

Abstract
Under the classical view, selection depends more or less directly on mutation: standing genetic variance is maintained by a balance between selection and mutation, and adaptation is fuelled by new favourable mutations. Recombination is favoured if it breaks negative associations among selected alleles, which interfere with adaptation. Such associations may be generated by negative epistasis, or by random drift (leading to the Hill–Robertson effect). Both deterministic and stochastic explanations depend primarily on the genomic mutation rate, U. This may be large enough to explain high recombination rates in some organisms, but seems unlikely to be so in general. Random drift is a more general source of negative linkage disequilibria, and can cause selection for recombination even in large populations, through the chance loss of new favourable mutations. The rate of species-wide substitutions is much too low to drive this mechanism, but local fluctuations in selection, combined with gene flow, may suffice. These arguments are illustrated by comparing the interaction between good and bad mutations at unlinked loci under the infinitesimal model.
Keywords: modifier, mutation load, evolution of recombination, Hill–Robertson effect
1. Introduction
Mutation is the ultimate source of all genetic variation, and is essential for evolution by natural selection: indeed, most of our genome has been shaped primarily by mutation and random drift. Following the rediscovery of Mendel's laws at the turn of the last century, the first geneticists emphasized major mutations as being responsible for the origin of species. In contrast, the biometricians who were establishing the first statistical studies of evolution emphasized the role of selection in shaping standing variation, by bringing together many slight variations into favourable combinations (Provine 1971). Two decades later, after the efficacy of selection on slight Mendelian variants had been established by both theory and by breeding experiments, different views on the role of mutation in evolution persisted in the contrast between the ‘classical’ and the ‘balance’ views (Lewontin 1974). Under the classical view, associated with Hermann Muller, variation around the wild-type is maintained by a short-term balance between mutation and selection. Thus, standing variation is mere noise, and adaptation is due to favourable mutations—either rare novelties, or alleles that become favourable following a change in environment. On this view, the pattern of standing variation is barely relevant, and adaptation is more or less directly dependent on mutation. In contrast, on the balance view, variation is maintained by complex processes such as overdominance, frequency-dependent selection and heterogeneous selection in structured populations. Although mutation ultimately provides variation, it has little influence on standing variation or on adaptation: a change in mutation rate would, on this view, have little influence.
Understanding the effects of sex and recombination, and why they are so widespread, depends on understanding the nature and causes of mutational variation. It seems most profitable to focus on the role of mutation, taking the classical view, simply because this is theoretically straightforward, and because mutation is a universal process, with a well-known molecular basis, which is open to empirical study using model organisms. In contrast, on the balance view, variation is due to complex interactions between ecological environment and population structure, which are hard to capture in laboratory studies. So, the key question is whether direct effects of good and bad mutations are sufficient to explain the prevalence of sexual reproduction and high rates of recombination. I will focus on eukaryotes with Mendelian inheritance, though many of the same issues arise with bacteria, archaea and viruses.
It seems most likely that sex and recombination evolved, and are maintained, because they generate variation which is the raw material for adaptation by natural selection. This idea was long ago set out by Weismann (1889; see Burt 2000), but it has taken a considerable theoretical effort to understand it clearly. Other theories exist, in which sex and recombination are side-effects of mechanisms for repairing double-stranded damage to DNA, or gain an advantage by impeding the response to fluctuating epistatic selection, or by reducing competition between siblings (Williams 1975; Bernstein et al. 1988; Kondrashov 1993; Hamilton 1996), However, these seem unlikely to provide a compelling explanation that applies across a broad range of organisms (Barton & Charlesworth 1998; Otto & Lenormand 2002; Agrawal 2006).
A population genetic advantage to recombination requires high levels of selected polymorphism, so that alleles under selection at different genetic loci have the opportunity to interact, and to be reshuffled by recombination. Recombination cannot be selected unless there are non-random associations between alleles (i.e. linkage disequilibria) that it can break up. Adaptation can be measured by the increase in mean fitness of the population caused by selection on allele frequencies, which is equal to the additive genetic variance in fitness (Fisher 1930). Thus, recombination will increase the rate of adaptation if it increases the additive variance in fitness, and it will only do that if the variance is depressed by negative associations between favourable alleles. In general, then, sex and recombination will be advantageous if there tend to be negative linkage disequilibria between favourable alleles (+ with −, and − with +) that perversely interfere with selection. Moreover, modifiers that increase the rates of sex and recombination will themselves gain a transient advantage through an association with the favourable combinations of alleles that they help to generate. What is crucial, then, is to understand how sufficiently strong and widespread negative linkage disequilibria can arise. Following Felsenstein (1974), I will distinguish between associations generated by deterministic selection versus random drift; following the theme of this issue, I focus on associations among mutations, good and bad.
Under the classical view, recombination allows deleterious mutations to be eliminated more efficiently, and increases the rate at which favourable alleles can be brought together, despite their association with deleterious alleles. First, I consider the effects of negative linkage disequilibria that are generated deterministically by negative epistasis, and in particular, by truncation selection. I will contrast three cases: asexual reproduction, unlinked loci and most extreme, a population that is forced into linkage equilibrium in every generation. Following Charlesworth (1990, 1993a) I will use the infinitesimal model, which neglects changes in allele frequency as being very slow, relative to changes in linkage disequilibrium among loosely linked loci. I then turn to the effects of random linkage disequilibria that are generated stochastically, by sampling drift. Here, considerable progress can be made by following the probability of fixation of a single favourable allele within a very large population, modelled as a branching process. Together, these theoretical models give a rather general understanding of the effects of free recombination, which can be related to observable features of spontaneous mutations.
As will be clear from the reference list, Brian Charlesworth has played a key role in shaping the research described here—both empirical and theoretical. He showed how ‘background selection’ owing to deleterious mutations could explain patterns of neutral diversity in Drosophila (Charlesworth et al. 1995), and the degeneration of non-recombining regions (Bachtrog & Charlesworth 2002; Kaiser & Charlesworth 2008; Betancourt et al. 2009); how negative epistasis causes selection for sex and recombination (Charlesworth 1990, 1993a,b); and helped give the first direct estimates of genomic mutation rate in Drosophila (Haag-Liautard et al. 2007), and estimates of their effects on fitness (Loewe & Charlesworth 2006).
2. Deterministic associations
In an asexual population, subject to unidirectional deleterious mutation away from the wild-type, at a rate U per genome per generation, the mean fitness is reduced by a factor of exp (−U) below the maximum possible (Kimura & Maruyama 1966). Remarkably, this classical result is independent of how selection acts. It can be understood by realizing that at equilibrium, each wild-type individual has to produce one wild-type offspring, yet the chance that an offspring escapes any mutation is exp (−U), assuming a Poisson distribution of numbers of mutations. So, the wild-type must have fitness exp (U) higher than the mean fitness, which in the long run is one in an asexual population. This mutation load imposes a constraint on the genome-wide rate, which may have been especially severe in the first reproducing organisms, and is now, for those organisms with the largest functional genomes.
If the effects of different mutations on fitness multiply together, then a sexual population will remain at linkage equilibrium, and so recombination will have no effect: thus, mean fitness will be exp (−U) regardless of the mode of reproduction or the pattern of genetic linkage. However, negative epistasis together with recombination allows a far higher mutation rate to be tolerated (Kimura & Maruyama 1966). This can be understood using a graphical argument (figure 1a). If the effects of deleterious mutations on fitness increase as their number accumulates (i.e. if there is negative or synergistic epistasis), then the marginal selection on each additional allele can be much higher for a given genetic load, allowing the equilibrium load to be reduced.
Figure 1.

(a) The curve shows the log mean fitness as a function of the mean number of deleterious mutations. At equilibrium, this number equals U/s, where the average selection coefficient, s, is the gradient of the curve,  . Therefore, if the effects of deleterious mutations are multiplicative (shown by a straight line on this log scale), the mutation load (defined as the difference in log fitness between the fittest genotype and the population mean) is L = U, as indicated at left. With negative epistasis (shown by the curve), the distance at left between the tangent and the mean fitness is still equal to U, but the mutation load, L, is much smaller. In this example, truncation selection acts on the number of deleterious mutations, with a fraction L = 0.2 surviving, and genomic mutation rate U = 10; dots indicate the equilibrium point. The population is assumed to cluster around the mean, and to be at linkage equilibrium. (b) The log mean fitness at equilibrium as a function of U, keeping genotype fitnesses the same as in (a). This decreases steeply with further increases of U.
. Therefore, if the effects of deleterious mutations are multiplicative (shown by a straight line on this log scale), the mutation load (defined as the difference in log fitness between the fittest genotype and the population mean) is L = U, as indicated at left. With negative epistasis (shown by the curve), the distance at left between the tangent and the mean fitness is still equal to U, but the mutation load, L, is much smaller. In this example, truncation selection acts on the number of deleterious mutations, with a fraction L = 0.2 surviving, and genomic mutation rate U = 10; dots indicate the equilibrium point. The population is assumed to cluster around the mean, and to be at linkage equilibrium. (b) The log mean fitness at equilibrium as a function of U, keeping genotype fitnesses the same as in (a). This decreases steeply with further increases of U.
