

Abstract
Corpora amylacea (CA) have long been described in aging brains and in patients with neurodegenerative conditions, but their origins have been debated. It has been proposed that CA represent collections of nervous system breakdown products that accumulate within astrocytic cytoplasm. In support of this, studies have shown that CA include glycosylated material, ubiquitin, and an assortment of proteins derived from neuronal cytoplasm. On the other hand, many of these proteins are not specifically localized to neurons or astrocytes; some components of CA, such as complement proteins, are most abundantly expressed outside the central nervous system. The characteristic predilection for CA to accumulate near vessels and ependyma suggests that proteins extravasated from blood or transudated from CSF may form a component of these structures. In this study, we report the immunohistochemical localization of blood and platelet proteins thrombospondin1 and ADAMTS13 in CA from aged individuals and patients with vascular dementia. Thrombospondin1 localized to neurons, but was most prominently localized to CA. An independent serum and platelet expressed protein, ADAMTS13, was found in CA in the same brain regions. In vitro analysis shows that thrombospondin1 and ADAMTS13 form complexes together in cells and in direct protein binding assays. We speculate that CA could result from a conglomeration of interacting proteins from degenerating neurons and from extravasated blood elements released after transient breakdown of the blood–brain barrier.
Keywords: ADAMTS13, corpora amylacea, thrombospondin, vascular dementia, vascular proteins
INTRODUCTION
Corpora amylacea (CA) were described by Purkinje in 1837 in the brains of elderly patients. CA are classically recognized in normal aging brains, but can also be seen in a variety of neurological conditions, including multiple sclerosis, hippocampal sclerosis and medically refractory epilepsy.
CA are typically symmetrical spheroid bodies 10–50 um in diameter. The bodies are avidly PAS-positive and react strongly with iodide. These characteristics are consistent with biochemical studies of CA which indicate that they are rich in sugars,1 possibly conjugated into glycoproteins. The protein content of CA was estimated to be 4%,2 and a number of investigators have discovered specific protein components within CA, including ubiquitin,2,3 complement,2,4 NeuN,5 tau,3,6 the extracellular domain of amyloid precursor protein, Hsp proteins,2,7-9 S100,10 myelin basic protein, proteolipid protein, ferritin,11 nestin,12 Bcl-2, Bax, c-Jun,13 advanced glycosylation end products,14 proteoglycans,15 and anion exchange proteins.16 A recent proteomic analysis of CA from patients with multiple sclerosis has identified a large number of cytosolic proteins which are normally expressed in neurons.17 These studies suggest that CA may represent the accumulation of neuronal degenerative products that have been endocytosed and partitioned by astrocytes. CA have also been shown to react with digoxygenin labeled oligonucleotide probes in a sequence-preferential manner.18 CA are typically located in the subpial and subependymal regions of the brain which are rich in blood vessels and astrocytic processes. In addition, there is a perivascular predilection for CA accumulation.19 The reasons for the apparent proximity to vessels and CSF are not known.
In this study, we provide immunohistochemical evidence that two proteins expressed abundantly in the vascular system, thrombospondin1 and ADAMTS13, are components of CA in brains from normal subjects and patients with cerebrovascular disease. We confirmed the frequent occurrence of CA near vascular structures. In light of these new findings and prior reports, we speculate that proteins derived from the blood and platelets may contribute to CA formation.
