suggests the existence of modifier genes (genes that affect the expression of another gene), which modulate the phenotypic expression of the disease and incomplete penetrance of the disease (not all mutation carriers are clinically affected).
De novo mutations (newly arisen mutations) and germline mosaicism (mutation present in (part of) the germ cells) are rare in HCM.47-51 Because most mutations are unique for a family, many of the identified mutations have therefore not been described before. In certain countries and populations, however, founder mutations have been identified, arising from a common ancestor many generations ago. These founder mutations often comprise a large part (10 to 25%) of the detected mutations in these countries. Founder mutations for HCM have been found in the Netherlands,52,53 South Africa,54 Finland,55 Italy,56 Japan,57 South Asia58 and in the Amish population of the United States.59
Table 1.
Prevalence of sarcomeric genes associated with HCM.21,22,25,26,74-80
| Gene | Name | Detection rate (%) |
| Sarcomeric | ||
| Myofilament | ||
| - MYBPC3 | Myosin-binding protein C | 13-32 |
| - MYH7 | Beta myosin heavy chain | 4-25 |
| - TNNT2 | Troponin T2 | 0.5-7 |
| - TNNI3 | Cardiac troponin I | <5 |
| - MYL2 | Myosin light chain 2 | <5 |
| - MYL3 | Myosin light chain 3 | <1 |
| - TPM1 | Alpha tropomyosin | <1 |
| - ACTC | Alpha actin | <1 |
| - TNNC1 | Troponin C | <1 |
Founder mutations in the Netherlands
DNA diagnostics for HCM have been available in the Netherlands since 1996. In about 50% of the index patients a disease-causing mutation is detected. The majority of mutations are located in the MYBPC3 gene (20 to 35% of index patients). This can be explained by the occurrence of three Dutch founder mutations in the MYBPC3 gene, the c.2373_2374insG (p.Trp792fsX17) or alternatively c.2373dup, c.2864_2865delCT (p.Pro955fsX95) and c.2827C>T (p.Arg943X).
In a previous survey the c.2373_2374insG mutation was detected in almost a quarter of all HCM patients in the Netherlands, which is unique for cardiogenetic diseases. The mutation was described to be predominantly present in the north-western part of the country. The recombination frequency of the haplotype associated with this founder mutation suggested a common ancestor of at least 25 generations ago.52 The other two founder mutations were each detected in about 5% of the Dutch HCM patients.60
More recently the combined data on DNA diagnostics from all Dutch DNA laboratories screening HCM genes (Academic Medical Centre Amsterdam, Erasmus Medical Centre Rotterdam, Academic Hospital Maastricht) showed that between 1996 and 2006 one of the three prevalent founder mutations was detected in 157 out of 735 HCM index patients (21%). In 126 patients the c.2373_2374insG mutation was detected (17%). The c.2827C>T mutation and the c.2864_2865delCT in the MYBPC3 gene were less prevalent with 19 (2.6%) and 12 (1.6%) patients carrying this mutation, respectively.
Figure 1 shows the distribution of the three founder mutations in the Netherlands (per 1,000,000 inhabitants). We obtained these data by adjusting the different number of index patients carrying a founder mutation per postal code for the number of inhabitants per postal code available at the National Institute Statistics Netherlands (Centraal Bureau voor de Statistiek, CBS). In the Netherlands there are approximately 430,000 postal codes. The analysis was based on the PC2 code (the first two numbers of the postal code), which comprises 90 postal codes. Data were visualised with MapInfo Professional (MapInfo, Toronto, Canada).
Figure 1.

Distribution of the c.2373_2374insG mutation (A), the c.2827C>T mutation (B) and the c.2864_2865delCT mutation (C). Different colours refer to the number of patients with the mutation per 1,000,000 inhabitants in a specific postal code area. Between brackets the number of postal code areas.
The c.2373_2374insG mutation was predominantly present in the north and north-western part of the Netherlands (as described previously), the c.2827C>T mutation predominantly in the south and south-western part of the Netherlands and patients with the c.2864_2865delCT mutation were located in the south-west and middle of the country. This region-specific distribution of patients can explain the higher frequency of specific Dutch founder mutations in literature by DNA laboratories from that region.52,60
Although the relative distribution of the founder mutations in HCM is presented here, the frequency of founder mutations may be biased in those areas where no or very few patients were referred for DNA diagnostics. A study on referral for long-QT syndrome showed regional differences in referral of patients for DNA diagnostics.61 This can also be the case for HCM. However, less than one patient was referred in only four of 90 postal areas (mean number of referrals per region was 8.2), possibly interfering with reliable estimations of the frequency of founder mutations in these specific postal areas.