(a). Mutation load with truncation selection
The most efficient way to eliminate deleterious mutations is by truncation selection, allowing only the fittest fraction θ to reproduce (Crow & Kimura 1979); this is an extreme form of negative epistasis, in which a single additional mutation is lethal if it takes the individual above a threshold. The simple graphical argument of figure 1 does not apply directly, because fitness changes abruptly as a function of genotype. However, the marginal selection coefficient is easily calculated. If we imagine an underlying normally distributed quantitative trait that represents ‘genetic quality’, and which is subject to truncation selection, then the selection against an allele at locus i that reduces quality by αi is just si = αi f(θ)/σ, where f(θ) is the mean of the upper θth fraction of a normal distribution, measured in standard deviations, and σ2 is the variance in quality (Haldane 1930). A population at linkage equilibrium will have an allele frequency pi = μi/si at mutation selection balance, and so the genetic variance in quality will be 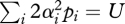
 (assuming diploidy and defining U = 2Σiμi). Therefore,
(assuming diploidy and defining U = 2Σiμi). Therefore,  —a result which can be seen directly, as a balance between the loss of quality due to mutation, U
—a result which can be seen directly, as a balance between the loss of quality due to mutation, U  , and the response to truncation selection on quality, σ f(θ). (Note that the scale for ‘quality’ is arbitrary—σ and α share the same dimensions.)
, and the response to truncation selection on quality, σ f(θ). (Note that the scale for ‘quality’ is arbitrary—σ and α share the same dimensions.)
In a sexual population, with a large number of unlinked loci, truncation selection will generate negative linkage disequilibrium, which will be halved in each generation by segregation of unlinked loci. Under the infinitesimal model, if selection reduces the genetic variance by a factor of ω < 1 (a function solely of the fraction selected, θ), then at equilibrium the genetic variance is reduced by a factor 1/(2 − ω) (Bulmer 1980), and so the genetic variance is given by 
It is not obvious how to judge the effect of the mutation load on absolute fitness under truncation selection, because only a fraction θ reproduces, regardless of how much genetic variance is there, or how many mutations have accumulated. Thus, under strict truncation selection there is no clear upper limit to the mutation rate that can be tolerated. I discuss this vexed question below.
(b). The mutational-deterministic hypothesis
Kondrashov (1988) made a forceful argument for the importance of deleterious mutations in driving the evolution of recombination, which did much to promote further research—both theoretical and empirical. Kondrashov (1984, 1988) showed by simulation that modifiers of sex and recombination could gain an advantage by alleviating the load, provided that the total mutation rate, U, is large. Crucially, he pointed out that a high genomic mutation rate (U > l, say) could only be tolerated if there were both negative epistasis and sexual reproduction. Thus, showing that U > 1 would necessarily imply both negative epistasis, and consequently, selection for sex and recombination.
An influential theoretical result states that in a population at equilibrium under selection alone, modifiers that reduce recombination are always favoured; this is known as the reduction principle (Feldman & Krakauer 1976; Feldman et al. 1996). For increased recombination to be selected, there must be either change through time (e.g. fluctuating epistasis, or negative associations between alleles that are increasing), or some other force such as mutation or migration, that can counterbalance change in allele frequency owing to directional selection. Indeed, models that include mutation or migration and so allow an equilibrium, have similar consequences for recombination as directional selection alone (Feldman et al. 1980; Lenormand & Otto 2000; Martin et al. 2006).
Charlesworth (1990) gave an elegant theoretical analysis that showed how Kondrashov's (1988) ‘mutational deterministic’ hypothesis leads to selection for modifiers of sex and recombination. He assumed that the number of deleterious mutations follows an approximately normal distribution, and that log fitness is a quadratic function of this number, so that selection maintains the normal distribution. Assuming that a large number of genes are involved (i.e. the infinitesimal model), allele frequencies change slowly, and recombination modifiers are affected mainly by changes in linkage disequilibrium owing to epistatic selection. Charlesworth (1990) compared three modes of reproduction: asexual; segregation of two non-recombining genomes in a diploid; and sex and recombination with multiple linear chromosomes. His analysis showed that with large U, and with parameters as estimated for larval viability in Drosophila, there could be substantial selection for recombination. However, most of the effect came from segregation, rather than from recombination, making it hard to explain how high recombination rates are maintained.
(c). A general quasi-linkage equilibrium approximation
Barton (1995a) gave a general analysis of selection on weak modifiers of recombination, which allowed for arbitrary interactions among multiple sites. The strength of selection for recombination was approximated by assuming that directional selection, s is weak, relative to recombination (s ≪ r), and that epistasis between any particular set of genes is very weak (ϵ ≪ s2). This allows a ‘quasi-linkage equilibrium’ (QLE) approximation, in which the selection for recombination can be related directly to the effects of selection and recombination on the mean and variance of log fitness:
 |
2.1 |
This is a simplified version of eqn (16) in Barton (1995a), which assumes that the modifier is flanked by the selected loci, and that it changes all recombination rates by the same factor; thus, selection for the modifier (δs) is proportional to this factor (δlog(r)). δV is the contribution of linkage disequilibria to the additive variance in log fitness, and must be negative for recombination to be favoured; 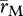 is the harmonic mean recombination rate between the modifier and each selected locus, and δlog(W) is the decrease in mean log fitness owing to recombination in each generation, as explained below. This is an approximation which assumes that s ≪ r ≪ 1; however, it is quite accurate for strong selection (cf. Charlesworth 1990, 1993a).
is the harmonic mean recombination rate between the modifier and each selected locus, and δlog(W) is the decrease in mean log fitness owing to recombination in each generation, as explained below. This is an approximation which assumes that s ≪ r ≪ 1; however, it is quite accurate for strong selection (cf. Charlesworth 1990, 1993a).
Selection increases mean fitness by precisely the genotypic variance in fitness, which includes both the additive component owing to the marginal effect of each allele, and also the non-additive component, because of epistasis and dominance interactions. Recombination causes an immediate loss of fitness, log(W), because of the break-up of gene combinations that had been favoured by epistasis, and similarly, segregation causes a loss owing to the break-up of associations between homologous genes in paternal and maternal genomes that had been generated by dominance components of the variance in fitness. If sex and recombination were to destroy all associations, leaving only the effect of changes in allele frequency, then mean fitness would fall back to an increase equal to the additive genetic variance. It is this immediate ‘recombination load’ (Charlesworth & Barton 1996) that drives the ‘reduction principle’. (Note that the recombination load is bounded above by the non-additive genetic variance in fitness).
If there is negative epistasis, then recombination also inflates the additive genetic variance by breaking- up the negative linkage disequilibria amongst favourable alleles, which increases mean fitness in future generations, if selection keeps acting in the same direction. To the extent that the modifier is linked to alleles that will increase under directional selection, it will tend to increase with them; this is expressed by the first term in equation (2.1), which involves the reciprocal of the harmonic mean recombination between the modifier and the selected loci. This QLE approximation describes Charlesworth's (1990) analysis well, though the latter extends to cover stronger selection. (Charlesworth's analysis is based primarily on the normal approximation, and does not require weak selection as such). Equation (2.1) also approximates Charlesworth's (1993a,b) analyses of directional and fluctuating selection, which give a similar advantage to recombination as does a mutation-selection balance. To summarize: equation (2.1) shows that the advantage of recombination depends primarily on how much negative linkage disequilibria reduce the variance in log fitness (V).
(d). Difficulties with negative epsitasis
The mutational-deterministic hypothesis (Kondrashov 1988) is attractive, because it relies on the deleterious mutations that afflict all organisms, and because it is open to a simple empirical test—if the genomic mutation rate is large, then negative epistasis must alleviate the mutation load, and there must necessarily be selection for recombination. However, it suffers from several difficulties, as indeed does the more general deterministic explanation, in which negative linkage disequilibria are built up by negative epistasis. Otto & Feldman (1997) point out that epistasis must not be too strong (otherwise, it imposes too high a recombination load), but also, must not vary much across different sets of genes. This is because epistasis of any sign contributes to the recombination load, through terms like  , whereas only negative epistasis contributes to the selection for recombination, through terms like siεi jsj < 0. Therefore, variance in epistasis (
, whereas only negative epistasis contributes to the selection for recombination, through terms like siεi jsj < 0. Therefore, variance in epistasis ( ) tends to select against recombination.
) tends to select against recombination.
A related difficulty is that it is hard to see why epistasis should tend to be systematically negative. It is true that negative epistasis (in the limit, truncation selection) tends to alleviate various kinds of genetic load (Kimura & Maruyama 1966; Sved 1968), but it is not at all clear that it should evolve to be negative. Metabolic models give mixed results, with no clear indication that they would cause negative epistasis (Keightley & Kacser 1987; Szathmary 1993). Selection for robustness to environmental and genetic perturbations may lead to negative epistasis, by analogy with arguments for the evolution of dominance: if some ‘safety margin’ has evolved, then moderate loss of function may have little effect on fitness, whereas larger numbers of deleterious mutations, especially when homozygous, may cause a substantial loss of fitness. However, though negative epistasis may evolve in this way in some models, there is again no clear theoretical support for its generality (Hansen 2006).