MATERIALS AND METHODS
Immunohistochemistry
Patient samples were obtained at autopsy at the University of Michigan Health System. Normal patient samples were obtained from the Alzheimer’s Disease Research Core at the University of Michigan; patients did not have known neurological diseases (average age of 62 years); eight of nine patients were male. Clinically diagnosed vascular dementia samples were obtained from males (age 80) and females (ages 64, 68 and 99). We analyzed frontal cortex with subcortical white matter from all normal and vascular dementia patients. In selected patients, we also examined sections from the hippocampus and choroid plexus. All tissues were fixed in 10% formaldehyde, embedded in paraffin, and cut into 5-micron thick serial sections. Sections were analyzed by immunohistochemical staining using standard techniques. Briefly, after deparaffinization and rehydration, antigen retrieval was performed using either microwave-assisted citrate or Tris buffer incubation; immunoperoxidase staining was conducted using a DAKO (Carpentaria, CA US) autostaining system. All sections were counterstained with hematoxylin. Digital images were captured using an Olympus system. Monoclonal antibody A6.1 against thrombospondin1 (Thermo) was used at 1:50 dilution. For some samples, anti-thrombospondin1 monoclonal SPM321 (Santa Cruz Laboratories, Santa Cruz, CA, US) was used at 1:50 dilution. ADAMTS13 mouse monoclonal (a kind gift from Dr. David Ginsburg) was used at 1:100. Two control monoclonal antibodies were used to ensure that CA staining was specific: 8G1 (anti-LRP1; Santa Cruz) and TRA1-85 (anti-CD147; Developmental Studies Hybridoma Bank at the University of Iowa) at 1:50 dilutions. Serial sections from the same tissue blocks were used to confirm CA by PAS staining.
Protein interaction studies
Immunoprecipitations were performed as described by Meng et al.20 Briefly, 293A cells were transfected with Flag-tagged ADAMTS13 (Accession AF414401;21) and/or thrombospondin1 (Accession NM_011580; Addgene plasmid 12405;22) cloned into CMV promoter-based expression vectors. Singly transfected cells served as negative controls. Lipofectamine 2000-mediated transfections were performed in 6-well plates using a total of 2 μg of DNA and 4 μL of lipid reagent per well. Cell lysates were prepared 24 h after transfection and then incubated with Flag antibody conjugated agarose beads (Sigma A1205) to pull down ADAMTS13. After washing, proteins from beads were boiled in sample buffer and analyzed by Western blotting. Primary antibodies to Flag (Sigma; F1804) and to thrombospondin1 (Thermo; D4.6) were applied at 1:1000 dilution.
In vitro binding studies
Recombinant ADAMTS13 (R&D Systems) was adsorbed to plastic wells overnight at 5.2 ug/mL in TBS with 2 mmol calcium chloride. We labeled purified thrombospondin1 (R&D Systems, Minneapolis, MN, US) using Alexa700 succinimide as described by Meng et al.20 Free label was quenched with Tris buffer and removed by gel filtration prior to incubation with ADAMTS13 coated plates at a series of concentrations in duplicate. After an overnight incubation, wells were washed three times with TBS with calcium. Bound protein was visualized and quantified on a flatbed scanner (Licor, Odyssey, Lincoln, NE, US). Labeled recombinant IgG Fc protein (R&D Systems) incubated with ADAMTS13 bound to plates was used in one set of negative control reactions. In a second set of negative controls, labeled thrombospondin1 was incubated with bovine serum albumin (BSA) adsorbed plates. Fc protein exhibited negligible binding, which was subtracted from values of bound thrombospondin1; labeled thrombospondin1 did not bind to BSA.
RESULTS
Thrombospondin1 expression in normal brain
The initial purpose of our study was to investigate the distribution of thrombospondin1 in normal and vascular dementia brain tissue. In normal control individuals, we observed light neuronal and occasionally astrocytic staining using a monoclonal antibody against thrombospondin1, A6.1. There was sporadic, moderate staining of blood vessels. In three of nine patients, there was strong spherical staining that appeared in the subpial cortex and periventricular regions. PAS staining of adjacent sections identified these structures as CA, which were found significantly around blood vessels. In most cases the peripheral rim (halo) of the CA stained well but the staining of the core was missing. In a minority of CA, both the periphery and the core stained well. This was most likely due to artefactual loss of core material during immunostaining. Representative photos of CA staining in subependymal, subpial, and perivascular regions of the frontal lobe of normal subjects is shown in Figure 1. In six of nine patients, we saw neither PAS-positive CA, nor spherical staining with thrombospondin1 antibodies.
Fig. 1.
