

Abstract
Due to its much slower deamidation rate comparing to that of asparagine (Asn), studies on glutamine (Gln) deamidation have been scarce, especially on the differentiation of its isomeric deamidation products: α- and γ-glutamic acid (Glu). It has been shown previously that electron capture dissociation (ECD) can be used to generate diagnostic ions for the deamidation products of Asn: aspartic acid (Asp) and isoaspartic acid (isoAsp). The current study explores the possibility of an extension of this ECD based method to the differentiation of the α- and γ-Glu residues, using three human crystallin peptides (αA (1-11), βB2 (4-14), and γS (52-71)) and their potentially deamidated forms as model peptides. It was found that the z•-72 ions can be used to both identify the existence and locate the position of the γ-Glu residues. When the peptide contains a charge carrier near its N-terminus, the c+57 and c+59 ions may also be generated at the γ-Glu residue. It was unclear whether formation of these N-terminal diagnostic ions is specific to the Pro-γ-Glu sequence. Unlike the Asp containing peptides, the Glu containing peptides generally do not produce diagnostic side chain loss ions, due to the instability of the resulting radical. The presence of Glu residue(s) may be inferred from the observation of a series of zn•-59 ions, although it was neither site specific, nor without interference from the γ-Glu residues. Finally, several interference peaks exist in the ECD spectra, which highlights the importance of using high performance mass spectrometers for confident identification of γ-Glu residues.
Introduction
Deamidation is one of the most important protein post-translational modifications (PTMs) which contributes to aging, diseases (such as celiac disease, urinary tract infection, cataract formation, cancer, and neurodegenerative diseases, e.g. Alzheimer's, Huntington's, and Parkinson's diseases), and affects the purity and shelf life of pharmaceutical products.1 Under physiological conditions, nonenzymatic deamidation of asparagine (Asn, N) residues may occur both in vivo and in vitro, which generates acidic isomers, aspartic acid (Asp) and isoaspartic acid (isoAsp), with + 0.984 Da mass shift.1,2 In cells, the deamidated proteins which generate an isoAsp residue can be partially repaired by protein L-isoaspartyl O-methyltransferase (PIMT) to change the isoAsp to Asp, but often the modified proteins are degraded by proteasomes.3,4 Inefficient degradation, caused by either increased deamidation products and decreased proteasome activity with age5 or proteasomal dysfunction,6 may lead to accumulation of the damaged or modified proteins and related diseases.
Like Asn, glutamine (Gln, Q) deamidates by direct hydrolysis under acidic conditions, and proceeds via a glutarimide intermediate at neutral or alkaline conditions,2,7 Scheme 1. The deamidation rate of Gln is much slower than that of Asn,1 except when Gln is located at the N-terminus, under which condition it deamidates more rapidly than Asn to form a pyroglutamic acid.2 For example, even the fastest deamidating QG sequence has a median deamidation half-life of about 660 days as measured in the pentapeptide GXQGG,8 as opposed to less than one day for Asn deamidation in the NG sequence.9 In general, the half-lives of the sequence-dependent Asn peptide deamidation at neutral pH and physiological temperature range from about 0.5 to 500 days, while those of Gln range from about 600 to 20,000 days.1 Consequently, most known Gln deamidation are present in long lived proteins, such as eye lens crystallins. Crystallins are highly soluble structural proteins and comprise 90% of lens proteins, which include three classes: alpha (α), beta (β), and gamma (γ). They undergo little turnover during their life spans, allowing accumulation of many kinds of modifications.10-12 Among these, deamidation is one of the most prevalent, which decreases crystallin solubility, alters lens transparency, and is associated with cataract formation, a leading cause of blindness.13 Extensive Gln deamidation has been reported in aged or diseased lens crystallin proteins. 12-21
Scheme 1.