Until a few years ago, the phenotype of MYBPC3 mutation carriers was considered to be mild and accompanied by a late onset of symptoms,62 but recent studies and also our own observations show that caution must be exercised with assigning prognostic significance to a particular mutation or mutations in a particular gene, since previous studies are performed in small cohorts involving larger families with variable disease penetrance and expression.23,27,67 Data from the Academic Medical Centre Amsterdam on disease penetrance in 88 probands and 213 relatives show that the age of clinical diagnosis is not different for various types of MYBPC3 gene mutations (figure 2). This also holds for the three Dutch founder mutations. Unpublished data on Dutch mutation carriers suggest that there is no association between the phenotype and the mutated gene. All HCM mutations show age dependent and incomplete disease penetrance.68,69 Many founder mutation carriers only show hypertrophy from adulthood, and some are still without significant hypertrophy in their eighties (figure 2).
Figure 2.
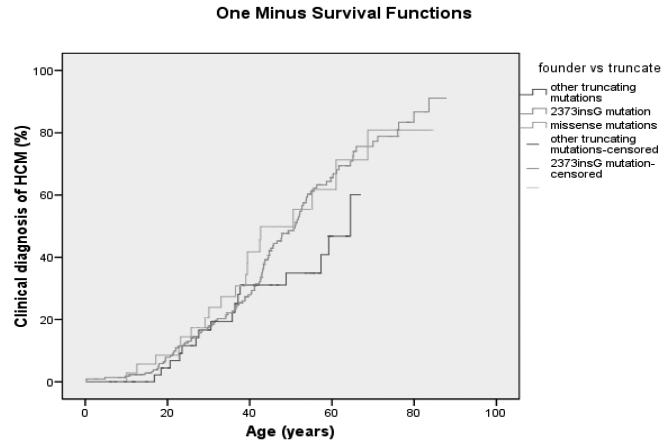
Disease penetrance in 301 HCM mutation carriers with different types of mutations in the MYBPC3 gene.
Screening of relatives: genetic counselling and testing
Consensus guidelines for HCM encourage screening of relatives, because of the risk of HCM-associated sudden cardiac death in these relatives and the availability of effective preventive options.7 Recently a multidisciplinary consensus on genetic testing and counselling in HCM has been developed by different scientific associations (Multidiscipline guideline: Genetic diagnostics and genetic counselling in Hypertrophic Cardiomyopathy (HCM); www.nvvc.nl).70 Identification of a disease-causing mutation in an HCM patient (the index) provides the opportunity to accurately identify the asymptomatic adult relatives who are at risk by means of predictive DNA testing. In the Netherlands diagnostic and predictive DNA testing is covered by standard health insurance, and DNA testing for HCM has increased since 1996. Specialised multidisciplinary cardiogenetics outpatient clinics are now present in all eight university hospitals and in some local hospitals. To detect mutation carriers, systematic screening of relatives in families with a disease-causing mutation, so called cascade screening, is performed in these centres.71 Cascade screening is performed in all HCM families with a detected disease causing mutation in one of the genes associated with HCM, including the Dutch founder mutations. Relatives without the familial HCM mutation can be reassured and discharged from cardiological follow-up. Relatives who carry the familial HCM mutation are, according to international guidelines, like affected HCM patients (with or without mutation), advised to have periodic cardiological screening for left ventricular hypertrophy and risk factors for SCD.18 If no mutation can be detected in an HCM index patient, a genetic form of HCM cannot be excluded and first-degree relatives are still at risk to develop HCM. Therefore they are advised to have periodic cardiological evaluations (annually between 12 and 18 years and once every five years >18 years) directed at diagnosing hypertrophy or ECG abnormalities associated with HCM.18
Predictive genetic testing for hereditary diseases in healthy relatives of index patients is known to be associated with psychosocial distress.72 Relatives tested for a mutation predisposing them to HCM have to deal with the risk of disease and its progression, the threat of SCD, and the possibility of having transmitted the disease to their offspring. In addition, advised lifestyle adjustments and the sudden death of relatives may cause anxiety. Literature shows that psychosocial distress for hereditary heart diseases is mostly concentrated around the period the testing is performed and returns to baseline levels after 18 months, comparable with the general population.72,73 Therefore it is recommended that predictive testing is performed only after genetic counselling, where these issues are discussed with the relatives. We recommend offering psychological support on a voluntary basis to all relatives before predictive genetic testing for HCM.
Acknowledgements
We gratefully acknowledge Y. Blauw (Boston Scientific, Guidant) for providing software (MAPinfo) to map the patients with a founder mutation in the MYBPC3 gene.