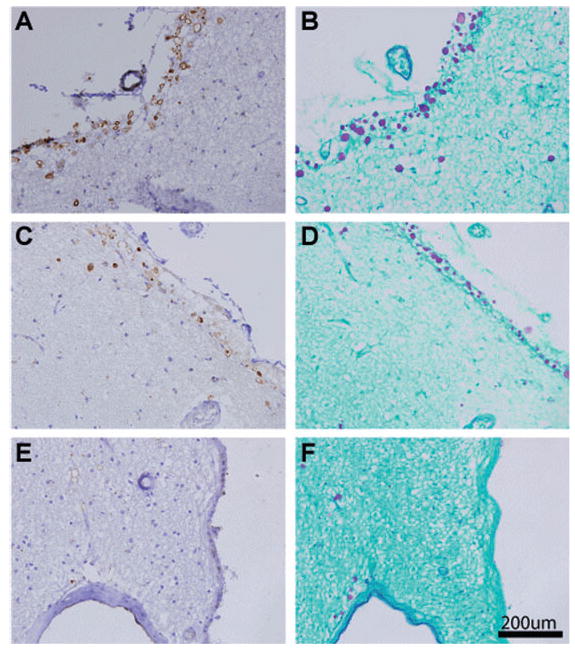
Thrombospondin1 localizes to corpora amylacea (CA) in normal aged human brain. Frontal lobe sections were stained with thrombospondin1 specific antibody A6.1 (A, C, E) or PAS (B, D, F). Abundant staining of spherical CA was observed in the subpial cortex (A–D) and around blood vessels abutting the ependyma (E, F). Scale bar represents 200 um.
Thrombospondin1 staining in vascular dementia cases
In all four cases of vascular dementia, we observed neuronal staining similar to normal controls.All four cases had abundant spherical thrombospondin1 staining in cortex and periventricular regions that was more prominent than in normal cases and corresponded to CA seen on PAS staining (Fig. 2). As in normal controls, thrombospondin1 staining of CA had a halo appearance in some regions, but a solid stain in other regions. In addition, in two cases, we observed amyloid plaques that were strongly reactive with thrombospondin1 antibodies (not shown). As in normal cases, vessels did not react strongly or uniformly with thrombospondin1 antibodies; however, halos of thrombospondin1 immunoreactivity were common in the substance around blood vessels. Overall, thrombospondin1 staining in CA was found in both normal and vascular dementia cases, but the stained CA were more frequent and more prominent in the latter.
Fig. 2.
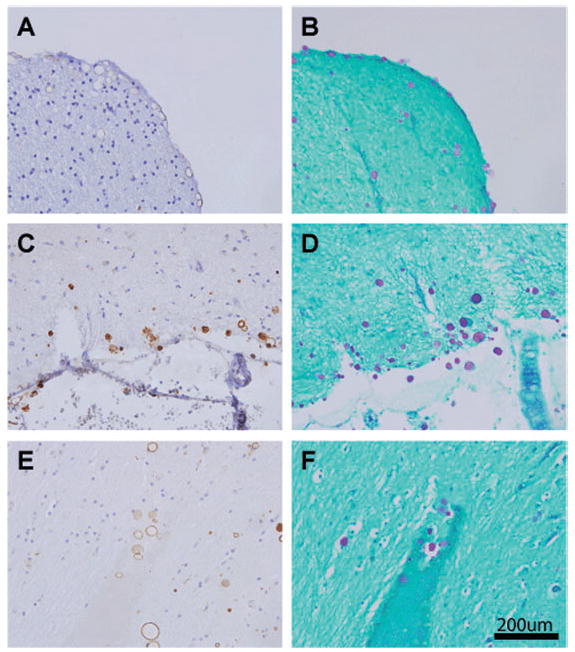
Thrombospondin1 localizes heavily to corpora amylacea (CA) in multi-infarct dementia. Sections were stained with A6.1 (A, C, E) or PAS (B, D, F), as in Fig. 1. A similar distribution of staining was noted, but in general, more CA were observed in multi-infact dementia cases. CA staining was heterogeneous, with some labeling evenly through the CA and some demonstrating a halo of reactivity. Strong staining for thrombospondin1 was observed in subpial cortex (A–D) and in perivascular spaces (E, F).