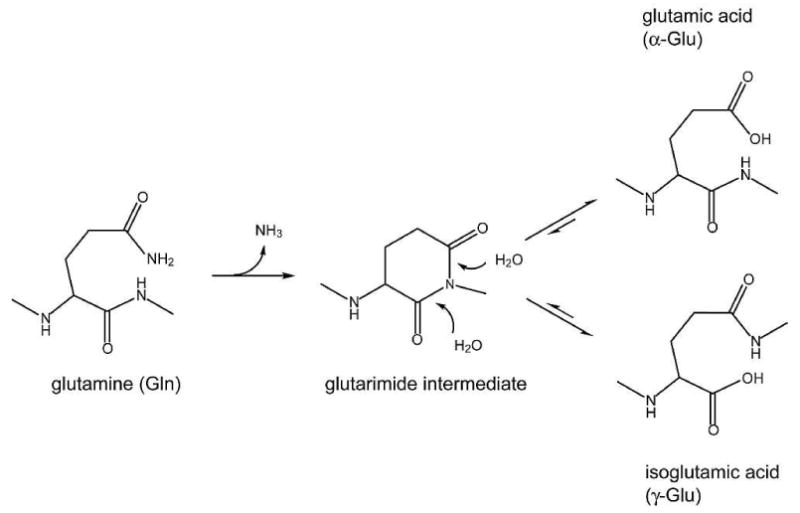
General mechanism for glutamine deamidation to form glutamic acid and isoglutamic acid via a glutarimide intermediate.
The Asn deamidation in peptides/proteins is extensively studied, both to determine the deamidation site(s) and to differentiate the isomeric deamidation products.22-25 Although the +0.984 Da mass shift caused by deamidation can be easily identified using mass spectrometry (MS), it is a much more challenging task to differentiate the isomeric deamidation products. A variety of methods have been developed to differentiate the Asp and isoAsp residues, including the nuclear magnetic resonance (NMR), Edman sequencing, HPLC, and antibody based detection methods.26 All these methods have their limitations, and they either require a relatively large quantity of peptide samples or are time consuming. The antibody based method also requires the use of specific antibodies. Mass spectrometry has become a powerful tool for deamidation studies, because of its small sample amount requirement, fast and accurate detection, as well as the more detailed information provided by the tandem MS experiments. Recently, a fast and accurate electron capture dissociation (ECD) based method has been developed to distinguish the Asp and isoAsp residues. ECD is based on the dissociative recombination of multiply protonated polypeptide molecules with low-energy (≤0.2 eV) electrons, which induces the homolytic cleavage of the peptide backbone N-Cα bond, yielding c and z• fragment ions.27-29 The additional methylene group inserted into the backbone of the isoAsp containing peptide creates a second fragmentation channel, which proceeds via direct cleavage of the Cα-Cβ bond, generating the diagnostic ions (c+57 and z•-57) for isoAsp containing peptides.22,23,25 In addition, this ECD based method can provide the quantitative measurement of the relative abundance of the isoAsp residues in deamidated proteins.24 The same diagnostic fragments have also been detected using the electron-transfer dissociation method (ETD),26 which has a similar backbone fragmentation mechanism to ECD.30-32
However, there have only been a limited number of Gln deamidation studies, even at the peptide level.8 The first Gln deamidation study was reported in 1991 on a dipeptide, which showed the formation of glutarimide intermediate and deamidation products, α- and γ-glutamate (Glu, E).7 Similar to the Asn deamidation study, it is relatively easy to determine the number of Gln deamidation site(s) in peptides/proteins based on the ∼+1 Da mass shift per site. The exact Gln deamidation site(s) can be identified by using either tandem MS methods, such as collision-induced dissociation (CID), or amino acid sequencing methods.33-35 Several methods, including hydrolysis, Edman degradation, and NMR, have been developed to distinguish the isomeric α- and γ-glutamyl peptides, but each has its own disadvantages.36,37 There have also been a number of MS studies on the differentiation of the α- and γ-glutamyl residues, including the electron impact (EI) mass spectrometry,38 chemical ionization (CI) mass spectrometry,36 and fast atom bombardment (FAB)/collisional activation (CA) tandem mass spectrometry.37 Capillary electrophoresis (CE) was also used for the separation of two α-glutamyl tripeptides and their potential degradation and isomerization products at pH 3 and pH 7, based on the different acidity and basicity between the α- and γ-isomers.39 Recently, CID was used to characterize the α- and γ-glutamyl dipeptides in negative ion mode.40 All of these methods were limited to small peptides with less than five amino acids, and most of these methods can only be applied to peptides with glutamyl residue located at the N-terminus to form the cyclic structure for identification.
Because of the importance of Gln deamidation in many pathological processes and the limitations of the existing methods discussed above, there is a clear need for a fast, sensitive and accurate method to differentiate the α- and γ-Glu residues in relatively large peptides. In this work, three human crystallin peptides in their potentially deamidated forms, with sizes ranging from eleven to twenty amino acid residues, were studied by ECD to develop an ECD based method for direct differentiation of the α- and γ-Glu residues in longer peptides, based on the knowledge of the Asp/isoAsp differentiation.
