

Abstract
Population genetics is fundamental to our understanding of evolution, and mutations are essential raw materials for evolution. In this introduction to more detailed papers that follow, we aim to provide an oversight of the field. We review current knowledge on mutation rates and their harmful and beneficial effects on fitness and then consider theories that predict the fate of individual mutations or the consequences of mutation accumulation for quantitative traits. Many advances in the past built on models that treat the evolution of mutations at each DNA site independently, neglecting linkage of sites on chromosomes and interactions of effects between sites (epistasis). We review work that addresses these limitations, to predict how mutations interfere with each other. An understanding of the population genetics of mutations of individual loci and of traits affected by many loci helps in addressing many fundamental and applied questions: for example, how do organisms adapt to changing environments, how did sex evolve, which DNA sequences are medically important, why do we age, which genetic processes can generate new species or drive endangered species to extinction, and how should policy on levels of potentially harmful mutagens introduced into the environment by humans be determined?
Keywords: deleterious and advantageous mutations, distribution of mutational effects, linkage, background selection, Muller's ratchet, evolution
1. Introduction
Mutations are one of the fundamental forces of evolution because they fuel the variability in populations and thus enable evolutionary change. Based on their effects on fitness, mutations can be divided into three broad categories: the ‘good’ or advantageous that increase fitness, the ‘bad’ or deleterious that decrease it and the ‘indifferent’ or neutral that are not affected by selection because their effects are too small. While this simplistic view serves well as a first rule of thumb for understanding the fate of mutations, research in recent decades has uncovered a complex web of interactions. For example, (i) the effects of mutations often depend on the presence or absence of other mutations, (ii) their effects can also depend on the environment, (iii) the fate of mutations may depend on the size and structure of the population, which can severely limit the ability of selection to discriminate among the three types (making all seem nearly ‘indifferent’), and (iv) mutations' fate can also depend on the fate of others that have more pronounced effects and are in close proximity on the same chromosome.
A major theoretical goal in the study of the population genetics of mutations is to understand how mutations change populations in the long term. To this end, we have to consider many features of evolution and extant populations at both the phenotypic and molecular level, and ask how these can be explained in terms of rates and kinds of mutations and how they are affected by the forces that influence their fates.
We have increasing amounts of information at our disposal to help us answer these questions. The continuous improvement of DNA sequencing technology is providing more detailed genotypes on more species and observations of more phenomena at the genomic level. We are also gaining more understanding of the processes that lead from changes at the level of genotypes through various intermediate molecular changes in individuals to new visible phenotypes. Use of this new knowledge presents both opportunities and challenges to our understanding, and new methods have been developed to address them.
Brian Charlesworth has been at the forefront of many of the developments in the population genetics of mutations, both in the collection and analysis of new data and in providing new models to explain the observations he and others have made. This themed issue of Phil. Trans. R. Soc. B is dedicated to him to mark his 65th birthday. The authors of the accompanying papers have individually made important contributions to the field and have been directly associated with or indirectly influenced by his work.
In this collection of papers, various aspects are considered in detail, and in this introduction, we aim to provide an overview as a basis for the in-depth treatments that follow. We outline some of the theories that serve as the quantitative basis for more applied questions and have been developed with the main aims of: (i) measuring the rates at which different types of mutations occur in nature, (ii) predicting quantitatively their subsequent fate in populations, and (iii) assessing how they affect some properties of populations and therefore could be used for inference. The subsequent papers are broadly arranged in a continuum from specific questions of basic parameter estimation (strength of mutation, selection, recombination), via those that contribute a combination of biological theories and data on these parameters, to those which mostly address broader biological theories.
There is an enormous range of mutational effects on fitness, and wide differences exist in the strength of other evolutionary forces that operate on populations. This generates an array of complex phenomena that continues to challenge our capacity to mechanistically understand evolution. To make problems tractable, theoreticians have divided the parameter space into smaller regions such that specific simplifying assumptions can be made. These typically comprise assuming the absence of particular events (e.g. no recombination) or the presence of particular equilibria (e.g. mutation-selection balance). Subsequently, new theories are often developed in which these assumptions are relaxed so as to narrow the gap to reality, typically including more interactions between various evolutionary forces, albeit at the cost of becoming less tractable to analysis.
The dynamics of mutations are dominated by chance, yet we search for general principles that are independent of particular random events. This tension is reflected in the models used. All mutations start out as single copies and most are lost again by chance, so we can at best predict probabilities of particular fates; but the stochastic models that can deal rigorously with randomness are often too complex to analyse for realistic scenarios. If we are interested only in the mean outcome of many individual random events, we may approximate the process by deterministic models that predict a precise outcome; but these approximations can break down if only few individuals or rare events are involved.
To facilitate concise descriptions, there is a long history in population genetics of using mathematical symbols as abbreviations for various parameters and observations, but unfortunately there is no unique nomenclature. To try to meet our two conflicting goals of conciseness and readability, we list some important evolutionary parameters and their common abbreviations in table 1. Even so, for good reasons of history or local convention, some of these symbols are defined differently in some papers in this collection.
Table 1.
Some parameters in the population genetics of mutations*.
| U | mutation rate per generation per genome; check context for effects of mutations |
| Ge, G | effective haploid genome size (all functional base pairs), total haploid genome size (with neutral sites) |
| μ, μ10, μ01 | mutation rate per locus or per site per generation, away from the preferred base, and back |
| κ, tn/tv | mutational bias: μ10/μ01, transition/transversion ratio |
| re, rco, rgc | effective recombination rate, cross over rate, gene conversion rate |
| s | selection coefficient; measures changes in fitness; check context for exact definitions (homozygous or heterozygous; positive or negative) |
| h | dominance coefficient so that sh is the effect of heterozygous mutations |
| DME (or DFE) | distribution of mutational effects on fitness |
| WA | Wrightian fitness of a genotype A (one of the many ways fitness can be defined) |
| ε | epistasis: interactions of mutational effects. If fitness is multiplicative, ε = WAB − WAWB |
| Ne, N | effective population size, census population size |
| m | migration rate |
| Pfix | probability of fixation of a (mutant) allele |
| Tfix, Tloss | time to fixation, loss in generations |
| KA, KS (or DN, DS) | rate of DNA divergence per site between two species corrected for multiple hits (see context for method); substitutions can be non-synonymous (change amino acids), or synonymous (or silent) (check context) |
| πA, πS, θWA, θWS | DNA diversity within a population per site: π is the average pairwise nucleotide diversity, θW is Watterson's estimate; for explanation of indices see KA, KS |
| D, D′, r2 | measures of linkage disequilibrium (LD) |
| VG | genetic variance in a quantitative trait |
| VM | increment in VG from new mutations per generation |
| VE | environmental variance in a quantitative trait |
*For historical reasons and due to the limitations of the alphabet, several symbols have different meanings in different contexts. Examples: h2 = heritability of a quantitative trait; r = rate of selfing; r = rate of population growth; D = ‘Tajima's D’, where D < 0 may indicate population expansion or directional selection and D > 0 a bottleneck or balancing selection.
Naturally, a review of this length cannot cover all aspects of the population genetics of mutations. For example, mutation plays a pivotal part in coalescent theory (Hein et al. 2005) and in the construction of genotype–phenotype maps that are at the core of some efforts to understand adaptive landscapes, which provide a paradigm for understanding many broader aspects of population genetics from the perspective of individual mutations (‘causes cancer or not’), as reviewed elsewhere (Loewe 2009). Here we focus almost entirely on how populations of individuals are changed by large numbers of mutations that have specified effects on fitness.
In §2 of this paper, we discuss what is known about the diversity of mutations, and here and subsequently we refer to other papers in this themed issue that provide more in-depth information. In §3, we review some of the relevant theory in population genetics, starting with (i) simple theories that treat the fate of individual mutations in isolation before turning to more complicated models that consider (ii) linkage, (iii) epistasis, (iv) quantitative genetics approaches, and (v) challenges faced when attempting to integrate all these. Subsequently, we provide an overview of several general questions that have been resolved and others that remain (§4) and finally some conclusions (§5).
2. Mutations
It has often been said that mutations are random, a statement that is simultaneously true and false: true because mutations do not originate in any way or at any time that is related to whether their effects are beneficial—one of the central tenets of Neodarwinism; and false because mutations are the result of complex biochemical reactions that result in non-uniformly distributed mutation frequencies, favouring some (random) changes over others.
(a). Types and rates of mutations
Mutations are caused by physical changes to the hereditary material and, because DNA is a long sequence of base pairs organized into physically unlinked chromosomes, there are many possible ways it can change. There are (i) point mutations that change only a single letter and lead to so-called ‘single nucleotide polymorphisms’ in populations, (ii) insertions and deletions of various sizes (also called ‘indels’ if it is difficult to decide which of the two actually happened; these can also lead to ‘copy number variants’), (iii) transpositions that move a sequence from one position to another, and can thereby cause mutations at the boundaries, (iv) inversions of various sizes that change the orientation of a stretch of DNA, (v) chromosome mutations that affect long enough pieces of DNA to become visible under the microscope and might even lead to the loss or duplication of a whole chromosome (also known as non-disjunctions), and (vi) changes in the ploidy level, where a whole copy of the genome is either gained or lost.
A special class of mutations is caused by transposable elements. As reviewed in this themed issue by Lee & Langley (2010), there are various types of these elements that can move around in the genome and can copy, insert or excise themselves, sometimes in response to conditions such as stress. Mechanisms exist to control the frequency of transposition events to limit the damage from resulting deleterious mutations.