ADAMTS13 staining of CA in normal and vascular dementia patients
Thrombospondin1 is most abundantly expressed in the blood and platelets. Thus, it is possible that proteins from the blood may contribute to CA. To further support the potential platelet/blood origin of CA, we examined brains for presence of other platelet/blood expressed proteins. ADAMTS13 is a protein involved in the cleavage of high molecular weight von Willebrand factor (vWF) multimers; regulation of vWF size has been suggested to be important in the prevention of thrombosis. ADAMTS13 is synthesized in the liver and by platelets, and relatively high concentrations (0.5–1.0 ug/mL) circulate within the blood,23 where ADAMTS13 regulates physiological vWF size.24 We used a monoclonal antibody against ADAMTS13 to examine whether this circulating protein could be found within CA of the brain. In all cases of both normal aged brain and vascular dementia examined, we found strong localization of ADAMTS13 to regions where we previously found strong PAS-positive CA and halo- or spherical-shaped thrombospondin1 deposits (Fig. 3). ADAMTS13 protein was found in a halo distribution, as was noted for thrombospondin1. We did not see any clear differences in the ADAMTS13 staining pattern of CA compared to thrombospondin1, although it appeared that ADAMTS13 may not be present in a minor subset of CA. As in the case of thrombospondin1, we found increased incidence of ADAMTS13 staining in CA of vascular dementia patients, which paralleled the higher amount of CA observed in this group.
Fig. 3.
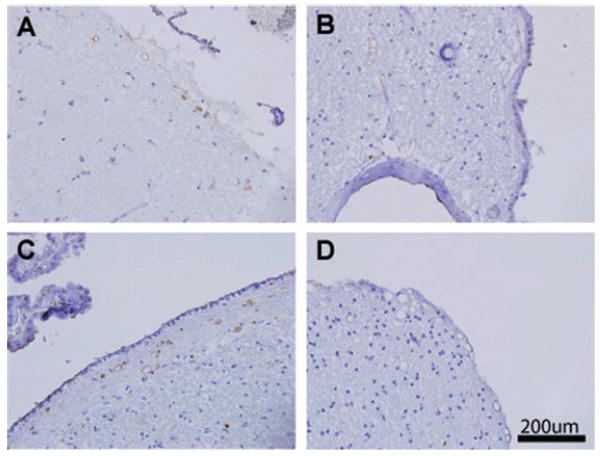
ADAMTS13 localizes to corpora amylacea (CA) in normal brain and multi-infarct dementia. Sections from normal (A, B) and vascular dementia (C, D) brains were stained for ADAMTS13, which showed a similar distribution of staining as thrombospondin1. Halo-shaped staining occurred in the cortex (A, D) and perivascular regions abutting the ependyma (B). Subependymal CA were strongly labeled as well (C).
Negative controls
To confirm the specificity of our studies, sections from normal and vascular dementia cases containing CA were also stained with monoclonal antibodies against LRP1 and CD147 (not shown). CA was not labeled by these antibodies; however, LRP1 was found in neurons, glia, and vascular smooth muscle, as has been observed in prior studies;25 in addition, CD147 was seen in neurons and microvessels, as has been described,26,27 but not in CA.
Molecular interactions between thrombospondin1 and ADAMTS13
Protein aggregates in the brain result from a variety of molecular processes, most prominently abnormal protein-protein interactions. It is likely that CA result from cumulative precipitation of molecules initially nucleated within a core; molecules that accumulate in CA would be predicted to directly interact in biochemical assays. Since both thrombospondin1 and ADAMTS13 are localized to CA, we tested whether the two proteins could form molecular complexes. First, we examined whether the two proteins could form complexes in cells expressing recombinant proteins. Expression constructs encoding thrombospondin1 or ADAMTS13 were transfected either singly or in combination and pulled down with monoclonal antibodies directed toward recombinant, epitope-tagged ADAMTS13 (Fig. 4A). ADAMTS13 was precipitated with monoclonal antibodies and immunoprecipitates were then analyzed by immunoblotting. Control vector transfected 293 cells did not express either of the proteins. In cells cotransfected with both ADAMTS13 and thrombospondin1, we detected both proteins in precipitates. However, when thrombospondin1 alone was transfected, no thrombospondin1 could be detected in precipitates. The presence of thrombospondin1 in ADAMTS13-containing immunoprecipitates indicated that coexpression of the two proteins in cells results in the formation of stable protein complexes.
Fig. 4.