(e). The cost of selection
As well as reducing the mutation load, recombination with negative epistasis also reduces the ‘cost of natural selection’. Haldane (1957) showed that the total loss of reproductive capacity required to raise an allele from a low frequency p0 is approximately log (1/p0); just as with mutation load, this result applies to asexuals and to sexual populations at linkage equilibrium, for any pattern of selection. (Of course, favourable mutations are not in themselves costly: the ‘cost’ is in slow evolution by natural selection, rather than instantaneous adaptation). However, negative epistasis allows any number of rare alleles to be fixed in a sexual population. This can be understood by thinking of the most favourable case of truncation selection. Then, any number of rare alleles can be picked up, and will increase in frequency by a factor 1/θ in each generation, where θ is the fraction selected. Once these alleles become common, recombination is needed to bring them together and fix the fittest genotype. However, since most of the cost of selection is incurred during the long time that favourable alleles are increasing from low frequency (approx. (1/S)log(1/p0) generations), this argument shows that almost all of the cost of selection can be avoided in a freely recombining population. Charlesworth (1993a) showed this deterministic advantage of recombination in the less extreme case of directional selection on a quantitative trait that also experiences stabilizing selection towards a moving optimum, and hence, negative epistasis (see also Burger (1999) and Waxman & Peck (1999)). Here, stabilizing selection reduces the additive genetic variance, and recombination restores it, hence speeding the response.
Recombination also speeds up the response to directional selection in the presence of a mutation load, provided that there are negative interactions between the genes involved. In an infinite population, the response to directional selection on an additive trait is independent of mutation load, if effects on log fitness add up. However, if truncation selection acts on the trait, plus some measure of the mutation load, then negative associations will build up that interfere with selection—individuals with higher trait values will tend to carry a higher load of deleterious mutations, because these will have been partially shielded from selection by the higher trait (figure 2).
Figure 2.

The association between a selected trait and the mutation load slows down the response to truncation selection. The upper line shows the response to truncation selection under the infinitesimal model of a trait with genetic variance at linkage equilibrium V1 = 20; 20 per cent survive in each generation, and linkage disequilibria reduce the variance to 11.6. The lower line shows the response when truncation selection acts on the sum of this trait, and the number of deleterious mutations. Because truncation selection is spread over two traits, and because there is a correlation of 21 per cent between mutation load and the favoured trait, the selection response is substantially reduced (U = 10, as in figure 1).
3. Random associations
(a). Reduced diversity in regions of reduced recombination
About 20 years ago, it was observed that regions of the Drosophila genome with low recombination have low nucleotide diversity at silent sites. This is not associated with any reduction in between-species divergence, and so cannot be explained by differences in mutation rate (Aguade et al. 1989; Stephan & Langley 1989; Begun & Aquadro 1992). Similar associations have been found in other groups (Nachman 2002), though the causes of the correlation seen in humans remain unclear (Hellman et al. 2005; Spencer et al. 2006; Cai et al. 2009). Similarly, exceptionally low diversity is seen in genomes, or regions of genome, with little or no recombination, such as Y chromosomes (Bachtrog & Charlesworth 2002; Charlesworth et al. 2009), dot chromosomes (Betancourt et al. 2009), obligate selfers (Charlesworth 2003) or endosymbiotic bacteria (Funk et al. 2001).
If such patterns are because of the population genetic effects of recombination, then they must be caused by selection at linked loci, and mediated by linkage disequilibria between selected loci, and the observed neutral markers. Moreover, such linkage disequilibria must be generated by random drift, since there can be no epistasis with neutral markers. Random associations that reduce diversity must also interfere with selection, reducing both the ability of populations to accumulate favourable mutations and to eliminate deleterious ones. This must lead, finally, to selection for modifiers that increase recombination. So, the simple observation of a correlation between neutral diversity and recombination implies the existence of interference among selected loci that must lead to selection for recombination. Here, I summarize the relevant theory, and in §4, return to the interpretation of the relation between diversity and recombination.
(b). The Hill–Robertson effect
As well as producing random fluctuations in allele frequency that reduce genetic diversity, genetic drift also produces random associations between alleles at different loci. These random linkage disequilibria tend to become negative, and so interfere with selection; this interference favours increased recombination, in exactly the same way as when negative linkage disequilibria are generated by epistasis (equation 2.1). It seems at first paradoxical that random drift should lead to negative associations between favoured alleles, because the immediate effect of drift is impartial: associations between any pair of alleles are, on average, zero and have the same distribution regardless of how the alleles affect fitness. Thus, the tendency for random associations between alleles to become negative is because of an interaction between drift and selection at the two loci. This can be understood in two ways. First, positive associations will accelerate selection, and will rapidly fix the fit ++ combinations that they produce. In contrast, negative associations shield alleles from selection, reducing the variance in fitness, and so will tend to persist. In the extreme case of asexuality, populations can fix for a mixture of +− and −+ combinations with the same fitness, and so will maintain negative linkage disequilibria indefinitely.
Another way to understand why random linkage disequilibria tend to interfere with directional selection was set out by Hill & Robertson (1966). The effect on fitness of alleles at one locus are obscured by association with random genetic backgrounds, each with their own effect on fitness. In other words, random association of one locus with other selected loci induce random perturbations that act in the same way as classical random drift, and interfere with selection. On this view, the fluctuations in allele frequency at the selected locus do average to zero, but their long-term effect is negative. To see this, think of a favourable mutation that increases relative fitness by s, and that starts at some low frequency p0 = 1/2N. Its chance of fixation is 2s(Ne/N), and so its expected frequency in the long term is 2s(Ne/N) = (4Nes)p0. Any reduction in the effective population size, Ne, reduces its long-term expected frequency in direct proportion; this effect is mediated by negative linkage disequilibria that arise during its passage to fixation, but the effect is most easily understood as an inflation of drift at the focal locus.
(c). Unlinked loci
Hill & Robertson's (1966) argument applies most directly to the effects of selection on unlinked loci, which causes rapid fluctuations that are precisely analogous to random drift. With no selection anywhere in the genome, the rate of sampling drift is proportional to the variance in genic fitness (i.e. to the variance in the number of copies left by each gene). With selection on unlinked loci, fitness is inherited and the correlation between the fitness of genetic backgrounds in successive generations is 1/2. Therefore, a gene that increases fitness by δ in one generation will on average cause an increase of δ/2 in the next, δ/4 in the next, and so on; the net increase is 2δ. Thus, a heritable variance in fitness V, owing to unlinked loci, has 22 = 4 times the effect of non-heritable variance (Robertson 1961). This heuristic argument is confirmed by analysis of the infinitesimal model (Barton 2009). If genes have multiplicative effects, we can write fitness as ez, where the log fitness, z, is an additive trait, which has variance v. Then, the diversity at a neutral locus is reduced by a factor e−4v and the probability of fixation of a favoured allele is reduced by the same factor. This formula applies to any source of fitness variance. The variance in log fitness owing to deleterious mutation is approximately U  , and the variance owing to selective sweeps at a rate Λ is approximately 2Λ s̄, where
, and the variance owing to selective sweeps at a rate Λ is approximately 2Λ s̄, where  in the mean effect of mutations on log fitness. Even if the genome-wide rates of mutation and of selective sweeps, U, Λ, are of order one,
in the mean effect of mutations on log fitness. Even if the genome-wide rates of mutation and of selective sweeps, U, Λ, are of order one,  is likely to be small, and so neither seems a plausible source of fitness variance. However, fluctuating selection might well contribute substantial further variance (Burt 2000; Merila & Sheldon 2000); then, unlinked loci could have significant effects on both diversity and adaptation.
is likely to be small, and so neither seems a plausible source of fitness variance. However, fluctuating selection might well contribute substantial further variance (Burt 2000; Merila & Sheldon 2000); then, unlinked loci could have significant effects on both diversity and adaptation.
Hitch-hiking effects are much stronger with linkage: using Robertson's (1961) argument, variance in log fitness v, owing to loci that recombine at rate r, inflates the rate of drift in proportion to v/r2, which diverges when averaged over a linear genetic map. However, the argument breaks down when recombination and selection are of the same order; this suggests that we should truncate the average at r ∼ s, so that the net effect on the rate of drift, averaged over a linear map of length R, is 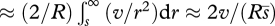 , where
, where  is the arithmetic mean selection on segregating variation. (The factor of 2 arising because the genetic map extends to either side of the locus of interest.) Since the variance in log fitness owing to either deleterious mutation or to sweeps at a given rate are both proportional to
is the arithmetic mean selection on segregating variation. (The factor of 2 arising because the genetic map extends to either side of the locus of interest.) Since the variance in log fitness owing to either deleterious mutation or to sweeps at a given rate are both proportional to  , this rough argument suggests that the net effect may be independent of
, this rough argument suggests that the net effect may be independent of  . More detailed analysis shows that this argument holds for deleterious mutations, but not for selective sweeps (Barton 2009). With linkage, the effects of fitness variance depends on its source (positively or negatively selected alleles), and on its target (neutral, deleterious or favourable alleles). These cannot be described by a single parameter, the additive variance in fitness, as was the case with no linkage. So, to understand the effects of linkage, we first consider the extreme case of asexuality.