At each level, biochemical factors are such that some types of changes occur more frequently than others. For example, in many species there are many more transitions than transversions, the methylation of CpG sites in mammals leads to about tenfold higher mutation rates at these sites (Rosenberg et al. 2003) and the ratio of insertions to deletions can differ among species (Gregory 2004; Grover et al. 2008).
Mutation rates are difficult to measure because the events are so rare that it is like measuring the frequency of needles in haystacks. Historically, this has been accomplished mainly by finding single genes or groups of genes that lead to phenotypic changes that can easily be observed in populations with known descent (Drake et al. 1998) and extrapolating to the level of genomes. As Kondrashov & Kondrashov (2010) point out in their contribution to this issue, the recent advances in post-genomic sequencing technology have led to breakthroughs that now allow direct determination of mutation rates in species with sequenced genomes, work which Charlesworth has stimulated by his developments of theory and to which he has contributed directly (Haag-Liautard et al. 2007). Future work in this area is important because accurate estimates of mutation rates at different sites and in different species can be important for testing alternative theories.
Mutations are frequently classified as non-synonymous or synonymous according to whether or not they change the amino acid sequence, which depends entirely on the function of the mutated base pair. They are easy to recognize and so are frequently used in population genetic tests. They provide useful rules of thumb: e.g. synonymous sites often evolve neutrally or under weak selection and non-synonymous sites are often under strong purifying selection, even if its strength is difficult to quantify.
(b). The distribution of mutational effects on fitness (DME)
What is the distribution of mutational effects on fitness? Charlesworth (1996b) once described this as the first and most difficult question he would ask the fairy godmother of evolutionary genetics. It specifies the probability distribution of selection coefficients for spontaneous mutations of a given genome. Thus, it could be argued that the DME is highly dependent on the genotype; but all organisms are complex functional networks and have ‘learned’ to live with a flux of new mutations, such that powerful normalizing forces might cause the DMEs we can observe today to share important properties. For example, the total of fitness degrading and fitness increasing effects that get fixed might be in equilibrium such that there is no unbounded change in fitness in most populations.
In their paper in this issue, Keightley & Eyre-Walker (2010) show that one of the most robust findings from research on DMEs is that the effects span many orders of magnitude. It is well known that some deleterious mutations are lethal while others appear to be effectively neutral in all population genetic tests, implying that heterozygous selection coefficient s of mutants ranges from −1 (lethal) to more neutral than −10−7 (effectively neutral for some Drosophila species). It is hard to see why any particular range of intermediate selection coefficients should not exist, raising problems for many population genetic theories tailored towards dealing only with mutations of a particular effect. Different types of mutations typically have different DMEs. For example, conservative amino acid changes usually have much smaller effects than frame-shift mutations that disfigure the rest of a protein (Sunyaev et al. 2001).
In diploids, the selection coefficients of heterozygous mutations are modulated by their dominance h. The correlation between h and s is typically strongly negative, which suggests that the properties of underlying biochemical reaction networks are usually pivotal for determining dominance (Charlesworth 1979; Kacser & Burns 1981; Phadnis & Fry 2005).
Of course not all mutations are harmful, and the occasional fitness increasing mutations drive adaptive evolution. In this issue Orr (2010) points out how some intriguing statements can be made about advantageous mutations beyond the fact that they are usually rare and difficult to observe. They include (i) back mutations that occur if a large enough number of slightly deleterious mutations was previously fixed, possibly at a time when the effective population size was smaller (Charlesworth & Eyre-Walker 2007), (ii) compensatory mutations that at least partially repair some harmful effects at the molecular level (e.g. Burch & Chao 1999; Innan & Stephan 2001; Kern & Kondrashov 2004), (iii) quantitative trait mutations that can either increase or decrease the value of a trait with an impact on fitness (e.g. Keightley & Halligan 2009), (iv) resistance mutations that are part of biological arms races between hosts and parasites (Hamilton et al. 1990), and (v) mutations that enable a species to start expanding into a new ecological niche (e.g. Elena & Lenski 2003; Bergthorsson et al. 2007). The frequencies and DMEs of these groups are probably very different and their prediction and estimation are likely to be fruitful fields for further research.
Our knowledge of DMEs has come from laboratory experiments like that of Trindade et al. (2010) and from population genetics approaches as used by Keightley & Eyre-Walker (2010), both of which are reported in this issue. Experimental approaches for inferring DMEs are based on mutation accumulation experiments pioneered by Mukai in Drosophila (Mukai et al. 1972; Keightley & Eyre-Walker 1999; Lynch et al. 1999), and one of the most extensive experiments of this type was completed by Charlesworth et al. (2004). Their strength is in the direct observation of the consequences of mutations that have relatively large effects of around 1 per cent, but they require great care to control for potential confounding factors and mutations of small effects cannot be detected. Thus, many researchers have recently used population genetics models and DNA sequence data to infer DMEs (Loewe & Charlesworth 2006; Keightley & Eyre-Walker 2010). This is done by assuming an analytic DME (e.g. lognormal or gamma), using a population genetics model to predict from the DME observable DNA sequence patterns, and adjusting the assumed DME until it predicts the data. Such DME estimates are most reliable for slightly deleterious mutational effects (|s| slightly above 1/Ne, where Ne is the effective population size). Their limitations have inspired a third approach for estimating DMEs, which builds on what are intended as realistic computational systems biology models to infer the effects of mutations in relatively well understood systems such as a circadian clock or a signal transduction pathway (Loewe 2009). While this approach cannot lead to general statements except by testing many different systems, it may provide more precise statements on the DME of advantageous mutations.
3. Theories on the population genetics of mutations
Important questions to be addressed include (i) predicting the fate of individual mutations such as their fixation probability Pfix and times to loss Tloss or fixation Tfix in a population, (ii) how a given flux of mutations will impact properties of a population such as nucleotide diversity (πA, πS), divergence (KA, KS), survival or the rate of evolution of quantitative traits, (iii) how the fates of different mutations will affect each other, (iv) how quantitative genetic variation is maintained, and (v) the estimation of evolutionary parameters of populations and species from DNA sequence patterns (e.g. recombination rate Ne, etc.).
Theories to investigate some of these questions can be categorized by the complexity of the models assumed and by their general approach: those restricted to single sites, in which all mutations are treated as completely independent of each other; those invoking linkage, in which changes in the frequency of mutations are no longer independent, even if their effects are independent; and those invoking epistasis in which the effects of mutations depend on which others are present.
In each case, the overall effects of mutations can be studied in two ways. The analysis may focus on individuals and their mutations explicitly, track their fate and later summarize the behaviour of many mutations in a population to compute quantities like DNA sequence diversity. Alternatively it may focus directly on quantitative traits in which mutations are not individually identified but considered more implicitly as components of the total effect on either individual or population mean phenotype.
(a). Single site theories
One of the successes of twentieth century population genetics was the development of single site analyses that describe the fate of a mutation at one site independent of all others in the genome (Kimura 1962; Kimura & Ohta 1969a,b; Crow & Kimura 1970). This site may be affected by mutation and back mutation, directional selection and balancing selection, migration and drift, but is assumed to be unaffected by linkage or changes at other loci, including those due to selection. Once the behaviour of a single site is understood, the behaviour of many such sites can be predicted, again assuming they all evolve independently. These models have been used to investigate topics such as codon bias (Bulmer 1991; McVean & Charlesworth 1999), reviewed in this themed issue by Sharp et al. (2010), who consider particularly the factors determining this bias. This approach has also helped estimate the strength of selection on amino acid changes (Loewe et al. 2006).
In the simplest models, few evolutionary forces are assumed to act in the population. For example mutation-selection balance models exclude drift (e.g. Crow & Kimura 1970) and the Neutral Theory excludes selection (Kimura 1983), as in much of the recent work on coalescent theory (Hein et al. 2005). Charlesworth and colleagues expanded Kimura's single site theory of mutation-selection-drift equilibrium to accommodate the presence of back mutations (McVean & Charlesworth 1999). This is an important step towards more realism because the infinite sites assumption of earlier work fails over very long periods of time as back mutations become increasingly probable. It is elegant because the model requires only one additional parameter. Substitution rates (KA/KS) and DNA sequence diversity (πA/πS) can then be predicted under the assumption of a given DME (Loewe et al. 2006) by computing the fixation probabilities Pfix, the resulting fluxes of mutations that take Tfix generations to get fixed or Tloss generations to get lost, and taking genome wide weighted averages over all these classes of mutations.
(b). Linkage theories
More complex models are needed to understand the response to selection in systems where recombination is limited between neighbouring sites.
The long term response and limits to the selective increase of quantitative traits influenced by many loci depend on the proportion of trait-increasing alleles that are fixed by selection. Robertson's (1960) theory of limits, in which loci were assumed to be independent, was extended by including the impact of linkage between pairs of advantageous trait genes (Hill & Robertson 1966). The analysis showed that, especially if one of the loci was at low frequency and had a large effect, it interfered with selection at the other such that the latter's fixation probability was greatly reduced. The authors did not consider new mutants, but obviously should have done so for effects are then most extreme. This observation stimulated the development of what might be called linkage theories, as they describe the consequences of linkage between different sites on the same chromosome (also called the ‘Hill–Robertson effect’ by Felsenstein (1974), a term that stuck; see McVean & Charlesworth (2000), Keightley & Otto (2006), Roze & Barton (2006), Coméron et al. (2008) and Kaiser & Charlesworth (2009)).