References
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785-9. [DOI] [PubMed] [Google Scholar]
- 2.Morita H, Larson MG, Barr SC et al. Single-gene mutations and increased left ventricular wall thickness in the community: the Framingham Heart Study. Circulation. 2006;113:2697-705. [DOI] [PubMed] [Google Scholar]
- 3.Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-8. [DOI] [PubMed] [Google Scholar]
- 5.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270-6. [DOI] [PubMed] [Google Scholar]
- 6.Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26:1699-708. [DOI] [PubMed] [Google Scholar]
- 7.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807-16. [DOI] [PubMed] [Google Scholar]
- 8.McKenna WJ, Spirito P, Desnos M, Dubourg O, Komajda M. Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult members of affected families. Heart. 1997;77:130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charron P, Komajda M. Molecular genetics in hypertrophic cardiomyopathy: towards individualized management of the disease. Expert Rev Mol Diagn. 2006;6:65-78. [DOI] [PubMed] [Google Scholar]
- 10.Cecchi F, Olivotto I, Montereggi A, Squillatini G, Dolara A, Maron BJ. Prognostic value of non-sustained ventricular tachycardia and the potential role of amiodarone treatment in hypertrophic cardiomyopathy: assessment in an unselected non-referral based patient population. Heart. 1998;79:331-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adabag AS, Casey SA, Kuskowski MA, Zenovich AG, Maron BJ. Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45:697-704. [DOI] [PubMed] [Google Scholar]
- 12.Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK, Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-5. [DOI] [PubMed] [Google Scholar]
- 13.Kofflard MJ, Waldstein DJ, Vos J, Ten Cate FJ. Prognosis in hypertrophic cardiomyopathy observed in a large clinic population. Am J Cardiol. 1993;72:939-43. [DOI] [PubMed] [Google Scholar]
- 14.Cannan CR, Reeder GS, Bailey KR, Melton LJI, Gersh BJ. Natural history of hypertrophic cardiomyopathy. A population-based study, 1976 through 1990. Circulation. 1995;92:2488-95. [DOI] [PubMed] [Google Scholar]
- 15.Kyriakidis M, Triposkiadis F, Anastasakis A, et al. Hypertrophic cardiomyopathy in Greece: clinical course and outcome. Chest. 1998;114:1091-6. [DOI] [PubMed] [Google Scholar]
- 16.Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation. 2000;102:858-64. [DOI] [PubMed] [Google Scholar]
- 17.Epstein AE, Dimarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: Executive Summary. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices). Circulation. 2008;117:e350-e408. [DOI] [PubMed] [Google Scholar]
- 18.Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-713. [DOI] [PubMed] [Google Scholar]
- 19.Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385-e484. [DOI] [PubMed] [Google Scholar]
- 20.Ackerman MJ, Van Driest SL, Ommen SR, et al. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002;39:2042-8. [DOI] [PubMed] [Google Scholar]
- 21.Girolami F, Olivotto I, Passerini I, et al. A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2006;7:601-7. [DOI] [PubMed] [Google Scholar]
- 22.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Driest SL, Vasile VC, Ommen SR, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903-10. [DOI] [PubMed] [Google Scholar]
- 24.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:739-44. [DOI] [PubMed] [Google Scholar]
- 25.Erdmann J, Daehmlow S, Wischke S, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet. 2003;64:339-49. [DOI] [PubMed] [Google Scholar]
- 26.Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227-32. [DOI] [PubMed] [Google Scholar]
- 27.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:463-9. [DOI] [PubMed] [Google Scholar]
- 28.Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation. 2000;102:1950-5. [DOI] [PubMed] [Google Scholar]
- 29.Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43:829-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richard P, Isnard R, Carrier L, et al. Double heterozygosity for mutations in the beta-myosin heavy chain and in the cardiac myosin binding protein C genes in a family with hypertrophic cardiomyopathy. J Med Genet. 1999;36:542-5. [PMC free article] [PubMed] [Google Scholar]
- 31.Richard P, Charron P, Leclercq C, et al. Homozygotes for a R869G mutation in the beta -myosin heavy chain gene have a severe form of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1575-83. [DOI] [PubMed] [Google Scholar]