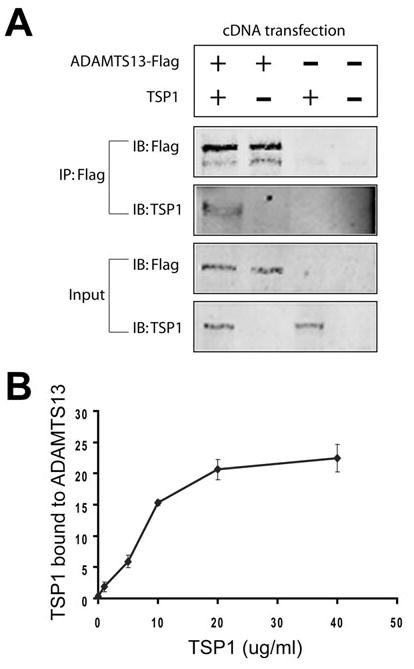
Molecular interaction between thrombospondin1 (TSP1) and ADAMTS13. (A) To test for interactions between thrombospondin1 and ADAMTS13 in cells, we transfected 293A cells with cDNAs for thrombospondin1 and ADAMTS13, either singly or in combination.We immunoprecipitated ADAMTS13 using the Flag antibody from cell lysates and probed precipitated proteins by immunoblotting. Thrombospondin1 protein coprecipitated with ADAMTS13 only in cotransfected cells. (B) To test for interaction in vitro, we adsorbed ADAMTS13 or bovine serum albumin (BSA) to plastic dishes. Labeled thrombospondin1 was applied to dishes at increasing doses and then detected using a scanner. Dose-dependent binding was observed. Thrombospondin1 does not appreciably bind to BSA. These experiments were replicated twice.
Second, we tested whether the two proteins could interact directly, in the absence of other cellular proteins (Fig. 4B). Purified proteins were testing in in vitro binding reactions to quantify interactions. We found that labeled thrombospondin1 was capable of directly binding to surface-immobilized ADAMTS13 specifically and with high affinity. In sum, two independent biochemical methods demonstrate that thrombospondin1 and ADAMTS13 are capable of forming specific protein-protein interactions, which may play a role in the formation of CA in human brains.
DISCUSSION
Although not specific for a disease process, CA are common in brains of aged individuals and patients with neurodegenerative conditions. The biochemical components of CA may reveal their origins and the mechanisms by which they are formed. In this report, we made two novel findings. First, we identified robust immunolocalization of thrombospondin1 and ADAMTS13 to CA in normal brains and in vascular dementia patients. Second, we demonstrated that thrombospondin1 and ADAMTS13 form protein complexes together, providing a possible mechanistic clue to their colocalization in CA.
Our original purpose was to investigate whether thrombospondin1 is differentially expressed in vessels in vascular dementia.We found thrombospondin1 reactivity in neurons and astrocytes in both normal and vascular dementia patients. In some patients, the protein was also localized to senile plaques. These findings match a previous report of thrombospondin1 immunostaining performed to compare normal control brains and Alzheimer’s disease patients.28 As in the previous work, we also observed reactivity within the microvasculature, though our vascular staining was not prominent.The discrepancy may be attributed to the significant difference in the thickness of the sections used in our study and the prior work: 5 microns in our study versus 40 microns in the work of Buee et al.28 In any case, we were unable to conclude that vascular dementia is associated with significant primary changes in vascular thrombospondin1. Rather, we have made a novel observation that thrombospondin1 is a prominent component of brain CA in both normal and vascular dementia patients.
Thrombospondin1 plays multiple biological roles through protean mechanisms. As a secreted signaling protein, thrombospondins bind to a large number of transmembrane receptors such as integrins, LRP, CD36, and CD47 triggering a multitude of cell-specific effects.29-32 In addition, thrombospondins serve as extracellular adapter proteins that bind to protein complexes and facilitate their clearance.33 As such, thrombospondin1 is the first example of a signaling molecule found in CA and the first regulator of the extracellular scavenging system found in CA by immunostaining. It is quite possible that the uptake of thrombospondin1 into CA is a consequence of its function as a target for endocytosis, given the appearance of multiple other proteins in CA which seem destined for catabolism.