The phenomena that result from linkage are complex because a great variety of processes are at work, described by evolutionary parameters that span many orders of magnitude. Thus we cannot point to a single ‘linkage theory’ for a general solution, but instead to a wide range of models developed (table 2). Each describes the consequences of linkage and selection for a particular range of recombination rates and selection coefficients, but we hope that future research will enable these to be combined as special cases of a more comprehensive theory. A common feature is the reduction in Ne as a result of linkage and selection, which can lead to competing explanations for similar phenomena as Stephan (2010) and Barton (2010) show in this issue. The resulting Ne can be used in some of the single site approaches above to predict observables such as the DNA sequence diversity in a population or codon bias.
Table 2.
Linkage theories.
| name | valid range | recombination | back mutations | mutations of largest effect |
|---|---|---|---|---|
| background selection (BGS) | removal of deleterious mutations always works | ≥0 | irrelevant | −−−− |
| Muller's ratchet (MR; slow regime) | removal of slightly deleterious mutations fails rarely (‘bad luck’) | 0a, ≥0b | usually neglected | −−− |
| Muller's ratchet (MR; fast regime) | removal of slightly deleterious mutations is expected to fail | 0a, ≥0c | 0a, ≥0c | −− |
| nearly neutral theory (NN) | very slightly deleterious and positive mutations accumulate (selection too weak to discern) | ≥0 | yes | +/0/ − |
| clonal interference (CI) | several advantageous clones interfere with each others' selection | 0 | ++/+++ | |
| interference selection (IS) | several advantageous mutations interfere with each other under partial linkage | >0d | ++/+++ | |
| hitch-hiking (HH) | strongly selected mutations drag less strongly selected (potentially deleterious) mutations to fixation. | ≥0 | ++++ |
+= advantageous, 0 = neutral and −= deleterious mutations, where repeated symbols indicate stronger selection.
aClassical theory of Muller's ratchet (Haigh 1978).
b‘Hill-Robertson Interference’ (Kaiser & Charlesworth 2009).
c‘Weak selection Hill–Robertson effect’ (McVean & Charlesworth 2000).
d‘Hill–Robertson effect’ (Roze & Barton 2006).
The magnitude of selection coefficients and recombination rates play pivotal roles in specifying which linkage theory can be applied. As we discuss below, some can be ordered on a continuum according to the selection coefficients of the most strongly selected mutations that they consider. More work on combining them is needed to deal with the findings of extreme diversity of mutational effects and recombination rates. In his paper in this issue McVean (2010) describes how recent experimental and population wide DNA sequence analyses show that previous rather uniform estimates of recombination rates can stem from averaging over very broad scales and that the majority of recombination events, at least in humans, can occur in hotspots.
(i). Background selection
Most mutations for which selection is effective are strongly deleterious (e.g Keightley & Eyre-Walker 2010; Trindade et al. 2010). Therefore, we start with the theory of background selection (BGS), developed primarily by Charlesworth et al. (1993a, 1995, 1996a), and reviewed here by Stephan (2010). It is based on the reduction in Ne at a neutral site that results from the deterministic selective removal of strongly deleterious mutations at linked sites. Kimura's single site mutation-selection-drift analysis shows that deleterious mutations with effects beyond a certain size have no realistic chance of ever getting fixed, but the process of their removal generates BGS. For deterministic removal, the selection coefficient s needs to be only slightly larger than 1/Ne in recombining populations (Nordborg et al. 1996). In the absence of recombination, the situation is more complicated, because it also depends on the number of deleterious mutations that simultaneously hit the non-recombining region. The critical s can be up to several orders of magnitude larger for some realistic cases; more precise statements require determining s by finding the corresponding location of the ‘wall of BGS’ using Muller's ratchet (MR) theory (Loewe 2006).
(ii). Muller's ratchet
If the mutational effects are only slightly deleterious or there are too many mutations that simultaneously segregate in a population, deterministic purifying selection can be overwhelmed by random effects, leading to a ‘slow regime’ of mutation accumulation, where fixations of slightly deleterious mutations occasionally happen by bad luck. If such events are repeated over long periods of time, they can have substantial consequences on fitness and may degrade the corresponding genetic system. Examples are seen in Y-chromosomes (Charlesworth 1978; Charlesworth & Charlesworth 2000), or in the predicted genomic decay of a population (Lynch et al. 1995b; Loewe 2006). Much research on this scenario has concentrated on cases where no recombination occurs, as described by the classical theory of MR on which there is an extensive literature (Loewe 2006). Classically, MR describes the inevitable accumulation of slightly deleterious mutations that results from solely deleterious mutation pressure in the absence of any recombination that could repair some of the damage by recombining mediocre genotypes into good and bad ones. The unexpected stochastic loss of individuals with the least mutations causes mutation accumulation (Muller 1964; Felsenstein 1974), and diffusion theory has been used to estimate the relevant rates (Stephan et al. 1993; Gordo & Charlesworth 2000a,b; Etheridge et al. 2009). We consider the essence of MR to be in the deleterious mutation accumulation caused by the random loss from the population of the least loaded class of individuals. Hence, it would be possible to explore models of MR with rare recombination where deleterious mutations are still accumulated, but the decay of fitness is effectively reduced by processes that occasionally produce a less loaded class (e.g. recombination, back mutations or advantageous mutations). Indeed Charlesworth et al. (1993b), has explored related scenarios with low levels of recombination under the label ‘mutation accumulation’ and has also used the classical theory of MR without recombination to quantify processes that can degenerate Y-chromosomes (Charlesworth 1978; Charlesworth & Charlesworth 2000; Gordo & Charlesworth 2001).
If selection coefficients are further reduced with all else being equal, mutations will accumulate faster, eventually switching from the slow regime to the ‘fast regime’, where mutation accumulation is the norm rather than the rare exception. This represents a qualitative change from the slow regime above in which the removal of all deleterious mutations is expected and mutation accumulation occurs as a result of rare random failures. Not so in the fast regime, where values of the evolutionary parameters (e.g. U, s, re, see table 1) are such that the expected critical number of optimal genotypes falls below one and any move of the population towards equilibrium will also bring the best existing genotype closer to extinction or push it over the edge. While the resulting mutation accumulation is still a random process, it is now deterministically unavoidable and hence it has also been called ‘quasi-deterministic’ (Gordo & Charlesworth 2001). This regime was first described explicitly in the context of the classical theory of MR (Gessler 1995) and subsequently in settings that also allow for advantageous mutations (Desai & Fisher 2007; Rouzine et al. 2008). It is also relevant for the weak-selection linkage theory developed under the label ‘weak-selection Hill-Robertson effect’ (McVean & Charlesworth 2000), which is a special case of an extended theory of MR that describes the accumulation of slightly deleterious mutations under a wide range of conditions that include the potential for recombination and beneficial mutations. Special cases of this MR theory have already been developed under various labels and are important for understanding patterns of DNA diversity and divergence in regions of low recombination (McVean & Charlesworth 2000; Coméron et al. 2008; Kaiser & Charlesworth 2009) and the evolution of sex (Keightley & Otto 2006) as also discussed by Barton (2010).
(iii). Effective neutrality
If the strength of purifying selection is even weaker, Ohta's nearly neutral theory (NN) (Ohta 1992) and in the extreme Kimura's neutral theory become increasingly relevant. The upper limit to effective neutrality is at about |Nes| ≈ ¼, above which the dynamics differ substantially (Kimura 1983). If mutations with smaller effects are linked, they will still ‘interfere’ with each other's fixation by genetic drift, as fixation only happens in bundles, but they no longer interfere with their selection, which is effectively absent. As it is unlikely that mutations occur where s = 0 exactly, a large class of NN mutations must have very slightly increasing effects on fitness because, once a significant fraction of sites are fixed by genetic drift for mildly deleterious mutations, an appreciable rate of advantageous back mutations occur. Eventually an equilibrium κ is expected that characterizes the mutational flux between these types of sites and that is governed by mutational biases (McVean & Charlesworth 1999). It is hard to see how these mutational fluxes could be so skewed that the ratio of possible deleterious to advantageous mutations becomes as large as in the rest of the genome, where the fixation of advantageous mutations provides ample opportunities for deleterious mutations to occur. A consequence of the ‘blind’ mutational fluxes under NN is that many deleterious mutations can be fixed if Ne decreases for any reason (Kondrashov 1995). Correspondingly, the rate of adaptive substitutions increases after a population expansion and thus strongly depends on the history of Ne (Charlesworth & Eyre-Walker 2007). Thus, historic bottlenecks might increase the apparent rate of adaptive substitutions: if a reduction of Ne increases the fraction of sites occupied by mutations that otherwise would have been effectively deleterious, then as Ne increases again, back mutations can lead to subsequent waves of fixation of advantageous mutations, potentially affecting the interpretation of estimates of the fraction of adaptive substitutions in an evolutionary line. Such reasoning is based on assuming that the DME is not bimodal and there are enough sites for each relevant mutational effect.