. More detailed analysis shows that this argument holds for deleterious mutations, but not for selective sweeps (Barton 2009). With linkage, the effects of fitness variance depends on its source (positively or negatively selected alleles), and on its target (neutral, deleterious or favourable alleles). These cannot be described by a single parameter, the additive variance in fitness, as was the case with no linkage. So, to understand the effects of linkage, we first consider the extreme case of asexuality.
(d). Asexual populations
In an asexual population, at a balance between mutation to deleterious alleles at a rate U, and mean selection  against them, the number of mutations carried by an individual is Poisson, with mean
against them, the number of mutations carried by an individual is Poisson, with mean  . Hence, the fittest class will be very rare if
. Hence, the fittest class will be very rare if  , with frequency
, with frequency  . Yet, at a steady state under one-way mutation, the whole population must trace its ancestry back to this fittest class (Fisher 1930). Neutral diversity will equal that within this small subpopulation of 2N
. Yet, at a steady state under one-way mutation, the whole population must trace its ancestry back to this fittest class (Fisher 1930). Neutral diversity will equal that within this small subpopulation of 2N  genes, plus whatever diversity has built up by mutation since descent from the fittest class, approximately
genes, plus whatever diversity has built up by mutation since descent from the fittest class, approximately  generations ago. Thus,
generations ago. Thus,  , which will be very much less than 4N μ. Similarly, an allele with advantages less than
, which will be very much less than 4N μ. Similarly, an allele with advantages less than 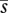 can only establish if it arises in the fittest class, and so its fixation probability is reduced by
can only establish if it arises in the fittest class, and so its fixation probability is reduced by  —a similar, although larger, reduction as for neutral diversity. Alleles with a large advantage can fix if they arise in a wider range of backgrounds, but will carry deleterious alleles to fixation (Peck 1994; Johnson & Barton 2002). In summary, deleterious mutations in an asexual population drastically reduce both neutral diversity and the rate of adaptation, by a factor of approximately
—a similar, although larger, reduction as for neutral diversity. Alleles with a large advantage can fix if they arise in a wider range of backgrounds, but will carry deleterious alleles to fixation (Peck 1994; Johnson & Barton 2002). In summary, deleterious mutations in an asexual population drastically reduce both neutral diversity and the rate of adaptation, by a factor of approximately  . Kaiser (2009) has recently shown that when there is a high mutation rate in a non-recombining region, the reduction is weaker than this formula, because of the intereference between deleterious mutations.)
. Kaiser (2009) has recently shown that when there is a high mutation rate in a non-recombining region, the reduction is weaker than this formula, because of the intereference between deleterious mutations.)
Selective sweeps through an asexual population also have a drastic effect, causing ‘periodic selection’ in which all variation is eliminated when a single favourable mutation fixes. Neutral diversity can only build up over the time since the most recent sweep (approx. 1/Λ), and so average pairwise diversity is π ∼ 2μ/Λ. As Fisher (1930) and Muller (1932) first pointed out, advantageous alleles can only fix if they arise within a background that is already on its way to fixation—unless they themselves have an advantage that is large enough to out-compete a previously established selective sweep. Either way, complete linkage drastically reduces the efficiency with which selection can accumulate adaptive mutations (see Rouzine et al. 2008, for a summary of recent theory for asexual populations).
(e). A linear map
The effects of mutations that are scattered over a linear genetic map lie somewhere between these two extremes of no linkage versus complete linkage. Surprisingly, though, some simple approximations are available, which depend on the density of deleterious mutations, U/R or of selective sweeps, Λ/R. Deleterious mutations with effect s reduce diversity at a linked locus by a factor  (Hudson & Kaplan 1995; eqn 3); averaging over a linear map, and assuming multiplicative selection, diversity is reduced by exp(−U/R) (Hudson & Kaplan 1995; Nordborg et al. 1996). With a heterogeneous density of mutations on the genetic map, the pattern of diversity does depend on selection, with lower diversity in regions where mutation and selection are strong relative to recombination: for example, Loewe & Charlesworth (2007) show that patterns of diversity within genes can be explained by ‘background selection’. However, the average over a linear map does not depend on the average selection strength, but rather, on the density of mutations, U/R. The net effects of background selection on the chance of fixation of a favoured allele are also reduced by a similar amount (Barton 1995b; eqn 22).
(Hudson & Kaplan 1995; eqn 3); averaging over a linear map, and assuming multiplicative selection, diversity is reduced by exp(−U/R) (Hudson & Kaplan 1995; Nordborg et al. 1996). With a heterogeneous density of mutations on the genetic map, the pattern of diversity does depend on selection, with lower diversity in regions where mutation and selection are strong relative to recombination: for example, Loewe & Charlesworth (2007) show that patterns of diversity within genes can be explained by ‘background selection’. However, the average over a linear map does not depend on the average selection strength, but rather, on the density of mutations, U/R. The net effects of background selection on the chance of fixation of a favoured allele are also reduced by a similar amount (Barton 1995b; eqn 22).
Maynard Smith & Haigh (1974) showed that a selective sweep of strength S reduces neutral diversity at a linked locus by (on average) 2N (S)−2r/S (see Stephan et al. 1992). This can be interpreted as the chance that two lineages coalesce in the unique genome that carried the original positive mutation, rather than recombining away to some more distant ancestry (figure 3). The net rate of coalescence between two lineages because of the rate of recurrent sweeps, Λ, scattered over a map of length R, averages 2(Λ/R) (S/log(2N S)), compared with a rate 1/(2N) due to a sampling drift. Thus, neutral diversity is reduced by a factor:
 |
3.1 |
which depends on both the density of sweeps (Λ/R) and on the strength of selection relative to drift (2N S).
Figure 3.

The different effects of a selective sweep on neutral diversity (a) and on a weakly favoured allele (b). Neutral lineages will only coalesce if they trace right back to near the origin of the sweep. Diagram (a) shows two lineages (black, grey) that both trace back into the fitter background, but both then recombine away into the ancestral background, and so remain unrelated. Such recombination, allowing the lineages to escape coalescence, can occur throughout the long time taken for the new mutation to increase from one copy (shown by disc at lower left). Diagram (b) shows how a weakly favoured allele is knocked back by a sweep. To survive, it must recombine onto the new background doing the brief duration of the sweep—giving less scope for recombination than for neutral diversity.
A linked selective sweep has a more severe effect on the survival of an advantageous mutation than it does on neutral diversity. While a rare allele with some small advantage, s, is struggling to increase from low frequency, it is vulnerable to being knocked back by the substitution of a strongly selected allele at a linked locus: the effect is as if its frequency were suddenly reduced by a random factor, which averages 1 − (s/S)r/S (Barton 1995b), and which will be substantial if linkage is tighter than the advantage of the strongly selected allele (r ≪ S). (Throughout this section, s refers to the advantage of the allele that is increasing from low numbers, while S refers to the selection on sweeps that are already established.) In contrast, the effect on neutral diversity is restricted to a narrower region of the genetic map: as we trace a lineage back through the sweep, it can recombine away, onto the ancestral background, at any time back until the sweeping allele originated (t ∼ (1/S) log(4NeS)) (figure 3). Therefore, the effect of sweeps on neutral diversity is significant over a map length of r ∼ log(4NeS), which is much less than r ∼ S for strongly selected sweeps (log(4NeS) ≫ 1).
(f). Multiple sweeps
The expected effect of multiple selective sweeps, occurring at random times and at random locations on the genetic map, can be found by approximating their effect as a series of random catastrophes that each reduce allele frequency by some fraction, averaging (s/S)−r/S. On average, these will knock back any rare allele at a rate scrit, and so its chance of fixation is just 2(s − scrit). This result seems puzzling at first, because random hitch-hiking events do not alter the expected frequency of an allele. However, almost all sweeps originate on the common background, and so knock the rare allele back. These are countered by extremely rare events where the favourable mutation arises in coupling with the rare allele, and give it an extremely large boost. However, such rare events can do no more than making fixation certain, and so overall, have negligible effect on fixation probability. Thus, selective sweeps set a threshold selection coefficient, below which an adaptive allele has negligible chance of fixation in a large population. (Strictly speaking, the probability of fixation tends to zero as N tends to infinity, if s < scrit; Barton 1994). This critical threshold is proportional to the variance in log fitness due owing to sweeps, per unit map length (v/R; Barton 1994):
 |
3.2 |
It is remarkable that multiple selective sweeps have a substantially stronger effect on adaptation than on neutral diversity—in effect, preventing adaptation via weakly selected mutations. Presumably, the effect on very weakly selected alleles (Nes ∼ 1) is intermediate between those for neutral alleles and for strongly selected alleles (Nes ≫ 1), which are sure to be established once above some low frequency. A weakly selected allele experiences a series of random fluctuations, which can be approximated as a diffusion with a rate of drift approximately 1/(2Ne). Thus, we expect the probability of fixation to be given by the classical formula, 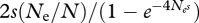 ; this tends to 1/(2Ne) for a strictly neutral allele, and to
; this tends to 1/(2Ne) for a strictly neutral allele, and to  for a deleterious allele. Whether fixation of alleles with such small effects is significant, either for degradation by random drift or for weakly selected adaptations such as codon usage bias, is an open question (Kondrashov 1995).
for a deleterious allele. Whether fixation of alleles with such small effects is significant, either for degradation by random drift or for weakly selected adaptations such as codon usage bias, is an open question (Kondrashov 1995).