From where is thrombospondin1 originating? The most abundant source of thrombospondin1 is platelets; thrombospondin1 makes up 3% of the total platelet protein and up to 25% of thrombin-activated platelet secretory protein. Other cells in the brain which stain for thrombospondin1 include neurons and glial cells,28 which contain much less thrombospondin1 than platelets. It is not clear whether these cells make thrombospondin1 in situ or internalize it from sources such as platelets and serum, but the latter is theoretically possible since these cells express receptors for thrombospondin uptake.25,34,35 Similarly, CA thrombospondin1 could result from endocytosis of platelet proteins that have crossed into the brain. Platelet proteins have never been implicated in CA pathogenesis, but longstanding observations suggest that this could be the case. It is intriguing to note that CA are most commonly seen in regions of the brain that are in close proximity to high concentrations of vessels. Thus, extravasated plasma protein or platelet components which leach out of vessels over time could contribute to CA. This would provide an explanation for why CA are localized to subpial and subependymal regions more commonly than deeper layers of the cortex, which are formed predominantly by neurons and astrocytes. It is also noteworthy that CA were found in patients with vascular dementia, a disorder which is predicted to compromise the blood-brain barrier. Other conditions that lead to CA formation, including aging, Alzheimer’s disease, multiple sclerosis, and temporal lobe sclerosis, are associated with chronic vascular compromise. If indeed platelets are the source of thrombospondin1 in CA, we predict that vascular injury and blood-brain barrier dysfunction would be strong risk factors for CA formation. In further support for this notion, complement proteins, which are largely serum-derived, have been consistently reported in CA.2,4
In additional support of a potential contribution of platelet/blood proteins to CA, we show that another platelet/blood protein, ADAMTS13, is found in CA. ADAMTS13 is found in high abundance in the serum,23 and also is expressed in the liver, platelets, and endothelial cells. In aggregate, our new observations that CA include multiple proteins found in high abundance in the blood support the possibility that CA are in part composed of blood protein products.
Finally, we show that thrombospondin1 and ADAMTS13 form protein complexes using two complementary experimental approaches. It is conceivable that the two proteins circulate together, and during breaches of the blood-brain barrier, transit into the brain parenchyma where they are endocytosed and eventually deposited into CA. It is also possible that each protein reaches the brain independently, and that protein-protein interactions cause the two proteins to aggregate within the cell together and participate in the formation of CA in situ. Further experiments will be required to differentiate these possibilities, but these experiments provide evidence that CA proteins could result from protein-protein interactions, molecular forces that form the basis for many pathological markers of neurodegenerative conditions (e.g. senile plaques, tangles, Lewy bodies).
In summary, we show that thrombospondin1 and ADAMTS13 accumulate and are partitioned into CA of the brain in both normal aged and vascular dementia patients. Since these proteins are expressed highly in the blood, their progressive, pathological extravasation into the brain during aging or neurodegeneration may participate in the genesis of CA which form around blood vessels. The direct protein interactions we describe here for the first time suggest that thrombospondin1 and ADAMTS13 are paired either in the blood or during the formation of CA.
Acknowledgments
We thank Drs. Jimo Borjigin for helpful discussions. Drs. David Ginsberg and Karl Desch generously provided antibodies and expression clones for ADAMTS13. Dr.Andrew Lieberman and the Michigan Alzheimer’s Disease Research Center provided critical patient samples, and we thank the patients and families who have donated precious tissue resources that were used in this study. The ADRC is supported by grant P50-AG08671. Nancy McAnsh and the University of Michigan Comprehensive Cancer Center provided technical support for experiments. NIH grants NS052681 and NS054724 support MMW. Antibody TRA1-85 was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa.