(iv). Fixing beneficial mutations
An increase in advantageous selection coefficients leads to two important effects. First, fixation probability increases (Pfix ≈ 2s for a single site), although an excessive supply of advantageous mutations with little recombination can lead to diminishing returns as mutations compete with each other instead of with the inferior wild-type (de Visser et al. 1999). Second, if such mutations occur regularly, equilibrium DNA diversity is affected because advantageous mutations have an increased chance of fixation so more of them stay as polymorphisms in the population on their way to fixation, even if the majority is still lost eventually. As mutations with large s are presumably rare, many properties of the population will be affected only temporarily and no stable equilibrium occurs. The dynamics of these processes merit further study and indeed important insights have recently been achieved (Stephan et al. 2006). Various specific models have been developed to describe how advantageous mutations interact with each other and with other mutations. Models of clonal interference (CI) have been used to understand the evolution of bacteria as shown by Sniegowski & Gerrish (2010) in this themed issue. CI occurs when various asexual clones with different advantageous mutations compete against each other for fixation, for example in cells with different successful adaptations of metabolism to a new carbon source. To incorporate low levels of recombination, models of interference selection (IS) have been developed to explain how weak but frequent adaptive evolution might shape the features of genomes that recombine rarely, using Drosophila as an example (Coméron & Kreitman 2002; Coméron et al. 2008). Such frequent advantageous mutations might come from codon-bias adaptation, and their fixation could be eased if recombination rates increase through the presence or lengthening of introns. Related models also help in understanding the origin of sex (Roze & Barton 2006), as also discussed by Barton (2010). Finally models of Hitchhiking (HH) deal with strong selective sweeps that occur once or repeatedly, such as could be generated by adaptations to parasites or changing environments (Hamilton et al. 1990). These usually drag to fixation a multitude of linked mutations with weak or no effects (Maynard Smith & Haigh 1974). HH causes a reduction of Ne, particularly in regions of low recombination rates, but as Stephan (2010) discusses further, it is difficult to distinguish this from the reduction of Ne due to BGS, despite the vast difference in the underlying selection. Much future work will be required to properly quantify the various contributions of the theories above to patterns of genetic diversity.
(c). Epistatic interactions between mutational effects
In the models discussed above, all mutational effects are assumed to be mutually independent, whereas a wide range of interactions, or epistasis, between them have been reported frequently. Such findings have stimulated the desire to build more realistic models of evolution which incorporate epistasis (Wolf et al. 2000; Phillips 2008). There are several types of epistasis and the literature is littered with non-intuitive adjectives intended to describe them (table 3). Ultimately, they are defined by the values of related fitness effects, so precise definitions have to be checked for each study. Table 3 provides definitions using a multiplicative model to define the absence of epistasis (i.e. WAB = WAWB/W); but an additive model (WAB = WA + WB − W), a log transform of the multiplicative case, could have been used. The definitions apply to haploids, and further complexities can be introduced when considering diploid genotypes, multi-locus models or longer sequences of mutations. When thinking about epistasis, we can either consider consequences for fitness values (e.g. ‘negative’/‘positive’ epistasis) or the relative size of mutational effects (e.g. ‘synergistic’/‘antagonistic’ epistasis), but unfortunately the mapping between these terms is not identical for positive and negative mutations (table 3). Next we consider some categories of epistatic interactions and some of their potential implications.
Table 3.
Types of epistasis, compared to a multiplicative model*.
| effects of mutations on a trait relative to wild-type value W |
synergistic epistasis |
antagonistic epistasis |
meaning | ||
|---|---|---|---|---|---|
| WAB/W | WA/W | WB/W | 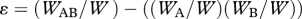 |
||
| <1 | <1 | <1 | ε < 0 | ε > 0 | only magnitude of epistasis is affected |
| <1 | <1 | >1 | ε < 0 | ε > 0 | not all mutational paths decrease the trait |
| <1 | >1 | >1 | ε < 0 | multiple valleys or ‘compensatory’ mutations | |
| signs of mutational effects change the mapping of positive/negative epistasis | |||||
| >1 | <1 | <1 | ε > 0 | multiple peaks or ‘compensatory’ mutations | |
| >1 | >1 | <1 | ε > 0 | ε < 0 | not all mutational paths increase the trait |
| >1 | >1 | >1 | ε > 0 | ε < 0 | only magnitude of epistasis is affected |
*ε < 0 is negative epistasis, ε > 0 is positive epistasis. For intuitive explanations see text. Synergistic epistasis is also known as ‘aggravating epistasis’ and corresponding mutations can be called ‘enhancers’, ‘synthetic interactions’ or ‘synthetically sick’. Antagonistic epistasis is also termed ‘buffering epistasis’, ‘diminishing returns’ and corresponding mutations can be called ‘partial suppressors’ and ‘alleviating’ (Segre et al. 2005). Epistasis that generates fitness valleys is also known as ‘compensatory epistasis’ (Phillips 2008), ‘multiple peaks’ (Weinreich et al. 2005) or ‘reciprocal sign epistasis’ (Poelwijk et al. 2007).
Synergistic epistasis: the absolute effect of the second mutation is bigger than that of the first. At the extreme, a long series of such mutations would eventually increase the effects of slightly deleterious mutations on fitness to a degree that may stop them accumulating (Kondrashov 1994). However, synergistic epistasis cannot stop MR if some of the mutations have very small effects (Butcher 1995). Synergistic epistasis could also play a role in the evolution of sex, if genomic mutation rates are high enough (U > 1; Kondrashov 1988; Haag-Liautard et al. 2007; Barton 2010).
Antagonistic epistasis: the absolute effect of the second mutation is smaller than that of the first. In this case, at the extreme, a long series of such mutations in a population with sufficiently high fitness would eventually lead to an effectively neutral rate of mutation accumulation with no further measurable fitness changes. In more realistic settings and on a more immediate timescale, antagonistic epistasis may counter the overall increases in the strength of selection that are caused by synergistic epistasis.
Epistasis that limits the paths of evolution: if mutation A is deleterious and mutation B is beneficial, but the combination AB is even more beneficial, then the evolutionary path towards AB can only proceed in the sequence B → A (and not A → B, unless random drift overrides selection). This has been termed ‘sign epistasis’ and was found in the context of the protein evolution that leads to some antibiotic resistance (Weinreich et al. 2005, 2006; Poelwijk et al. 2007). By comparing mutations fixed in homologous proteins in different species, Kondrashov et al. (2002) estimated that sign epistasis affected about 10 per cent of amino acid substitutions.
Epistasis that generates selective valleys: this occurs when two deleterious mutations A and B are beneficial if they appear together, either because B compensates for A (e.g. by restoring base-pairing in RNA structures (Parsch et al. 1997; Innan & Stephan 2001)) or because A and B traverse a fitness valley that leads to another local optimum with a different fitness. An assumption of Wright's Shifting Balance Theory of evolution is that such epistatic interactions are common; but evidence for this theory is lacking (Coyne et al. 1997, 2000), even though compensatory evolution may play an important role in molecular evolution (Kimura 1985). Further work is required on the frequency and depth of selective valleys.
The complexity and interdependence of biological systems are such that interactions among gene products are an essential requirement for life. Yet most population genetic analysis, including that relating to mutations, has proceeded using either multiplicative or additive non-epistatic models, which differ little unless effects are large or an additive model leads to negative values (by ‘fixing’ too many deleterious mutations). These models are attractive because they are simple to use, do not require extensive lists of (usually unknown) parameters, and offer the potential opportunity to obtain general results that do not depend on specific parameters.
The question is therefore the extent to which theories based on the simplification of non-epistatic models are relevant in nature. In this issue Crow (2010) concludes that many models of evolution do not depend on the magnitude of epistasis. Although there are potentially many epistatic terms describing many loci, these are likely to contribute very little if the functional relationship between genotype and phenotype is continuous. They are thus hard to measure except for genes of large effect. Molecular biologists may be seeing so many epistatic interactions in part due to ascertainment bias: to elucidate molecular pathways, genes may be knocked out, thereby highlighting potential interactions within the system investigated.
Analyses in various contexts have shown that multiplicative/additive models provide adequate descriptions of variance components and other properties for many complex traits, including complex heritable diseases (Keightley & Kacser 1987; Risch 1990; Hill et al. 2008; Slatkin 2008). For example, many current evolutionary rates depend on additive genetic rather than epistatic components, and Kimura's analysis of pseudo-linkage equilibrium shows that even in the presence of epistasis, rates of evolution stabilize to those described by additive variances (Kimura 1965; Nagylaki 1993; Crow 2010).
Epistasis has been included in population genetic models mainly by using simple approaches, typically as a mathematical function without knowledge of the underlying complexity. For example, synergistic epistasis has been defined by a quadratic function of numbers of deleterious alleles (Charlesworth 1990; Dolgin & Charlesworth 2006). There is a long debate in population genetics about whether there is more synergistic or antagonistic epistasis and hence on the shape of the functions. Unfortunately, it has been difficult to determine from experiments which dominates, because measuring enough mutational interactions at sufficient accuracy is so much work and because there is a large variability in epistatic effects, with about as many synergistic as antagonistic effects found. This wide distribution of epistatic effects (Elena & Lenski 1997; Segre et al. 2005; Sanjuan & Elena 2006) leads to qualitatively different results for many models that include only one type of epistasis or none: for example, a model of slightly deleterious mutation accumulation could no longer rely on synergistic epistasis to stop new mutations getting fixed. More work is needed to explore the consequences of realistic systems of epistatic interactions on long-term evolution.
Where next? It may be possible to explore epistatic interactions using computational systems biology models, as described in a recent framework for evolutionary systems biology, that could help construct genotype–phenotype maps (Loewe 2009). Cheap automatable simulations on a massive scale for a wide range of parameters then become possible, once a system has been properly modelled. Nevertheless, such results will apply only to the particular model being investigated; and the high levels of complexity might render intractable any such models that have too many interactions and too many unknown parameters with potentially large effects.
Even if multiplicative/additive models are not refuted by the data, work on epistasis is important for understanding the population genetics of mutations, particularly to address questions about long-term evolution. Each species presumably lives on or near some local adaptive peak; epistatic interactions determine how many ways there are to evolve between peaks. An increased understanding of the fitness consequences of prolonged mutation accumulation may help to predict risks to endangered species. Increases of mutation rates that may be caused by use of technology by humans might dangerously upset potentially finely balanced natural long-term equilibria between fitness increasing and fitness decreasing processes that are influenced by many evolutionary factors including epistasis (Loewe 2006).