- 32.Andersen PS, Havndrup O, Bundgaard H, et al. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: total or partial haploinsufficiency. Eur J Hum Genet. 2004;12:673-7. [DOI] [PubMed] [Google Scholar]
- 33.Carrier L, Bonne G, Bahrend E, et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. 1997;80:427-34. [PubMed] [Google Scholar]
- 34.Richard P, Villard E, Charron P, Isnard R. The Genetic Bases of Cardiomyopathies. J Am Coll Cardiol. 2006;48:A79-A89. [Google Scholar]
- 35.Marian AJ, Mares A Jr, Kelly DP, et al. Sudden cardiac death in hypertrophic cardiomyopathy. Variability in phenotypic expression of beta-myosin heavy chain mutations. Eur Heart J. 1995;16:368-76. [DOI] [PubMed] [Google Scholar]
- 36.Anan R, Greve G, Thierfelder L, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest. 1994;93:280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang TH, Lee WH, Kimura A, et al. Early expression of a malignant phenotype of familial hypertrophic cardiomyopathy associated with a Gly716Arg myosin heavy chain mutation in a Korean family. Am J Cardiol. 1998;82:1509-13. [DOI] [PubMed] [Google Scholar]
- 38.Marian AJ, Roberts R. Molecular genetic basis of hypertrophic cardiomyopathy: genetic markers for sudden cardiac death. J Cardiovasc Electrophysiol. 1998;9:88-99. [DOI] [PubMed] [Google Scholar]
- 39.Watkins H, Rosenzweig A, Hwang DS, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1108-14. [DOI] [PubMed] [Google Scholar]
- 40.Roberts R, Sigwart U. New concepts in hypertrophic cardiomyopathies, part II. Circulation. 2001;104:2249-52. [DOI] [PubMed] [Google Scholar]
- 41.Fananapazir L, Epstein ND. Genotype-phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation. 1994;89:22-32. [DOI] [PubMed] [Google Scholar]
- 42.Consevage MW, Salada GC, Baylen BG, Ladda RL, Rogan PK. A new missense mutation, Arg719Gln, in the beta-cardiac heavy chain myosin gene of patients with familial hypertrophic cardiomyopathy. Hum Mol Genet. 1994;3:1025-6. [DOI] [PubMed] [Google Scholar]
- 43.Coviello DA, Maron BJ, Spirito P, et al. Clinical features of hypertrophic cardiomyopathy caused by mutation of a ‘hot spot’ in the alpha-tropomyosin gene. J Am Coll Cardiol. 1997;29:635-40. [DOI] [PubMed] [Google Scholar]
- 44.Havndrup O, Bundgaard H, Andersen PS, et al. The Val606Met mutation in the cardiac beta-myosin heavy chain gene in patients with familial hypertrophic cardiomyopathy is associated with a high risk of sudden death at young age. Am J Cardiol. 2001;87:1315-7. [DOI] [PubMed] [Google Scholar]
- 45.Epstein ND, Cohn GM, Cyran F, Fananapazir L. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the beta-myosin heavy chain gene. A 908Leu----Val mutation and a 403Arg----Gln mutation. Circulation. 1992;86:345-52. [DOI] [PubMed] [Google Scholar]
- 46.Van Driest SL, Ackerman MJ, Ommen SR, et al. Prevalence and severity of ‘benign’ mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation. 2002;106:3085-90. [DOI] [PubMed] [Google Scholar]
- 47.Rai TS, Ahmad S, Bahl A, et al. Genotype phenotype correlations of cardiac beta-myosin heavy chain mutations in Indian patients with hypertrophic and dilated cardiomyopathy. Mol Cell Biochem. 2009;321:189-96. [DOI] [PubMed] [Google Scholar]
- 48.Forissier JF, Richard P, Briault S, et al. First description of germline mosaicism in familial hypertrophic cardiomyopathy. J Med Genet. 2000;37:132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watkins H, Thierfelder L, Hwang DS, McKenna W, Seidman JG, Seidman CE. Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest. 1992;90:1666-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watkins H, Anan R, Coviello DA, Spirito P, Seidman JG, Seidman CE. A de novo mutation in alpha-tropomyosin that causes hypertrophic cardiomyopathy. Circulation. 1995;91:2302-5. [DOI] [PubMed] [Google Scholar]
- 51.Cuda G, Perrotti N, Perticone F, Mattioli PL. A previously undescribed de novo insertion-deletion mutation in the beta myosin heavy chain gene in a kindred with familial hypertrophic cardiomyopathy. Heart. 1996;76:451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alders M, Jongbloed R, Deelen W et al. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J. 2003;24:1848-53. [DOI] [PubMed] [Google Scholar]
- 53.Michels M, Solima OII, Kofflard MJ, et al. Diastolic Abnormalities as the First Feature of Hypertrophic Cardiomyopathy in Dutch Myosin-Binding Protein C Founder Mutations. J Am Coll Cardiol Imaging. 2009;2:58-64. [DOI] [PubMed] [Google Scholar]