References
- 1.Sakai M, Austin J, Witmer F, Trueb L. Studies of corpora amylacea. I. Isolation and preliminary characterization by chemical and histochemical techniques. Arch Neurol. 1969;21:526–544. doi: 10.1001/archneur.1969.00480170098011. [DOI] [PubMed] [Google Scholar]
- 2.Cisse S, Perry G, Lacoste-Royal G, Cabana T, Gauvreau D. Immunochemical identification of ubiquitin and heat-shock proteins in corpora amylacea from normal aged and Alzheimer’s disease brains. Acta Neuropathol. 1993;85:233–240. doi: 10.1007/BF00227716. [DOI] [PubMed] [Google Scholar]
- 3.Singhrao SK, Neal JW, Newman GR. Corpora amylacea could be an indicator of neurodegeneration. Neuropathol Appl Neurobiol. 1993;19:269–276. doi: 10.1111/j.1365-2990.1993.tb00437.x. [DOI] [PubMed] [Google Scholar]
- 4.Singhrao SK, Morgan BP, Neal JW, Newman GR. A functional role for corpora amylacea based on evidence from complement studies. Neurodegeneration. 1995;4:335–345. doi: 10.1016/1055-8330(95)90024-1. [DOI] [PubMed] [Google Scholar]
- 5.Korzhevskii DE, Giliarov AV. Demonstration of nuclear protein neun in the human brain corpora amylacea. Morfologiia. 2007;131:75–76. [PubMed] [Google Scholar]
- 6.Loeffler KU, Edward DP, Tso MO. Tau-2 immunore-activity of corpora amylacea in the human retina and optic nerve. Invest Ophthalmol Vis Sci. 1993;34:2600–2603. [PubMed] [Google Scholar]
- 7.Erdamar S, Zhu ZQ, Hamilton WJ, Armstrong DL, Grossman RG. Corpora amylacea and heat shock protein 27 in Ammon’s horn sclerosis. J Neuropathol Exp Neurol. 2000;59:698–706. doi: 10.1093/jnen/59.8.698. [DOI] [PubMed] [Google Scholar]
- 8.Gati I, Leel-Ossy L. Heat shock protein 60 in corpora amylacea. Pathol Oncol Res. 2001;7:140–144. doi: 10.1007/BF03032581. [DOI] [PubMed] [Google Scholar]
- 9.Martin JE, Mather K, Swash M, Garofalo O, Leigh PN, Anderton BH. Heat shock protein expression in corpora amylacea in the central nervous system: clues to their origin. Neuropathol Appl Neurobiol. 1991;17:113–119. doi: 10.1111/j.1365-2990.1991.tb00702.x. [DOI] [PubMed] [Google Scholar]
- 10.Hoyaux D, Decaestecker C, Heizmann CW, et al. S100 proteins in Corpora amylacea from normal human brain. Brain Res. 2000;867:280–288. doi: 10.1016/s0006-8993(00)02393-3. [DOI] [PubMed] [Google Scholar]
- 11.Singhrao SK, Neal JW, Piddlesden SJ, Newman GR. New immunocytochemical evidence for a neuronal/ oligodendroglial origin for corpora amylacea. Neuropathol Appl Neurobiol. 1994;20:66–73. doi: 10.1111/j.1365-2990.1994.tb00958.x. [DOI] [PubMed] [Google Scholar]
- 12.Buervenich S, Olson L, Galter D. Nestin-like immunoreactivity of corpora amylacea in aged human brain. Brain Res Mol Brain Res. 2001;94:204–208. doi: 10.1016/s0169-328x(01)00166-8. [DOI] [PubMed] [Google Scholar]
- 13.Botez G, Rami A. Immunoreactivity for Bcl-2 and C-Jun/AP1 in hippocampal corpora amylacea after ischaemia in humans. Neuropathol Appl Neurobiol. 2001;27:474–480. doi: 10.1046/j.1365-2990.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- 14.Kimura T, Takamatsu J, Miyata T, Miyakawa T, Horiuchi S. Localization of identified advanced glycation end-product structures, N epsilon(carboxymethyl)lysine and pentosidine, in age-related inclusions in human brains. Pathol Int. 1998;48:575–579. doi: 10.1111/j.1440-1827.1998.tb03953.x. [DOI] [PubMed] [Google Scholar]
- 15.Liu HM, Anderson K, Caterson B. Demonstration of a keratan sulfate proteoglycan and a mannose-rich glycoconjugate in corpora amylacea of the brain by immunocytochemical and lectin-binding methods. J Neuroimmunol. 1987;14:49–60. doi: 10.1016/0165-5728(87)90100-7. [DOI] [PubMed] [Google Scholar]