(d). Analysis at the level of quantitative traits
Parameters for quantitative traits such as means, variances and correlations among relatives can be estimated from phenotypic data, but provide little information about the underlying genotypes or the distribution of their effects on traits and on fitness. The same parameters can also estimate the impacts of mutations, but with similar limitations, although new techniques increasingly enable individual mutants to be identified, their effects on traits to be measured, and their mode of action to be determined. There is no all-embracing theory of mutation for quantitative traits; rather there are models that explain to some degree the observed phenomena.
A fundamental parameter in models of quantitative traits is the rate of increase in genetic variation due to mutation per generation. It can be estimated from mutation accumulation experiments starting from an inbred or isogenic base, for which the increase in variability among unselected lines or the response to artificial selection is measured. The result is typically scaled as the ‘mutational heritability’ VM/VE the ratio of the increment VM in the genetic variance per generation to the environmental variance VE (table 1). Estimates of this quantity (summarized by e.g. Houle et al. 1996; Keightley & Halligan 2009) are typically in the range 0.0001–0.01, centred on VM/VE ≈ 0.001. They are similar for different traits and species, with some indication of an increase with generation time. For a typical trait with a coefficient of variation (CV) of 10 per cent, this represents an increment in CV of about 0.3 per cent per generation. How the mutational variance is controlled and why it is rather homogeneous is not understood at any mechanistic level, but presumably reflects past conflicting evolutionary pressures.
The distribution of effects of natural mutations on quantitative traits can be assessed from mutation accumulation studies, but information is limited because small effects are hard to detect. Inferences depend on the assumptions made about the distribution, which appears to be less leptokurtic than the exponential, implying that for many traits mutations of moderate effect are not rare, in contrast to the more leptokurtic distributions found for fitness in such experiments (Keightley & Halligan 2009). In selection experiments, mutants of large effect are frequently detected for quantitative traits, many with strongly deleterious effects on fitness (e.g. López & López-Fanjul 1993). Effects of mutations on the mean can be highly asymmetric, as exemplified by observed excesses of fitness-decreasing mutations.
More complete information can be obtained from insertional mutagenesis experiments which enable individual mutants to be identified and their effects on any number of traits to be estimated (Mackay et al. 1992; Mackay 2009). In this issue Mackay (2010) summarizes results and shows that the mutants have a wide range of effects, typically affect many traits at the same time (pleiotropy), often influence fitness directly (for example through reduced viability), and usually modulate the effects of other mutations (epistasis).
Mathematical models used in the analysis of the effects of mutations on quantitative traits are usually simplistic, in part so they are tractable and in part due to lack of detailed information. There is therefore some gap between the modelling and the real world but, as Crow (2010) illustrates, approximations such as an assumed lack of epistasis may not be critical. Linkage is also typically ignored, partly justified because segregating mutants relating to a trait are probably scattered across the genome.
A point of reference is an additive model where mutants are assumed to be neutral with respect to fitness. The variance within populations stabilizes at 2NeVM, and, assuming a random walk model, the rate of population divergence due to drift each generation between unselected lines approaches 2VM (Lynch & Hill 1986). Under directional selection, if mutant effects on the trait are infinitesimally small or have a symmetric distribution around zero, the rate of response of the trait is proportional to the genetic variance at equilibrium, 2NeVM, independent of the selection intensity (Hill & Keightley 1988).
The standing genetic variance observed within populations is not simply proportional to Ne, however, and must depend on the influence of selection. A major activity in theoretical research has therefore been to assess the roles of mutation, selection and other factors in explaining the high levels of variation maintained in quantitative traits in natural populations. One class of model is based on a balance between mutation and stabilizing selection, whereby fitness depends on the phenotype for the trait and mutants are at a selective disadvantage because they cause deflection from the optimum. If selection is assumed to act solely on the target trait, and it is determined by few loci, the predicted variance maintained is less than observed for a ‘typical’ strength of stabilizing selection (Turelli 1984). One inadequacy of this model is that stabilizing selection acting on other traits through pleiotropic effects is ignored, for with pleiotropy more loci are likely to influence the trait but the aggregate selection is stronger.
In an alternative model the mutant's effects on fitness are assumed not to depend at all on its effect on the target trait, but only through other pleiotropic effects. Under this purely pleiotropic model high levels of genetic variance can be maintained, but population means are unstable and there is little apparent stabilizing selection (Keightley & Hill 1990). While variants of these models still do not fully explain both the stabilizing selection and the genetic variances observed (Bürger 2000; Johnson & Barton 2005; Zhang & Hill 2005), comprehensive surveys indicate that the strength of stabilizing selection on any trait may be much weaker than has been assumed (Kingsolver et al. 2001). Thus there remains much uncertainty about the mechanisms whereby levels of genetic variation in quantitative traits are maintained and to what extent forces are involved that maintain heterozygosity other than by a mutation-drift-selection balance.
Recent genome-wide association studies indicate that variation maintained in quantitative traits is contributed by many segregating loci. For example almost 50 loci have been shown to contribute to standing variation in height (Weedon & Frayling 2008), yet together these contribute only about 5 per cent of the variance of this highly heritable trait. There must therefore be many more segregating, each contributing a very small amount of genetic variance. Similarly, multiple pleiotropic contributing factors explaining only part of the variation were inferred for a disease trait, schizophrenia susceptibility (Purcell et al. 2009). Such analyses miss genes of very low frequency, however, even if they have a large effect, and it has been argued that deleterious rare variants are contributing much of the variation in disease (Goldstein 2009). Incorporating such data into models for maintenance of variation will itself be a challenge.
Substantial changes in quantitative traits solely from the input of mutations can be achieved in laboratory experiments from isogenic base populations, and such is the case, for example in Escherichia coli (Lenski & Travisano 1994), D. melanogaster (e.g. Caballero et al. 1991; Mackay et al. 1994) and mice (Keightley 1998). Because of sampling, however, the patterns of response can be quite erratic, particularly if some mutations have a large effect on the trait. Molecular analysis of long term selection experiments started from an isogenic founder also provides a way to identify the mutations which contributed to the response and to assess their pleiotropic effects and interactions among them (Barrick et al. 2009).
Over very long time periods, divergence among unselected lines has tended to increase more slowly than neutral predictions (Mackay et al. 1995). Similarly, over experiments spanning hundreds of generations or more, such an attenuation of responses have been observed in selection lines started from an isogenic base in E. coli (Lenski & Travisano 1994; Elena & Lenski 2003) and Drosophila (Mackay et al. 2005). Such a plateau can occur because there is a limited potential number of functional alleles and no new useful mutations occur, or because back mutations dominate, or because fitness effects are limiting, e.g. segregating mutants with highly deleterious pleiotropic effects on fitness (López & López-Fanjul 1993). Continuing rapid responses in animal breeding programmes show that mutation plays an increasing role in long continued selection, and also that the influence of unfavourable pleiotropic effects on fitness traits can be minimized by direct selection on such traits (Hill 2010).
A topic of concern is the potential rate of deterioration of the fitness of small populations in the presence of recurrent mutations and the consequent risk to its survival (Lynch et al. 1995a). Calculations depend critically on the effects and rates of mutation (deleterious mutations increase risk, particularly those of intermediate s, while advantageous mutations reduce risk), with risk increasing if population size is small, recombination rate is low, and the population is fragmented (for additional factors see Loewe 2006). Most of these dependencies are not linear and interact with each other. To predict the evolution of fitness in these complex systems and how the survival of species might be influenced by increases in mutation rates caused by humans is a major task.
(e). The future challenge: integrating and testing theories
Many simple theories have been developed and a major challenge for future research is to integrate them with the aim of accurately predicting the behaviour of systems that lie near boundaries. These can be very interesting biologically but be poorly predicted as their parameter combinations stretch the assumptions of locally good models (e.g. the transition between regimes of MR (Loewe 2006)).
We need to know how all relevant processes interact to influence mutation accumulation and patterns of diversity. To do so, it is important to integrate the linkage theories that deal with different mutational effects (table 2), as these usually span many orders of magnitude in real organisms. Charlesworth has contributed to such integration by considering the effects of BGS in combination with MR (Gordo & Charlesworth 2001) and investigating a wide range of selection coefficients in simulation models with back mutation and recombination (McVean & Charlesworth 2000; Kaiser & Charlesworth 2009). In principle, general multi-locus models provide a way of integrating all the aspects of evolution discussed above (Kirkpatrick et al. 2002), but these need large numbers of parameters to be specified. This problem affects all complex models and provides motivation to develop simple models, but these may be of limited reliability when extrapolating beyond testable conditions. Massively parallel computer simulations could help by facilitating the forward simulation of complex models, highlighting by parameter sensitivity analyses those parameters that are most worthwhile to determine experimentally, and estimating parameters via Approximate Bayesian Computation so as to avoid the need to know the likelihood function of complex models (Beaumont & Rannala 2004). The cumbersome experimental work needed for estimating sensitive parameters with precision implies that such modelling work can benefit greatly from being based on a model organism. Brian Charlesworth has always maintained that Drosophila is ideal in this respect and the large body of research on fruit flies in the last 100 years supports this view. Papers in this themed issue, including those by Hughes, Lee & Langley, Mackay, McDermot & Noor, and Stephan, highlight some of the power of the Drosophila model.
4. General questions and applications
An insight into the population genetics of mutations can contribute to a deeper understanding of many practical and theoretical challenges that we face today. Here we highlight just some of them.