(g). Interference due to weakly selected alleles
These results assume that the alleles that cause Hill–Robertson interference (whether selected positively or negatively) evolve deterministically. Then, neutral diversity can be found using the structured coalescent (Wakeley 2008), in which lineages trace back through different genetic backgrounds, whose frequencies change in a known way. Similarly, multitype branching processes give the probability of fixation of a single favourable allele, which depends on the genetic background in which it finds itself. What if drift and selection have comparable strength (Nes ∼ 1), so that the genetic backgrounds responsible for interference fluctuate randomly?
The effect of weakly selected alleles on neutral diversity can be found using the structured coalescent, but allowing for the fluctuating frequencies of the genetic backgrounds (Hudson & Kaplan 1988; Barton & Etheridge 2004). If these fluctuations mirror those due purely to drift (as when the backgrounds are defined by neutral alleles), there can be no effect on linked loci. If they fluctuate less (as with balanced polymorphism), then neutral diversity increases, though only in a narrow region of the map (r ∼ μ; Hudson & Kaplan 1988 ; Kaplan et al. 1988). Conversely, if background frequencies change systematically, as with selective sweeps or deleterious mutations, diversity is reduced. When selection is weak (Nes ∼ 1), random fluctuations due to drift greatly reduce hitch-hiking effects; for example, the effect of background selection with no recombination is roughly halved when Nes ∼ 3, compared with the large Nes limit (Barton & Etheridge 2004; fig. 12). Nevertheless, because a very large number of sites may be under weak selection, their cumulative effects can be significant (McVean & Charlesworth 2000).
The main outstanding theoretical problem is to understand how selection over a large number of loci generates Hill–Robertson interference, and how that in turn selects for recombination. The infinitesimal model provides a simple approximation for unlinked loci that identifies the additive variance in fitness as the key parameter. However, linked loci can have a much stronger effect—especially, on selection for recombination, since modifiers must remain linked to the fitter combinations that they help produce. The selection for recombination owing to two selected loci can be found theoretically, for both fluctuations in established polymorphisms (Barton & Otto 2005), and for the stochastic increase of a single favourable mutation (Roze & Barton 2006). In both cases, simple extrapolation from two selected loci to many implies that selection for recombination should be very weak—proportional to the square of the heritable fitness variance—unless that variance is very high. Yet, simulations of large numbers of loci show much stronger effects than expected by extrapolation from two selected loci. Iles et al. (2003) simulate selection on standing variation, and show that for fixed variance in fitness, Hill–Robertson interference, and the consequent selection for recombination, increases with the number of loci. Keightley & Otto (2006) simulate deleterious mutation at many loci, and show a similar increase in Hill–Robertson interference with the number of genes. The challenge is to find a theoretical approximation that can explain these patterns.
4. Discussion
This condensed summary of the theory relating to interference between selected loci, and its consequences for the evolution of recombination shows the considerable progress that has been made in understanding the theoretical issues—primarily, in laying out a taxonomy of the distinct issues, and in identifying the importance of key parameters such as the density of mutations and of selective sweeps on the genetic map. In the following discussion, I focus on two issues: the limits to the amount of selection that may be acting, and the evidence as to its actual extent.
(a). Evidence on the extent and consequences of Hill–Robertson interference
The correlation between recombination and diversity, first seen in Drosophila, has driven a large research program—both empirical and theoretical—that aims to answer (at least) four questions. What kind of selection is responsible for reducing neutral diversity? Does this also interfere with selection itself, reducing adaptation as well as neutral diversity in region of low recombination? Are such effects also important across the bulk of the genome, in regions of typical recombination? What is the net selection for recombination?
Most attention has been given to finding whether reduced diversity is due mainly to the flux of favourable mutations, sweeping through to fixation, or to background selection, owing to elimination of deleterious mutations. These have the advantage of providing clear alternative hypotheses, described by simple and observable parameters: the rate of species-wide selective sweeps, Λ, and the genomic mutation rate, U. However, as discussed below, neither are likely to be sufficient explanations, either for reduced diversity and adaptation in regions of low recombination, or for the maintenance of recombination. Selection that fluctuates in time and space may be more important, but cannot be summarized by a few simple parameters. A distinct aspect of this question is whether weakly selected loci (Nes ∼ 1) have a significant influence on linked loci; such alleles have distinct effects that are harder to analyse or to measure than those evolving deterministically with negligible influence from random drift.
To a first approximation, a reduction in neutral diversity can be seen as due to an increased rate of random drift, described by a reduced effective population size, Ne. This is expected to reduce the chance of fixation of favourable alleles, by a ratio Ne/N, and to increase the chance that deleterious alleles will fix if Nes is small. However, as explained above, random linkage disequilibria can have substantially different effects on selected alleles than on neutral: on the one hand, fixation probability of favourable mutations can be reduced much more than neutral diversity, but on the other hand, linkage to weakly selected alleles can have much smaller effect than linkage to strongly selected alleles.
Evidence that linkage to selected loci reduces adaptation as well as diversity comes from weaker codon usage bias in regions of low recombination (Kliman & Hey 1993), and from lower rates of non-synonymous substitution and higher frequencies of rare (presumably deleterious) alleles in regions of low recombination (Betancourt & Presgraves 2002; Presgraves 2005; Betancourt et al. 2009). If Hill–Robertson interference is extensive, then there should be a positive correlation between levels of neutral diversity and the rate of adaptive substitution. However, Macpherson et al. (2007) found that in regions of the Drosophila simulans genome with a higher rate of non-synonymous divergence from D. melanogaster, silent-site diversity is both lower and more heterogeneous and that diversity is also more heterogeneous—consistent with the effect of selective sweeps. Similarly, Cai et al. (2009) found that silent-site diversity is lower in regions with higher divergence and functional density, and with lower recombination. On the other hand, Bullaughey et al. (2008) found no correlation between recombination and non-synonymous divergence in humans. Generally, interpreting correlations between rates of amino acid divergence and recombination is difficult: for example, hominids show a higher rate of divergence in gene expression and in 5′-noncoding sequences than murids, which has been interpreted as owing to accumulation of weakly deleterious substitutions as a result of a lower hominid effective population size (Keightley et al. 2005). By a similar argument, a higher rate of non-synonymous divergence could be seen as being due to Hill–Robertson interference, rather than as causing it, as assumed above: the direction of causation depends on the distribution of selection coefficients.
The correlation between neutral diversity and recombination that is seen in Drosophila does not directly show whether interference from linked loci is significant across the bulk of the genome, in regions of high recombination as well as low. However, it does demonstrate the existence of a source of random drift that could be the main process that shapes neutral variation, and that limits the effectiveness of selection across the whole genome. The key observation that even the most abundant species have only moderately high genetic variation (Lewontin 1974; Nevo et al. 1984; Lynch & Conery 2000) shows that random drift cannot be simply due to sampling, which would give a negligible rate inversely proportional to census numbers, ∼1/N. In their original analysis of hitch-hiking, Maynard Smith & Haigh (1974) argued that selective sweeps must necessarily be the dominant source of drift in any sufficiently large population; Gillespie (2000, 2001) has elaborated this view, that random drift is primarily due to fixation of favourable mutations. The observation of reduced silent-site diversity in regions of low recombination is consistent with this, and if it is explained by selective sweeps, then Maynard Smith & Haigh's (1974) argument implies that it must be the main source of drift in abundant species. However, there are two caveats. First, background selection reduces effective population size by a constant factor, independent of actual numbers, and so would have the same proportionate effect, however large the population. Second, diversity in abundant species may be limited by sporadic bottlenecks, rather than by selective sweeps. Thus, the two observations of a correlation between diversity and recombination, and of modest diversity in even abundant species could be explained either by a predominant effect of selective sweeps, or by a combination of background selection with population bottlenecks (figure 4)—or, of course, by some combination of these three processes.
Figure 4.

The relation between neutral diversity, recombination rate R, and population size (small, medium, large, reading upwards), for population (a) bottlenecks, (b) deleterious mutation and (c) selective sweeps. With bottlenecks (a), diversity is independent of recombination rate, but reaches an upper limit as census numbers increase. With ‘background selection’ (b), diversity increases with recombination but is strictly proportional to census numbers. With selective sweeps (c), diversity increases to an upper limit with both population size and recombination.