- 16.Renkawek K, Bosman GJ. Anion exchange proteins are a component of corpora amylacea in Alzheimer disease brain. Neuroreport. 1995;6:929–932. doi: 10.1097/00001756-199504190-00026. [DOI] [PubMed] [Google Scholar]
- 17.Selmaj K, Pawlowska Z, Walczak A, Koziolkiewicz W, Raine CS, Cierniewski CS. Corpora amylacea from multiple sclerosis brain tissue consists of aggregated neuronal cells. Acta Biochim Pol. 2008;55:43–49. [PubMed] [Google Scholar]
- 18.Balea IA, Illes P, Schobert R. Affinity of corpora amylacea for oligonucleotides: sequence dependency and proteinaceous binding motif. Neuropathology. 2006;26:277–282. doi: 10.1111/j.1440-1789.2006.00695.x. [DOI] [PubMed] [Google Scholar]
- 19.Cavanagh JB. Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev. 1999;29:265–295. doi: 10.1016/s0165-0173(99)00003-x. [DOI] [PubMed] [Google Scholar]
- 20.Meng H, Zhang X, Hankenson KD, Wang MM. Thrombospondin2 potentiates notch3/jagged1 signaling. J Biol Chem. 2009;284:7866–7874. doi: 10.1074/jbc.M803650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 22.Bornstein P, Alfi D, Devarayalu S, Framson P, Li P. Characterization of the mouse thrombospondin gene and evaluation of the role of the first intron in human gene expression. J Biol Chem. 1990;265:16691–16698. [PubMed] [Google Scholar]
- 23.Soejima K, Nakamura H, Hirashima M, Morikawa W, Nozaki C, Nakagaki T. Analysis on the molecular species and concentration of circulating ADAMTS13 in Blood. J Biochem. 2006;139:147–154. doi: 10.1093/jb/mvj013. [DOI] [PubMed] [Google Scholar]
- 24.Levy GG, Motto DG, Ginsburg D. ADAMTS13 turns 3. Blood. 2005;106:11–17. doi: 10.1182/blood-2004-10-4097. [DOI] [PubMed] [Google Scholar]
- 25.Wolf BB, Lopes MB, VandenBerg SR, Gonias SL. Characterization and immunohistochemical localization of alpha 2-macroglobulin receptor (low-density lipoprotein receptor-related protein) in human brain. Am J Pathol. 1992;141:37–42. [PMC free article] [PubMed] [Google Scholar]
- 26.Albrecht U, Seulberger H, Schwarz H, Risau W. Correlation of blood-brain barrier function and HT7 protein distribution in chick brain circumventricular organs. Brain Res. 1990;535:49–61. doi: 10.1016/0006-8993(90)91823-y. [DOI] [PubMed] [Google Scholar]
- 27.Sameshima T, Nabeshima K, Toole BP, et al. Expression of emmprin (CD147), a cell surface inducer of matrix metalloproteinases, in normal human brain and gliomas. Int J Cancer. 2000;88:21–27. doi: 10.1002/1097-0215(20001001)88:1<21::aid-ijc4>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 28.Buee L, Hof PR, Roberts DD, Delacourte A, Morrison JH, Fillit HM. Immunohistochemical identification of thrombospondin in normal human brain and in Alzheimer’s disease. Am J Pathol. 1992;141:783–788. [PMC free article] [PubMed] [Google Scholar]
- 29.Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 30.Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawler J. The functions of thrombospondin-1 and -2. Curr Opin Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Strickland DK, Bornstein P. Extracellular matrix metalloproteinase 2 levels are regulated by the low density lipoprotein-related scavenger receptor and thrombospondin 2. J Biol Chem. 2001;276:8403–8408. doi: 10.1074/jbc.M008925200. [DOI] [PubMed] [Google Scholar]
- 34.Husemann J, Silverstein SC. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer’s disease brain. Am J Pathol. 2001;158:825–832. doi: 10.1016/S0002-9440(10)64030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ricciarelli R, D’Abramo C, Zingg JM, et al. CD36 overexpression in human brain correlates with beta-amyloid deposition but not with Alzheimer’s disease. Free Radic Biol Med. 2004;36:1018–1024. doi: 10.1016/j.freeradbiomed.2004.01.007. [DOI] [PubMed] [Google Scholar]