The extent to which deleterious, (near)-neutral or advantageous mutations shape DNA diversity (Stephan 2010), codon bias (Sharp et al. 2010) and repetitive element distribution (Lee & Langley 2010) is fundamental to our understanding of genomic structure, and to develop this beyond models of individual loci often requires understanding how much mutations interact epistatically (Crow 2010; Lee & Langley 2010; Mackay 2010).
Many mutations reduce fitness too much to accumulate in a population, but some maladaptive DNA changes have effects small enough to spread (Keightley & Eyre-Walker 2010). A species could be driven to extinction by their accumulation unless there are sufficiently many adaptive mutations. Although other important non-genetic processes (e.g. habitat fragmentation, hunting, pollution) and genetic factors (e.g. mutation, linkage, inbreeding) can contribute to species extinction, one can argue that extinctions are always caused by a lack of mutations that enable adaptation to new or rapidly changing situations. The nature of adaptive mutations is therefore important (Orr 2010). Indeed a finely tuned long-term balance may exist between fitness decreasing and fitness increasing processes, as reducing fitness decreasing processes (like DNA replication errors) becomes exceedingly costly. Since a large fraction of mutations is deleterious, any anthropogenic increase in mutation rates from mutagenic pollution will have manifold debilitating effects. These include obvious ones such as Mendelian genetic diseases and cancers, but also many more with a smaller impact that escape natural selection with potentially disastrous long-term consequences. Therefore, a better understanding of related long-term processes is important for developing reasonable policies that limit the release of mutagenic chemicals into the environment.
Evolutionary models are important for understanding a range of problems fundamental to biology and to other applications to health and welfare. For example, in this themed issue Hughes (2010) discusses the role of mutation in the evolution of ageing. This is a controversial subject on which Charlesworth (2000) has made significant contributions. Models of the evolution of pathogenic microbes (Sniegowski & Gerrish 2010) and their mutations (Trindade et al. 2010) are important for medical applications, such as optimizing the use of antibiotics to minimize resistance evolution and developing vaccines that might anticipate and neutralize simple evolutionary changes a pathogen is expected to produce. The wealth of data that can be obtained for these systems makes them attractive subjects for basic research on evolution. Some mutations that would be deleterious in natural populations provide the opportunity for improvement of crops and livestock in the farm environment. Understanding their pleiotropic effects, for example, is fundamental to long-term increase in food production.
At the fundamental level, broad questions such as the origin of species and their extinctions are influenced by the accumulation of mutations, and various models have accordingly been developed. In this issue, McDermott & Noor (2010) review recent advances regarding a particular mechanism of speciation, but there are many others (Coyne & Orr 2004). Long-term models of evolution, involving mutation, selection and chance that are required to answer such questions are also required to address the evolution of sex (Barton 2010), an example of a deceptively simple problem requiring a deep analysis.
5. Conclusions
We have provided an overview of the nature of mutations and theories that describe their fate once they have entered a population. Much work remains to be done, however, in order to integrate existing theories more fully and to better understand their implications. We have shown that such work is important for questions of practical interest, such as how fast species can adapt to new environments, how genetic factors can contribute to their extinction and what consequences follow from man-made technology driven increases in mutation rates that may unintentionally increase genetic diseases in humans as well as threaten the survival of endangered species. Mutations, however, also provide the raw material for the improvement of plants and animals for food production, and we need to know how best to use them. The population genetics of mutations is undoubtedly central to many theoretical and applied questions in biology.
Acknowledgements
We dedicate this paper to Brian Charlesworth in honour of his many profound contributions to population genetics. LL thanks BC for many inspiring discussions, stimulating thoughts and a long series of wonderful lab dinners. WGH is supported by USS and looks forward to being joined by BC. We thank Fedya Kondrashov, Mohamed Noor, Allen Orr and Wolfgang Stephan for suggestions that improved this manuscript. Our colleagues at the Institute of Evolutionary Biology and its predecessor departments (ICAPB et al.) in Edinburgh have provided an excellent working environment over many years, for which we are most grateful. The Centre for Systems Biology at Edinburgh is a Centre for Integrative Systems Biology (CISB) funded by BBSRC and EPSRC, reference BB/D019621/1.
Footnotes
One contribution of 16 to a Theme Issue ‘The population genetics of mutations: good, bad and indifferent’ dedicated to Brian Charlesworth on his 65th birthday.
References
- Barrick J. E., Yu D. S., Yoon S. H., Jeong H., Oh T. K., Schneider D., Lenski R. E., Kim J. F.2009Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461, 1243–1247 (doi:10.1038/nature08480) [DOI] [PubMed] [Google Scholar]
- Barton N. H.2010Mutation and the evolution of recombination. Phil. Trans. R. Soc. B 365, 1281–1294 (doi:10.1098/rstb.2009.0320) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont M. A., Rannala B.2004The Bayesian revolution in genetics. Nat. Rev. Genet. 5, 251–261 (doi:10.1038/nrg1318) [DOI] [PubMed] [Google Scholar]
- Bergthorsson U., Andersson D. I., Roth J. R.2007Ohno's dilemma: evolution of new genes under continuous selection. Proc. Natl Acad. Sci. USA 104, 17 004–17 009 (doi:10.1073/pnas.0707158104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M.1991The selection-mutation-drift theory of synonymous codon usage. Genetics 129, 897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch C. L., Chao L.1999Evolution by small steps and rugged landscapes in the RNA virus phi 6. Genetics 151, 921–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürger R.2000The mathematical theory of selection, recombination, and mutation Wiley series in mathematical and computational biology Chichester, UK: Wiley [Google Scholar]
- Butcher D.1995Muller's ratchet, epistasis and mutation effects. Genetics 141, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A., Toro M. A., Lopez-Fanjul C.1991The response to artificial selection from new mutations in Drosophila melanogaster. Genetics 128, 89–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B.1978Model for evolution of Y chromosomes and dosage compensation. Proc. Natl Acad. Sci. USA 75, 5618–5622 (doi:10.1073/pnas.75.11.5618) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B.1979Evidence against Fisher's theory of dominance. Nature 278, 848–849 (doi:10.1038/278848a0) [Google Scholar]
- Charlesworth B.1990Mutation-selection balance and the evolutionary advantage of sex and recombination. Genet. Res. 55, 199–222 (doi:10.1017/S0016672300025532) [DOI] [PubMed] [Google Scholar]
- Charlesworth B.1996aBackground selection and patterns of genetic diversity in Drosophila melanogaster. Genet. Res. 68, 131–149 (doi:10.1017/S0016672300034029) [DOI] [PubMed] [Google Scholar]
- Charlesworth B.1996bOpen questions: the good fairy godmother of evolutionary genetics. Curr. Biol. 6, 220 (doi:10.1016/S0960-9822(02)00457-8) [DOI] [PubMed] [Google Scholar]
- Charlesworth B.2000Fisher, Medawar, Hamilton and the evolution of aging. Genetics 156, 927–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Charlesworth D.2000The degeneration of Y chromosomes. Phil. Trans. R. Soc. Lond. B 355, 1563–1572 (doi:10.1098/rstb.2000.0717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth J., Eyre-Walker A.2007The other side of the nearly neutral theory, evidence of slightly advantageous back-mutations. Proc. Natl Acad. Sci. USA 104, 16 992–16 997 (doi:10.1073/pnas.0705456104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Morgan M. T., Charlesworth D.1993aThe effect of deleterious mutations on neutral molecular variation. Genetics 134, 1289–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D., Morgan M. T., Charlesworth B.1993bMutation accumulation in finite outbreeding and inbreeding populations. Genet. Res. 61, 39–56 (doi:10.1017/S0016672300031086) [Google Scholar]
- Charlesworth D., Charlesworth B., Morgan M. T.1995The pattern of neutral molecular variation under the background selection model. Genetics 141, 1619–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Borthwick H., Bartolome C., Pignatelli P.2004Estimates of the genomic mutation rate for detrimental alleles in Drosophila melanogaster. Genetics 167, 815–826 (doi:10.1534/genetics.103.025262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coméron J. M., Kreitman M.2002Population, evolutionary and genomic consequences of interference selection. Genetics 161, 389–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coméron J. M., Williford A., Kliman R. M.2008The Hill-Robertson effect: evolutionary consequences of weak selection and linkage in finite populations. Heredity 100, 19–31 (doi:10.1038/sj.hdy.6801059) [DOI] [PubMed] [Google Scholar]
- Coyne J. A., Orr H. A.2004Speciation Sunderland, MA: Sinauer Associates [Google Scholar]
- Coyne J. A., Barton N. H., Turelli M.1997Perspective: a critique of Sewall Wright's shifting balance theory of evolution. Evolution 51, 643–671 (doi:10.2307/2411143) [DOI] [PubMed] [Google Scholar]