If selection on linked loci does reduce neutral diversity across the whole genome, then it must also interfere to some extent with selection, and hence must lead to some selection for sex and recombination. The problem is to find whether such selection is strong enough to outweigh the various costs. We now have direct estimates of the total rate of mutation (Haag-Liautard et al. 2007; Lynch et al. 2008; Keightley et al. 2009); of the distribution of their negative effects on fitness (Loewe & Charlesworth 2006); of the total rate of adaptive species-wide substitutions (Smith & Eyre-Walker 2002; Eyre-Walker & Keightley 2009); and (very roughly) of the strength of positive selection involved, inferred from the size of genomic regions of reduced diversity (Macpherson et al. 2007). These estimates are uncertain, largely because of the confounding effect of different kinds of selection, and of population structure. But, leaving these uncertainties aside, would knowledge of such global parameters be enough to tell us the strength of selection for recombination, through some simple relation such as equation (2.1), which applies to the effects of the linkage disequilibria built up deterministically by epistasis?
A consistent excess of divergence, relative to that expected from within-species polymorphism, indicates that around 30–50 per cent of amino acid substitutions in Drosophila were adaptive; estimates for humans are lower, but might also be substantial (Eyre-Walker & Keightley 2009). Moreover, a substantial fraction of divergence in non-coding regions may also have been adaptive (Halligan & Keightley 2006). The detection of selective sweeps via regions of reduced diversity is consistent with these estimates, and indicates that many substitutions are quite strongly selected—with s ∼ 1 per cent, say (Macpherson et al. 2007); the very fact that so many sweeps can be detected shows that they cause a substantial reduction in diversity (though, with the caveat that a complex demography may give false indications of selection). However, even taking the higher estimates, the overall rate of substitution is so slow that the density of sweeps per map length, Λ/R cannot be very high—certainly, far too low to cause significant selection for recombination via Hill–Robertson interference (Roze & Barton 2006). Estimates of genome-wide mutation rate are more encouraging: a direct estimate of the total rate of deleterious mutation over the diploid genome of D. melanogaster of U ∼ 1.2 (Haag-Liautard et al. 2007) would be enough to give substantial selection for recombination if epistasis is generally negative (Charlesworth 1990). However, while another direct estimate for yeast is also surprisingly high (Lynch et al. 2008), there it seems that the mutations involved are very weakly selected, and so may have little effect on recombination.
Neither deleterious mutations nor the fixation of favourable mutations through the whole species can contribute much heritable variance in fitness—specifically, not enough to cause much interference from unlinked loci. However, it is plausible that the heritable variance is high, as a result of fluctuating selection and local sweeps. If it is high enough for unlinked loci to cause significant interference, then linked loci may give a still larger contribution, though the theory here is undeveloped. The contrast between the ‘classical’ and the ‘balance’ views remains unresolved: it remains to be seen whether the mass of genomic data will tell us about locally fluctuating selection, or will remain limited to estimating global parameters.
(b). Limits to the genetic load
The neutral theory of molecular evolution was motivated by arguments that selection could not act on the whole genome: organisms could not have enough excess reproduction to eliminate deleterious mutations, to maintain balanced polymorphisms, and to fix adaptive substitutions, at an extremely large number of sites (Kimura 1968; King & Jukes 1969). Such arguments, framed in terms of various kinds of ‘genetic loads’, have been neglected since the 1970s, when it was shown that truncation selection on a sexual population allows selection to act much more efficiently (Sved et al. 1967; Sved 1968). Yet, we must still ask whether real organisms are likely to be selected in this way. What are the highest rates of deleterious mutation, and of adaptive substitution, that can be sustained by a freely recombining population?
If truncation selection acts on an underlying additive trait, determined by the sum of effects of deleterious mutations, then at equilibrium 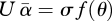 , where f(θ) is the mean of the θth fraction, measured in standard deviations, σ, and
, where f(θ) is the mean of the θth fraction, measured in standard deviations, σ, and  is the mean effect on log fitness of new mutations. Thus, an indefinitely high mutation rate can be sustained, provided the variance in vigour, σ2, is large enough. For example, if all mutations have the same effect (
is the mean effect on log fitness of new mutations. Thus, an indefinitely high mutation rate can be sustained, provided the variance in vigour, σ2, is large enough. For example, if all mutations have the same effect ( , say—the scale is arbitrary under truncation selection), then the variance in number of mutations is
, say—the scale is arbitrary under truncation selection), then the variance in number of mutations is 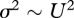 , or approximately 1000 if U ∼ 30 (taking f ∼ 1). With free recombination, numbers follow a Poisson distribution, with mean equal to the variance, and so each individual carries U2 ∼ 1000 deleterious mutations. The selection against each is
, or approximately 1000 if U ∼ 30 (taking f ∼ 1). With free recombination, numbers follow a Poisson distribution, with mean equal to the variance, and so each individual carries U2 ∼ 1000 deleterious mutations. The selection against each is  , or approximately 3 per cent—far larger than needed to overcome mutation or drift on a moderately large population. Selection on each mutation must be strong enough to resist degradation by drift and mutation pressure, and mutations with small effect may be fixed. Nevertheless, with truncation selection and free recombination, mutation load poses no serious limit in principle. However, in reality, selection coefficients may usually be much less than s ∼ 1 per cent, and so there may be a serious problem from the fixation of slightly deleterious mutations (Kondrashov 1995; Loewe 2006).
, or approximately 3 per cent—far larger than needed to overcome mutation or drift on a moderately large population. Selection on each mutation must be strong enough to resist degradation by drift and mutation pressure, and mutations with small effect may be fixed. Nevertheless, with truncation selection and free recombination, mutation load poses no serious limit in principle. However, in reality, selection coefficients may usually be much less than s ∼ 1 per cent, and so there may be a serious problem from the fixation of slightly deleterious mutations (Kondrashov 1995; Loewe 2006).
How might selection act on real organisms? There are two distinct issues: the relation between individual genotype and relative fitness (i.e. epistasis), and the effect of genotype on absolute fitness. Negative epistasis (in the extreme, truncation selection) reduces the mutation load by allowing much stronger marginal selection on each allele compared with what would be expected from the fitness of the optimal genotype (figure 1). In principle, we could measure the marginal effect of each of the deleterious alleles carried by an individual, and from this, predict the fitness of the ideal genotype by multiplying the marginal effects together. Theory predicts that at linkage equilibrium, this fitness must equal eU, relative to the mean fitness of one for a stable population. Plainly, the actual fitness of the optimal genotype is limited, implying that there must be negative epistasis if U is large (figure 1a; Kondrashov 1988). Is it plausible that marginal selection coefficients are in fact as large as is implied by this limit?
Negative epistasis could be owing to extensive redundancy, such that there must be many deleterious mutations before the organism degrades appreciably. This is suggested by carcinogenesis, which typically require multiple defects in the control of the cell cycle, and is the striking fact that a majority of genes in most eukaryotes can be deleted with little phenotypic effect. Yet, on this view, individuals must typically have already accumulated many defects, so that the marginal effect of an extra one is severe; then, a further increase in mutation rate would cause a disproportionate loss of fitness (figure 1b). Thus, to be plausible there must be some mechanism that would shift the fitness curve as the mutation rate changed, or that would limit the mutation rate itself. Under truncation selection, of course, there has to be some feedback such that a fixed fraction survives.
This brings us to the second issue of the relation between genotype and absolute fitness. Roughly, we can think of components of fitness that are required for individual survival and reproduction, regardless of the state of the rest of the population—development to adulthood, survival, fertility and so on. The total rate of mutation to genes involved in these components may be large only if the population evolves to have negative epistasis, such that each mutation has a large marginal effect. There may be other components of fitness that depend on competition between individuals, and do not alter the average number of offspring: for example, male secondary sexual traits, or female preferences for them. These could be under at least approximate truncation selection, and could sustain a high mutation rate. However, it seems likely that the sets of genes that affect the two kinds of trait would largely overlap, so that we cannot just add up the mutation rates for the two. If the effects of shared alleles on the two components of fitness are positively correlated, this will increase the strength of selection on them, and will reduce the load on the first component, that is under ‘hard’ selection (Agrawal 2001). (On the other hand, with negative correlations, so that sexual and natural selection are opposed, the first component of fitness may be depressed even with no mutation.) To raise yet further complications, female preferences may evolve for ‘good genes’ that are associated with increased male vigour. One way by which such preferences might evolve is through an epistatic handicap, in which only the most vigorous males can bear the cost of a signal trait. However, this leads to a positive correlation between fitness components that may increase mutation load.
We are left, then, with a theoretical upper limit under which truncation selection could allow extremely high mutation rates and (by similar arguments) rates of substitution. However, there seems to be no compelling reason why selection should evolve to act in such an efficient way, and so the traditional load arguments retain some force. Recent direct estimates of mutation rate and of the fraction of genome that is constrained by selection suggest that some species (including our own) may suffer a substantial mutation load, sufficient to cause significant selection for recombination (Charlesworth 1990). However, the rate of species-wide substitution in natural populations is too low to cause strong selection for recombination. Nevertheless, it remains possible that local populations experience far more directional selection, and that it is this which sustains widespread sex and recombination.