- 54.Moolman-Smook JC, De Lange WJ, Bruwer EC, Brink PA, Corfield VA. The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am J Hum Genet. 1999;65:1308-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaaskelainen P, Miettinen R, Karkkainen P, Toivonen L, Laakso M, Kuusisto J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann Med. 2004;36:23-32. [DOI] [PubMed] [Google Scholar]
- 56.Girolami F, Olivotto I, Passerini I, et al. A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2006;7:601-7. [DOI] [PubMed] [Google Scholar]
- 57.Kubo T, Kitaoka H, Okawa M et al. Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac Myosin-binding protein C gene among Japanese. J Am Coll Cardiol. 2005;46:1737-43. [DOI] [PubMed] [Google Scholar]
- 58.Dhandapany PS, Sadayappan S, Xue Y, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zahka K, Kalidas K, Simpson MA, et al. Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart. 2008;94:1326-30. [DOI] [PubMed] [Google Scholar]
- 60.Michels M, Hoedemaekers YM, Kofflard MJ, et al. Familial screening and genetic counselling in hypertrophic cardiomyopathy: the Rotterdam experience. Neth Heart J. 2007;15:184-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hofman N, Postema PG, van Langen IM, et al. Genetic identification of patients and families with a long-QT syndrome: large regional differences in the result of 10 years. Ned Tijdschr Geneeskd. 2007;151:644-8. [PubMed] [Google Scholar]
- 62.Charron P, Carrier L, Dubourg O, et al. Penetrance of familial hypertrophic cardiomyopathy. Genet Couns. 1997;8:107-14. [PubMed] [Google Scholar]
- 63.Charron P, Dubourg O, Desnos M, et al. Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation. 1998;97:2230-6. [DOI] [PubMed] [Google Scholar]
- 64.Maron BJ, Niimura H, Casey SA et al. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J Am Coll Cardiol. 2001;38:315-21. [DOI] [PubMed] [Google Scholar]
- 65.Moolman JA, Reith S, Uhl K et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation. 2000;101:1396-402. [DOI] [PubMed] [Google Scholar]
- 66.Niimura H, Bachinski LL, Sangwatanaroj S et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248-57. [DOI] [PubMed] [Google Scholar]
- 67.Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201-11. [DOI] [PubMed] [Google Scholar]
- 68.Christiaans I, Birnie E, van Langen IM et al. The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy Myosin-Binding Protein C gene mutation carriers: focus on predictive screening. Eur Heart J. 2009 Dec 16. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 69.Michels M, Soliman OI, Phefferkorn J et al. Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers. Eur Heart J. 2009;30:2593-8. [DOI] [PubMed] [Google Scholar]
- 70.van Langen IM, Arens Y, Baars H, et al., for the ICIN working group on Hereditary Heart Diseases. Multidiscipline guideline: Genetic diagnostics and genetic counselling in Hypertrophic Cardiomyopathy (HCM). Neth Heart J. 2010;18:144-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Langen IM, Hofman N, Tan HL, Wilde AA. Family and population strategies for screening and counselling of inherited cardiac arrhythmias. Ann Med. 2004;36(Suppl 1):116-24. [DOI] [PubMed] [Google Scholar]
- 72.Hendriks KS, Hendriks MM, Birnie E et al. Familial disease with a risk of sudden death: a longitudinal study of the psychological consequences of predictive testing for long QT syndrome. Heart Rhythm. 2008;5:719-24. [DOI] [PubMed] [Google Scholar]
- 73.Christiaans I, van Langen IM, Birnie E, Bonsel GJ, Wilde AA, Smets EM. Quality of life and psychological distress in hypertrophic cardiomyopathy mutation carriers: A cross-sectional cohort study. Am J Med Genet A. 2009;149A:602-12. [DOI] [PubMed] [Google Scholar]
- 74.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:463-9. [DOI] [PubMed] [Google Scholar]
- 75.Posch MG, Thiemann L, Tomasov P, et al. Sequence analysis of myozenin 2 in 438 European patients with familial hypertrophic cardiomyopathy. Med Sci Monit. 2008;14:CR372-CR374. [PubMed] [Google Scholar]
- 76.Osio A, Tan L, Chen SN, et al. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100:766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab. 2006;87:169-74. [DOI] [PubMed] [Google Scholar]
- 78.Theis JL, Bos JM, Bartleson VB, et al. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2006;351:896-902. [DOI] [PubMed] [Google Scholar]
- 79.Bos JM, Poley RN, Ny M, et al. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab. 2006;88:78-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayashi T, Arimura T, Itoh-Satoh M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2192-201. [DOI] [PubMed] [Google Scholar]