- Coyne J. A., Barton N. H., Turelli M.2000Is Wright's shifting balance process important in evolution? Evolution 54, 306–317 [DOI] [PubMed] [Google Scholar]
- Crow J. F.2010On epistasis: why it is unimportant in polygenic directional selection. Phil. Trans. R. Soc. B, 365, 1241–1244 (doi:10.1098/rstb.2009.0275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. F., Kimura M.1970An introduction to population genetics theory Edina, MN: Burgess International Group Incorporated [Google Scholar]
- Desai M. M., Fisher D. S.2007Beneficial mutation selection balance and the effect of linkage on positive selection. Genetics 176, 1759–1798 (doi:10.1534/genetics.106.067678) [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser J., Zeyl C. W., Gerrish P. J., Blanchard J. L., Lenski R. E.1999Diminishing returns from mutation supply rate in asexual populations. Science 283, 404–406 [DOI] [PubMed] [Google Scholar]
- Dolgin E. S., Charlesworth B.2006The fate of transposable elements in asexual populations. Genetics 174, 817–827 (doi:10.1534/genetics.106.060434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J. W., Charlesworth B., Charlesworth D., Crow J. F.1998Rates of spontaneous mutation. Genetics 148, 1667–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena S. F., Lenski R. E.1997Test of synergistic interactions among deleterious mutations in bacteria. Nature 390, 395–398 (doi:10.1038/37108) [DOI] [PubMed] [Google Scholar]
- Elena S. F., Lenski R. E.2003Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 4, 457–469 (doi:10.1038/nrg1088) [DOI] [PubMed] [Google Scholar]
- Etheridge A., Pfaffelhuber P., Wakolbinger A.2009How often does the ratchet click? Facts, heuristics, asymptotics. In Trends in stochastic analysis (eds Blath J., Mörters P., Scheutzow M.), pp. 365–390 Cambridge, UK: Cambridge University Press; (http://arxiv.org/abs/0709.2775v070) [Google Scholar]
- Felsenstein J.1974The evolutionary advantage of recombination. Genetics 78, 737–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessler D. D. G.1995The constraints of finite size in asexual populations and the rate of the ratchet. Genet. Res. 66, 241–253 (doi:10.1017/S0016672300034686) [DOI] [PubMed] [Google Scholar]
- Goldstein D. B.2009Common genetic variation and human traits. N. Engl. J. Med. 360, 1696–1698 (doi:10.1056/NEJMp0806284) [DOI] [PubMed] [Google Scholar]
- Gordo I., Charlesworth B.2000aOn the speed of Muller's ratchet. Genetics 156, 2137–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I., Charlesworth B.2000bThe degeneration of asexual haploid populations and the speed of Muller's ratchet. Genetics 154, 1379–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I., Charlesworth B.2001The speed of Muller's ratchet with background selection, and the degeneration of Y chromosomes. Genet. Res. 78, 149–161 [DOI] [PubMed] [Google Scholar]
- Gregory T. R.2004Insertion-deletion biases and the evolution of genome size. Gene 324, 15–34 (doi:10.1016/j.gene.2003.09.030) [DOI] [PubMed] [Google Scholar]
- Grover C. E., Yu Y., Wing R. A., Paterson A. H., Wendel J. F.2008A phylogenetic analysis of indel dynamics in the cotton genus. Mol. Biol. Evol. 25, 1415–1428 (doi:10.1093/molbev/msn085) [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C., Dorris M., Maside X., Macaskill S., Halligan D. L., Charlesworth B., Keightley P. D.2007Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445, 82–85 (doi:10.1038/nature05388) [DOI] [PubMed] [Google Scholar]
- Haigh J.1978The accumulation of deleterious genes in a population—Muller's ratchet. Theor. Popul. Biol. 14, 251–267 (doi:10.1016/0040-5809(78)90027-8) [DOI] [PubMed] [Google Scholar]
- Hamilton W. D., Axelrod R., Tanese R.1990Sexual reproduction as an adaptation to resist parasites (a review). Proc. Natl Acad. Sci. USA 87, 3566–3573 (doi:10.1073/pnas.87.9.3566) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein J., Schierup M. H., Wiuf C.2005Gene genealogies, variation and evolution: a primer in coalescent theory Oxford, UK: Oxford University Press [Google Scholar]
- Hill W. G.2010Understanding and using quantitative genetic variation. Phil. Trans. R. Soc. B 365, 73–85 (doi:10.1098/rstb.2009.0203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W. G., Keightley P. D.1988Interrelations of mutation, population size, artificial and natural selection. In Proc. 2nd Int. Conf. Quantitative Genetics (eds Weir B. S., Eisen E. J., Goodman M. M., Namkoong G.), pp. 57–70 Sunderland, MA: Sinauer [Google Scholar]
- Hill W. G., Robertson A.1966The effect of linkage on limits to artificial selection. Genet. Res. 8, 269–294 (doi:10.1017/S0016672300010156) [PubMed] [Google Scholar]
- Hill W. G., Goddard M. E., Visscher P. M.2008Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet 4, e1000008 (doi:10.1371/journal.pgen.1000008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle D., Morikawa B., Lynch M.1996Comparing mutational variabilities. Genetics 143, 1467–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes K. A.2010Mutation and the evolution of ageing: from biometrics to systems genetics. Phil. Trans. R. Soc. B 365, 1273–1279 (doi:10.1098/rstb.2009.0265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H., Stephan W.2001Selection intensity against deleterious mutations in RNA secondary structures and rate of compensatory nucleotide substitutions. Genetics 159, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T., Barton N.2005Theoretical models of selection and mutation on quantitative traits. Phil. Trans. R. Soc. Lond. B 360, 1411–1425 (doi:10.1098/rstb.2005.1667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H., Burns J. A.1981The molecular basis of dominance. Genetics 97, 639–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser V. B., Charlesworth B.2009The effects of deleterious mutations on evolution in non-recombining genomes. Trends Genet. 25, 9–12 (doi:10.1016/j.tig.2008.10.009) [DOI] [PubMed] [Google Scholar]
- Keightley P. D.1998Genetic basis of response to 50 generations of selection on body weight in inbred mice. Genetics 148, 1931–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Eyre-Walker A.1999Terumi Mukai and the riddle of deleterious mutation rates. Genetics 153, 515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Eyre-Walker A.2010What can we learn about the distribution of fitness effects of new mutations from DNA sequence data? Phil. Trans. R. Soc. B 365, 1187–1193 (doi:10.1098/rstb.2009.0266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Halligan D. L.2009Analysis and implications of mutational variation. Genetica 136, 359–369 (doi:10.1007/s10709-008-9304-4) [DOI] [PubMed] [Google Scholar]
- Keightley P. D., Hill W. G.1990Variation maintained in quantitative traits with mutation-selection balance: pleiotropic side-effects on fitness traits. Proc. R. Soc. Lond. B 242, 95–100 (doi:10.1098/rspb.1990.0110) [Google Scholar]
- Keightley P. D., Kacser H.1987Dominance, pleiotropy and metabolic structure. Genetics 117, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Otto S. P.2006Interference among deleterious mutations favours sex and recombination in finite populations. Nature 443, 89–92 (doi:10.1038/nature05049) [DOI] [PubMed] [Google Scholar]
- Kern A. D., Kondrashov F. A.2004Mechanisms and convergence of compensatory evolution in mammalian mitochondrial tRNAs. Nat. Genet. 36, 1207–1212 (doi:10.1038/ng1451) [DOI] [PubMed] [Google Scholar]
- Kimura M.1962On the probability of fixation of mutant genes in a population. Genetics 47, 713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M.1965Attainment of quasi linkage equilibrium when gene frequencies are changing by natural selection. Genetics 52, 875–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M.1983The neutral theory of molecular evolution Cambridge, UK: Cambridge University Press [Google Scholar]
- Kimura M.1985The role of compensatory neutral mutations in molecular evolution. J. Genet. 64, 7–19 (doi:10.1007/BF02923549) [Google Scholar]
- Kimura M., Ohta T.1969aThe average number of generations until extinction of an individual mutant gene in a finite population. Genetics 63, 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M., Ohta T.1969bThe average number of generations until fixation of a mutant gene in a finite population. Genetics 61, 763–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsolver J. G., Hoekstra H. E., Hoekstra J. M., Berrigan D., Vignieri S. N., Hill C. E., Hoang A., Gibert P., Beerli P.2001The strength of phenotypic selection in natural populations. Am. Nat. 157, 245–261 (doi:10.1086/319193) [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M., Johnson T., Barton N.2002General models of multilocus evolution. Genetics 161, 1727–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov A. S.1988Deleterious mutations and the evolution of sexual reproduction. Nature 336, 435–440 (doi:10.1038/336435a0) [DOI] [PubMed] [Google Scholar]
- Kondrashov A. S.1994Muller's Ratchet under epistatic selection. Genetics 136, 1469–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov A. S.1995Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? J. Theor. Biol. 175, 583–594 (doi:10.1006/jtbi.1995.0167) [DOI] [PubMed] [Google Scholar]
- Kondrashov F. A., Kondrashov A. S.2010Measurements of spontaneous rates of mutations in the recent past and the near future. Phil. Trans. R. Soc. B 365, 1169–1176 (doi:10.1098/rstb.2009.0286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov A. S., Sunyaev S., Kondrashov F. A.2002Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl Acad. Sci. USA 99, 14 878–14 883 (doi:10.1073/pnas.232565499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. C. G., Langley C. H.2010Transposable elements in natural populations of Drosophila melanogaster. Phil. Trans. R. Soc. B 365, 1219–1228 (doi:10.1098/rstb.2009.0318) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski R. E., Travisano M.1994Dynamics of adaptation and diversification: a 10 000-generation experiment with bacterial populations. Proc. Natl Acad. Sci. USA 91, 6808–6814 (doi:10.1073/pnas.91.15.6808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L.2006Quantifying the genomic decay paradox due to Muller's ratchet in human mitochondrial DNA. Genet. Res. 87, 133–159 (doi:10.1017/S0016672306008123) [DOI] [PubMed] [Google Scholar]