Acknowledgments
I would like to thank W. G. Hill and L. Loewe for organizing this special issue, and the Royal Society and Wolfson Foundation for their support. Also, A. Kondrashov and L. Loewe gave very helpful comments that helped improve the manuscript.
Footnotes
One contribution of 16 to a Theme Issue ‘The population genetics of mutations: good, bad and indifferent’ dedicated to Brian Charlesworth on his 65th birthday.
References
- Agrawal A. F.2001Sexual selection and the maintenance of sexual reproduction. Nature 411, 692–695 (doi:10.1038/35079590) [DOI] [PubMed] [Google Scholar]
- Agrawal A. F.2006Evolution of sex: why do organisms shuffle their genotypes? Curr. Biol. 16, 696–704 (doi:10.1016/j.cub.2006.07.063) [DOI] [PubMed] [Google Scholar]
- Aguade M., Miyashita N., Langley C. H.1989Reduced variation in the yellow-achaete-scute region in natural populations of Drosophila melanogaster. Genetics 122, 607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D., Charlesworth B.2002Reduced adaptation of a non-recombining neo-Y chromosome. Nature 416, 323–326 (doi:10.1038/416323a) [DOI] [PubMed] [Google Scholar]
- Barton N. H.1994The reduction in fixation probability caused by substitutions at linked loci. Genet. Res. 64, 199–208 (doi:10.1017/S0016672300032857) [Google Scholar]
- Barton N. H.1995aA general model for the evolution of recombination. Genet. Res. 65, 123–144 (doi:10.1017/S0016672300033140) [DOI] [PubMed] [Google Scholar]
- Barton N. H.1995bLinkage and the limits to natural selection. Genetics 140, 821–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. H. Why sex and recombination? Cold Spring Harbor Symp. Quant. Biol. 2009;74 doi: 10.1101/sqb.2009.74.030. ( doi:10.1101/sqb.2009.74.030) [DOI] [PubMed] [Google Scholar]
- Barton N. H., Charlesworth B.1998Why sex and recombination? Science 281, 1986–1990 (doi:10.1126/science.281.5385.1986) [PubMed] [Google Scholar]
- Barton N. H., Etheridge A. M.2004The effect of selection on genealogies. Genetics 166, 1115–1131 (doi:10.1534/genetics.166.2.1115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. H., Otto S. P.2005Evolution of recombination due to random drift. Genetics 169, 2353–2370 (doi:10.1534/genetics.104.032821) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun D. J., Aquadro C. F.1992Levels of naturally occurring DNA polymorphism correlate with recombination rate in Drosophila melanogaster. Nature 356, 519–520 (doi:10.1038/356519a0) [DOI] [PubMed] [Google Scholar]
- Bernstein H., Hopf F. A., Michod R. E.1988Is meiotic recombination an adaptation for repairing DNA, producing genetic variation, or both? In The evolution of sex (eds Michod R. E., Levin B. R.), pp. 139–160 Sunderland, MA: Sinauer Press [Google Scholar]
- Betancourt A. J., Presgraves D. C.2002Linkage limits the power of natural selection in Drosophila. Proc. Natl Acad. Sci. USA 99, 13 616–13 620 (doi:10.1073/pnas.212277199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt A. J., Welch J. J., Charlesworth B.2009Reduced effectiveness of selection caused by a lack of recombination. Curr. Biol. 19, 655–660 (doi:10.1016/j.cub.2009.02.039) [DOI] [PubMed] [Google Scholar]
- Bullaughey K., Przeworski M., Coop G.2008No effect of recombination on the efficacy of natural selection in primates. Genome Res. 18, 544–554 (doi:10.1101/gr.071548.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. G.1980The mathematical theory of quantitative genetics New York, NY: Oxford University Press [Google Scholar]
- Burger R.1999Evolution of genetic variability and the advantage of sex and recombination in changing environments. Genetics 153, 1055–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt A.2000Sex, recombination and the efficacy of selection—was Weissman right? Evolution 54, 337–351 [DOI] [PubMed] [Google Scholar]
- Cai J. J., Macpherson J. M., Sella G., Petrov D. A.2009Pervasive hitchhiking at coding and regulatory sites in humans. PLoS Genet. 5, e1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B.1990Mutation-selection balance and the evolutionary advantage of sex and recombination. Genet. Res. 55, 199–221 (doi:10.1017/S0016672300025532) [DOI] [PubMed] [Google Scholar]
- Charlesworth B.1993aDirectional selection and the evolution of sex and recombination. Genet. Res. 61, 205–224 (doi:10.1017/S0016672300031372) [DOI] [PubMed] [Google Scholar]
- Charlesworth B.1993bThe evolution of sex and recombination in a varying environment. J. Hered. 84, 345–350 [DOI] [PubMed] [Google Scholar]
- Charlesworth B., Barton N. H.1996Recombination load associated with selection for increased recombination. Genet. Res. 67, 27–41 (doi:10.1017/S0016672300033450) [DOI] [PubMed] [Google Scholar]
- Charlesworth B., Betancourt A. J., Kaiser V. B., Gordo I.2009Genetic recombination and molecular evolution. Cold Spring Harbor Symp. Quant. Biol. 74 (doi:10.1101/sqb.2009.74.015) [DOI] [PubMed] [Google Scholar]
- Charlesworth D.2003Effects of inbreeding on the genetic diversity of populations. Phil. Trans. R. Soc. Lond. B 358, 1050–1070 (doi:10.1098/rstb.2003.1296) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D., Charlesworth B., Morgan M. T.1995The pattern of neutral molecular variation under the background selection model. Genetics 141, 1619–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. F., Kimura M.1979Efficiency of truncation selection. Proc. Natl Acad. Sci. USA 76, 396–399 (doi:10.1073/pnas.76.1.396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A., Keightley P. D.2009Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol. Biol. Evol. 26, 2097–2108 (doi:10.1093/molbev/msp119) [DOI] [PubMed] [Google Scholar]
- Feldman M. W., Krakauer J.1976Genetic modification and modifier polymorphisms. In Population genetics and ecology (eds Karlin S., Nevo E.). New York, NY: Academic Press [Google Scholar]
- Feldman M. W., Liberman U.1986An evolutionary reduction principle for genetic modifiers. Proc. Natl Acad. Sci. USA 83, 4824–4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M. W., Christiansen F. B., Brooks L. D.1980Evolution of recombination in a constant environment. Proc. Natl Acad. Sci. USA 77, 4838–4841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M. W., Otto S. P., Christiansen F. B.1996Population genetic perspectives on the evolution of recombination. Ann. Rev. Genet. 30, 261–295 [DOI] [PubMed] [Google Scholar]
- Felsenstein J.1974The evolutionary advantage of recombination. Genetics 78, 737–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. A.1930The genetical theory of natural selection Oxford, UK: Oxford University Press [Google Scholar]
- Funk D. J., Wernegreen J. J., Moran N. A.2001Intraspecific variation in symbiont genomes: bottlenecks and the aphid—Buchnera association. Genetics 157, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. H.2000Genetic drift in an infinite population: the pseudo hitchhiking model. Genetics 155, 909–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. H.2001Is the population size of a species relevant to its evolution? Evolution 55, 2161–2169 [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C., Dorris M., Maside X., Macaskill S., Halligan D. L., Houle D., Charlesworth B., Keightley P. D.2007Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445, 82–85 (doi:10.1038/nature05388) [DOI] [PubMed] [Google Scholar]
- Haldane J. B. S.1930A mathematical theory of natural and artificial selection. VII. Selection intensity as a function of mortality rate. Proc. Camb. Phil. Soc. 27, 131–136 (doi:10.1017/S0305004100009427) [Google Scholar]
- Haldane J. B. S.1957The cost of natural selection. J. Genet. 55, 511–524 (doi:10.1007/BF02984069) [Google Scholar]
- Halligan D., Keightley P. D.2006Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison. Genome Res. 16, 875–884 (doi:10.1101/gr.5022906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton W. D.1996Narrow roads of gene land II Evolution of sex and sexual selection Oxford, UK: W. H. Freeman [Google Scholar]
- Hansen T. F.2006The evolution of genetic architecture. Annu. Rev. Ecol. Evol. Syst. 37, 123–157 (doi:10.1146/annurev.ecolsys.37.091305.110224) [Google Scholar]
- Hellmann I., Ebersberger I., Ptak S. E., Pääbo S., Przeworski M.2005A neutral explanation of the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 72, 1527–1535 (doi:10.1086/375657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W. G., Robertson A.1966The effect of linkage on limits to artificial selection. Genet. Res 8, 269–294 (doi:10.1017/S0016672300010156) [PubMed] [Google Scholar]
- Hudson R. B., Kaplan N. L.1988The coalescent process in models with selection and recombination. Genetics 120, 831–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R. R., Kaplan N. L.1995Deleterious background selection with recombination. Genetics 141, 1605–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles M. M., Walters J. R., Cannings C.2003Selection for recombination in large genomes. Genetics 165, 2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T., Barton N. H.2002The effect of deleterious alleles on adaptation in asexual populations. Genetics 162, 395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser V. B.2009Molecular evolution under low recombination. PhD thesis, University of Edinburgh, Edinburgh [DOI] [PubMed] [Google Scholar]