- Loewe L.2009A framework for evolutionary systems biology. BMC Syst. Biol. 3, 27 (doi:10.1186/1752-0509-3-27) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L., Charlesworth B.2006Inferring the distribution of mutational effects on fitness in Drosophila. Biol. Lett. 2, 426–430 (doi:10.1098/rsbl.2006.0481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L., Charlesworth B., Bartolomé C., Nöel V.2006Estimating selection on non-synonymous mutations. Genetics 172, 1079–1092 (doi:10.1534/genetics.105.047217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- López M. A., López-Fanjul C.1993Spontaneous mutation for a quantitative trait in Drosophila melanogaster. II. Distribution of mutant effects on the trait and fitness. Genet. Res. 61, 117–126 (doi:10.1017/S0016672300031220) [DOI] [PubMed] [Google Scholar]
- Lynch M., Conery J., Bürger R.1995aMutation accumulation and the extinction of small populations. Am. Nat. 146, 489–518 (doi:10.1086/285812) [Google Scholar]
- Lynch M., Conery J., Bürger R.1995bMutational meltdowns in sexual populations. Evolution 49, 1067–1080 (doi:10.2307/2410432) [DOI] [PubMed] [Google Scholar]
- Lynch M., Hill W. G.1986Phenotypic evolution by neutral mutation. Evolution 40, 915–935 (doi:10.2307/2408753) [DOI] [PubMed] [Google Scholar]
- Lynch M., Blanchard J., Houle D., Kibota T., Schultz S., Vassilieva L., Willis J.1999Perspective: spontaneous deleterious mutation. Evolution 53, 645–663 (doi:10.2307/2640707) [DOI] [PubMed] [Google Scholar]
- Mackay T. F. C.2009The genetic architecture of complex behaviors: lessons from Drosophila. Genetica 136, 295–302 (doi:10.1007/s10709-008-9310-6) [DOI] [PubMed] [Google Scholar]
- Mackay T. F. C.2010Mutations and quantitative genetic variation: lessons from Drosophila. Phil. Trans. R. Soc. B 365, 1229–1239 (doi:10.1098/rstb.2009.0315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay T. F. C., Lyman R. F., Jackson M. S.1992Effects of P element insertions on quantitative traits in Drosophila melanogaster. Genetics 130, 315–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay T. F. C., Fry J. D., Lyman R. F., Nuzhdin S. V.1994Polygenic mutation in Drosophila melanogaster: estimates from response to selection of inbred strains. Genetics 136, 937–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay T. F. C., Lyman R. F., Hill W. G.1995Polygenic mutation in Drosophila melanogaster: non-linear divergence among unselected strains. Genetics 139, 849–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay T. F. C., Lyman R. F., Lawrence F.2005Polygenic mutation in Drosophila melanogaster: mapping spontaneous mutations affecting sensory bristle number. Genetics 170, 1723–1735 (doi:10.1534/genetics.104.032581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J., Haigh J.1974The hitchhiking effect of a favorable gene. Genet. Res. 23, 23–35 [PubMed] [Google Scholar]
- McDermott S. R., Noor M. A. F.2010The role of meiotic drive in hybrid male sterility. Phil. Trans. R. Soc. B 365, 1265–1272 (doi:10.1098/rstb.2009.0264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G.2010What drives recombination hotspots to repeat DNA in humans? Phil. Trans. R. Soc. B 365, 1213–1218 (doi:10.1098/rstb.2009.0299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G. A. T., Charlesworth B.1999A population genetic model for the evolution of synonymous codon usage: patterns and predictions. Genet. Res. 74, 145–158 (doi:10.1017/S0016672399003912) [Google Scholar]
- McVean G. A. T., Charlesworth B.2000The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics 155, 929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T., Chigusa S. I., Mettler L. E., Crow J. F.1972Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. Genetics 72, 335–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H. J.1964The relation of recombination to mutational advance. Mutat. Res. 1, 2–9 [DOI] [PubMed] [Google Scholar]
- Nagylaki T.1993The evolution of multilocus systems under weak selection. Genetics 134, 627–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M., Charlesworth B., Charlesworth D.1996The effect of recombination on background selection. Genet. Res. 67, 159–174 (doi:10.1017/S0016672300033619) [DOI] [PubMed] [Google Scholar]
- Ohta T.1992The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23, 263–286 (doi:10.1146/annurev.es.23.110192.001403) [Google Scholar]
- Orr H. A.2010The population genetics of beneficial mutations. Phil. Trans. R. Soc. B 365, 1195–1201 (doi:10.1098/rstb.2009.0282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch J., Tanda S., Stephan W.1997Site-directed mutations reveal long-range compensatory interactions in the Adh gene of Drosophila melanogaster. Proc. Natl Acad. Sci. USA 94, 928–933 (doi:10.1073/pnas.94.3.928) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadnis N., Fry J. D.2005Widespread correlations between dominance and homozygous effects of mutations: implications for theories of dominance. Genetics 171, 385–392 (doi:10.1534/genetics.104.039016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P. C.2008Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9, 855–867 (doi:10.1038/nrg2452) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelwijk F. J., Kiviet D. J., Weinreich D. M., Tans S. J.2007Empirical fitness landscapes reveal accessible evolutionary paths. Nature 445, 383–386 (doi:10.1038/nature05451) [DOI] [PubMed] [Google Scholar]
- Purcell S. M., Wray N. R., Stone J. L., Visscher P. M., O'Donovan M. C., Sullivan P. F., Sklar P.2009Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (doi:10.1038/nature08185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N.1990Linkage strategies for genetically complex traits. I. Multilocus models. Am. J. Hum. Genet. 46, 222–228 [PMC free article] [PubMed] [Google Scholar]
- Robertson A.1960A theory of limits in artificial selection. Proc. R. Soc. Lond. B 153, 234–249 (doi:10.1098/rspb.1960.0099) [Google Scholar]
- Rosenberg M. S., Subramanian S., Kumar S.2003Patterns of transitional mutation biases within and among mammalian genomes. Mol. Biol. Evol. 20, 988–993 (doi:10.1093/molbev/msg113) [DOI] [PubMed] [Google Scholar]
- Rouzine I. M., Brunet E., Wilke C. O.2008The traveling-wave approach to asexual evolution: Muller's ratchet and speed of adaptation. Theor. Popul. Biol. 73, 24–46 (doi:10.1016/j.tpb.2007.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze D., Barton N. H.2006The Hill–Robertson effect and the evolution of recombination. Genetics 173, 1793–1811 (doi:10.1534/genetics.106.058586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan R., Elena S. F.2006Epistasis correlates to genomic complexity. Proc. Natl Acad. Sci. USA 103, 14 402–14 405 (doi:10.1073/pnas.0604543103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre D., Deluna A., Church G. M., Kishony R.2005Modular epistasis in yeast metabolism. Nat. Genet. 37, 77–83 [DOI] [PubMed] [Google Scholar]
- Sharp P. M., Emery L. R., Zeng K.2010Forces that influence the evolution of codon bias. Phil. Trans. R. Soc. B 365, 1203–1212 (doi:10.1098/rstb.2009.0305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M.2008Exchangeable models of complex inherited diseases. Genetics 179, 2253–2261 (doi:10.1534/genetics.107.077719) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski P. D., Gerrish P. J.2010Beneficial mutations and the dynamics of adaptation in asexual populations. Phil. Trans. R. Soc. B 365, 1255–1263 (doi:10.1098/rstb.2009.0290) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W.2010Genetic hitchhiking versus background selection: the controversy and its implications. Phil. Trans. R. Soc. B 365, 1245–1253 (doi:10.1098/rstb.2009.0278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W., Chao L., Smale J. G.1993The advance of Muller's ratchet in a haploid asexual population: approximate solutions based on diffusion theory. Genet. Res. 61, 225–231 (doi:10.1017/S0016672300031384) [DOI] [PubMed] [Google Scholar]
- Stephan W., Song Y. S., Langley C. H.2006The hitchhiking effect on linkage disequilibrium between linked neutral loci. Genetics 172, 2647–2663 (doi:10.1534/genetics.105.050179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev S., Ramensky V., Koch I., Lathe W., 3rd, Kondrashov A. S., Bork P.2001Prediction of deleterious human alleles. Hum. Mol. Genet. 10, 591–597 (doi:10.1093/hmg/10.6.591) [DOI] [PubMed] [Google Scholar]
- Trindade S., Perfeito L., Gordo I.2010Rate and effects of spontaneous mutations that affect fitness in mutator Escherichia coli. Phil. Trans. R. Soc. B 365, 1177–1186 (doi:10.1098/rstb.2009.0287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M.1984Heritable genetic variation via mutation-selection balance: Lerch's zeta meets the abdominal bristle. Theor. Popul. Biol. 25, 138–193 (doi:10.1016/0040-5809(84)90017-0) [DOI] [PubMed] [Google Scholar]
- Weedon M. N., Frayling T. M.2008Reaching new heights: insights into the genetics of human stature. Trends Genet. 24, 595–603 (doi:10.1016/j.tig.2008.09.006) [DOI] [PubMed] [Google Scholar]
- Weinreich D. M., Watson R. A., Chao L.2005Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution 59, 1165–1174 [PubMed] [Google Scholar]
- Weinreich D. M., Delaney N. F., Depristo M. A., Hartl D. L.2006Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312, 111–114 (doi:10.1126/science.1123539) [DOI] [PubMed] [Google Scholar]
- Wolf J. B., Brodie E. D., III, Wade M. J.2000Epistasis and the evolutionary process New York, NY: Oxford University Press [Google Scholar]
- Zhang X. S., Hill W. G.2005Genetic variability under mutation selection balance. Trends Ecol. Evol. 20, 468–470 (doi:10.1016/j.tree.2005.06.010) [DOI] [PubMed] [Google Scholar]