- Kaiser V., Charlesworth B.2008The effects of deleterious mutations on evolution in non-recombining genomes. Trends Genet. 25, 9–12 (doi:10.1016/j.tig.2008.10.009) [DOI] [PubMed] [Google Scholar]
- Kaplan N. L., Darden T., Hudson R. B.1988The coalescent process in models with selection. Genetics 120, 819–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Kacser H.1987Dominance, pleiotropy and metabolic structure. Genetics 117, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Otto S. P.2006Interference among deleterious mutations favours sex and recombination in finite populations. Nature 443, 89–92 (doi:10.1038/nature05049) [DOI] [PubMed] [Google Scholar]
- Keightley P. D., Lercher M. J., Eyre-Walker A.2005Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biology 3, e42 (doi:10.1371/journal.pbio.0030042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. D., Trivedi U., Thomson M., Oliver F., Kumar S., Blaxter M. L.2009Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 19, 1195–1201 (doi:10.1101/gr.091231.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M.1968Evolutionary rate at the molecular level. Nature 217, 624–626 (doi:10.1038/217624a0) [DOI] [PubMed] [Google Scholar]
- Kimura M., Maruyama T.1966Mutational load with epistatic gene interactions in fitness. Genetics 54, 1337–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. L., Jukes T. H.1969Non-Darwinian evolution. Science 164, 788–798 (doi:10.1126/science.164.3881.788) [DOI] [PubMed] [Google Scholar]
- Kliman R. M., Hey J.1993Reduced natural selection associated with low recombination in Drosophila melanogaster. Mol. Biol. Evol 10, 1239–1258 [DOI] [PubMed] [Google Scholar]
- Kondrashov A. S.1984Deleterious mutations as an evolutionary factor. I. The advantage of recombination. Genet. Res. 44, 199–218 (doi:10.1017/S0016672300026392) [DOI] [PubMed] [Google Scholar]
- Kondrashov A. S.1988Deleterious mutations and the evolution of sexual reproduction. Nature 336, 435–441 (doi:10.1038/336435a0) [DOI] [PubMed] [Google Scholar]
- Kondrashov A. S.1993Classification of hypotheses on the advantage of amphimixis. J. Hered. 84, 372–387 [DOI] [PubMed] [Google Scholar]
- Kondrashov A. S.1995Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over?. J. Theor. Biol. 175, 583–594 (doi:10.1006/jtbi.1995.0167) [DOI] [PubMed] [Google Scholar]
- Lenormand T., Otto S. P.2000The evolution of recombination in a heterogeneous environment. Genetics 156, 423–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin R. C.1974The genetic basis of evolutionary change New York, NY: Columbia University Press [Google Scholar]
- Loewe L.2006Quantifying the genomic decay paradox due to Muller's ratchet in human mitochondrial DNA. Genet. Res. 87, 133–159 (doi:10.1017/S0016672306008123) [DOI] [PubMed] [Google Scholar]
- Loewe L., Charlesworth B.2006Inferring the distribution of mutational effects on fitness in Drosophila. Biol. Lett. 2, 426–430 (doi:10.1098/rsbl.2006.0481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L., Charlesworth B.2007Background selection in single genes may explain patterns of codon bias. Genetics 175, 1381–1393 (doi:10.1534/genetics.106.065557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M., Conery J. S.2000The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (doi:10.1126/science.290.5494.1151) [DOI] [PubMed] [Google Scholar]
- Lynch M., et al. 2008A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl Acad. Sci. USA 105, 9272–9277 (doi:10.1073/pnas.0803466105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson J. M., Sella G., Davis J. C., Petrov D. A.2007Genome-wide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177, 2083–2099 (doi:10.1534/genetics.107.080226) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G., Otto S. P., Lenormand T.2006Selection for recombination in structured populations. Genetics 172, 593–609 (doi:10.1534/genetics.104.039982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J., Haigh J.1974The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23–35 (doi:10.1017/S0016672300014634) [PubMed] [Google Scholar]
- McVean G. A. T., Charlesworth B.2000The effects of Hill–Robertson interference between weakly selected sites on patterns of molecular evolution and variation. Genetics 155, 929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merila J., Sheldon B.2000Lifetime reproductive success and heritability in nature. Am. Nat. 155, 301–310 (doi:10.1086/303330) [DOI] [PubMed] [Google Scholar]
- Muller H. J.1932Some genetic aspects of sex. Am. Nat. 66, 118–138 (doi:10.1086/280418) [Google Scholar]
- Nachman M. W.2002Variation in recombination rate across the genome: evidence and implications. Curr. Opin. Genet. Dev. 12, 657–663 (doi:10.1016/S0959-437X(02)00358-1) [DOI] [PubMed] [Google Scholar]
- Nevo E., Beiles A., Ben-Schlomo R.1984The evolutionary significance of genetic diversity: ecological, demographic and life history correlates. In Measuring selection in natural populations (ed. Mani G. S.), pp. 13–213 Berlin, Germany: Springer [Google Scholar]
- Nordborg M., Charlesworth B., Charlesworth D.1996The effect of recombination on background selection. Genet. Res. 67, 159–174 (doi:10.1017/S0016672300033619) [DOI] [PubMed] [Google Scholar]
- Otto S. P., Feldman M. W.1997Deleterious mutations, variable epistatic interactions, and the evolution of recombination. Theor. Popul. Biol. 51, 134–147 (doi:10.1006/tpbi.1997.1301) [DOI] [PubMed] [Google Scholar]
- Otto S., Lenormand T.2002Resolving the paradox of sex and recombination. Nat. Rev. Genet. 3, 252–261 (doi:10.1038/nrg761) [DOI] [PubMed] [Google Scholar]
- Peck J. R.1994A ruby in the rubbish: beneficial mutations and the evolution of sex. Genetics 137, 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves D. C.2005Recombination enhances protein adaptation in Drosophila melanogaster. Curr. Biol. 15, 1651–1656 (doi:10.1016/j.cub.2005.07.065) [DOI] [PubMed] [Google Scholar]
- Provine W.1971The origins of theoretical population genetics Chicago, IL: University of Chicago Press [Google Scholar]
- Robertson A.1961Inbreeding in artificial selection programmes. Genet. Res. 2, 189–194 (doi:10.1017/S0016672300000690) [DOI] [PubMed] [Google Scholar]
- Rouzine I. M., Brunet E., Wilke C. O.2008The traveling-wave approach to asexual evolution: Muller's ratchet and speed of adaptation. Theor. Popul. Biol. 73, 24–46 (doi:10.1016/j.tpb.2007.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze D., Barton N. H.2006The Hill–Robertson effect and the evolution of recombination. Genetics 173, 1793–1811 (doi:10.1534/genetics.106.058586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. G. C., Eyre-Walker A.2002Adaptive protein evolution in Drosophila. Nature 415, 1022–1024 (doi:10.1038/4151022a) [DOI] [PubMed] [Google Scholar]
- Spencer C. C. A., Deloukas P., Hunt S., Mullikin J. C., Myers S., Silverman B., Donnelly P., Bentley D., McVean G.2006The influence of recombination on human genetic diversity. PLoS Genet. 2, e148 (doi:10.1371/journal.pgen.0020148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W., Langley C. H.1989Molecular genetic variation in the centromeric region of the X chromosome in three Drosophila ananassae populations I Contrasts between the vermilion and forked loci. Genetics 121, 89–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W., Wiehe T. H., Lenz M.1992The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor. Popul. Biol. 41, 237–254 (doi:10.1016/0040-5809(92)90045-U) [Google Scholar]
- Sved J. A.1968Possible rates of gene substitution in evolution. Am. Nat. 102, 283–293 (doi:10.1086/282542) [Google Scholar]
- Sved J. A., Reed T. E., Bodmer W. F.1967The number of balanced polymorphisms that can be maintained by natural selection. Genetics 55, 469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szathmary E.1993Do deletion mutations act synergistically? Metabolic control theory provides a partial answer. Genetics 133, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J.2008Coalescent theory: an introduction Englewood, CO: Roberts and Company [Google Scholar]
- Waxman D., Peck J. R.1999Sex and adaptation in a changing environment. Genetics 153, 1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismann A.1889The significance of sexual reproduction in the theory of natural selection. In Essays upon heredity and kindred biological problems (eds Poulton E. B., Schönland S., Shipley A. E.). Oxford, UK: Clarendon Press [Google Scholar]
- Williams G. C.1975Sex and evolution Princeton, NJ: Princeton University Press [Google Scholar]