Experimental
αA (1-11: MDVTIQHPWFK), βB2 (4-14: HQTQAGKPQSL), and γS (52-71: PNFAGYMYILPQGEYPEYQR) human crystallin peptides in their native and two potentially deamidated forms (synthesized by AnaSpec, Fremont, CA, USA) were analyzed by ECD at ∼10-5 M concentration in the spray solution containing 50:50 (v/v) methanol: water with 1% formic acid. ECD analysis was performed on a custom built qQq-Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS) equipped with a nano-spray source and a 7T actively shielded magnet.41,42 In ECD experiments, multiply charged precursor ions were isolated in the front end quadrupole (Q1) and externally accumulated in the collision cell (Q2). After that, ions were transmitted to and trapped in the cylindrical ICR cell to interact with low energy electrons (∼0.5 eV), generated by the indirectly heated dispenser cathode (Heatwave, Watsonville, CA, USA). A grid located in front of the cathode was set at +10 V to help guide the low energy electrons into the ICR cell. The time domain signals were Fourier transformed and the spectra were internally calibrated with the precursor and charge-reduced ions. The peak assignments were done manually.
Results and Discussion
Identification of Gln deamidation site(s)
ECD spectra of the three crystallin peptides and their corresponding potentially deamidated forms are shown in Figures 1, 2, and 3. The detailed peak lists are shown in the supplementary tables 1-9. The Gln deamidation site(s) can be easily and accurately identified by ECD, based on the ∼+1 Da mass shift per site caused by deamidation in the fragment ions containing the deamidated Gln residue. Figure 4 shows the comparison of several c ions of the βB2 crystallin peptide (4-14) in its Gln and γ-Glu forms. In the ECD spectrum of this γ-Glu containing peptide, c4 ion at m/z 514 shows a mass shift of ∼+2 Da, consistent with the substitution of the first two Gln residues with γ-Glu. Further, c8 ion at m/z 867 shows a ∼+2 Da mass shift, while c9 ion at m/z 996 shows a ∼+3 Da mass shift, unambiguously identifying the Gln residue at position 9 as the third Gln residue being replaced by γ-Glu.
Figure 1.
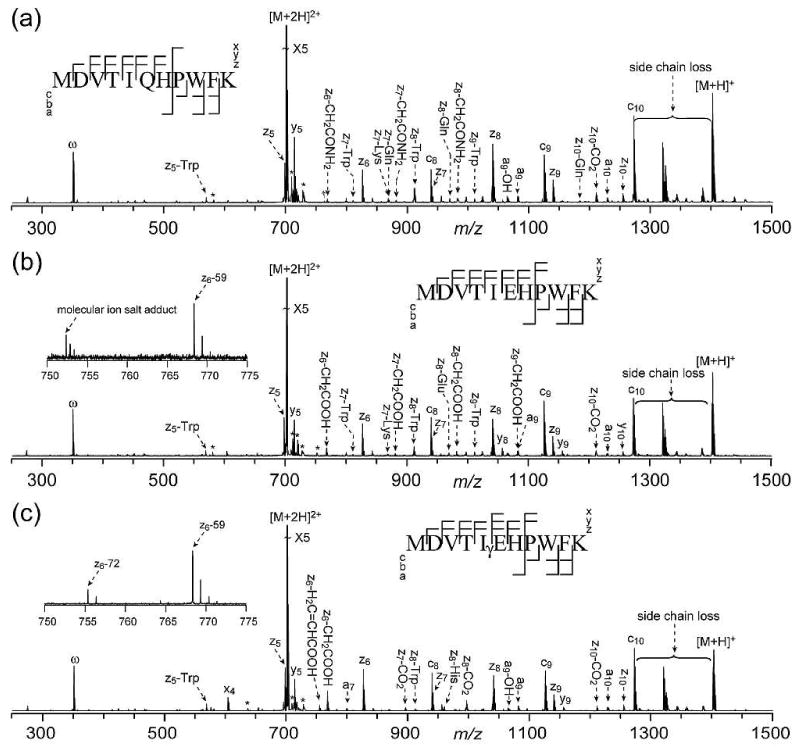
ECD spectra of αA crystallin peptide (1-11) in its Gln (a), Glu (b), and γGlu (c) forms. The insets show the enlarged regions of interest. Peaks marked with “-amino acid side residue” represent the entire side chain losses, and the partial side chain losses are represented by the molecular formulas of the departing group(s). ω: harmonic peaks. *: electronic noise peaks or salt adducts. †: internal fragments. Cleavage patterns are shown as the insets. (Figure 1a modified from reference 44 with permission)
Figure 2.
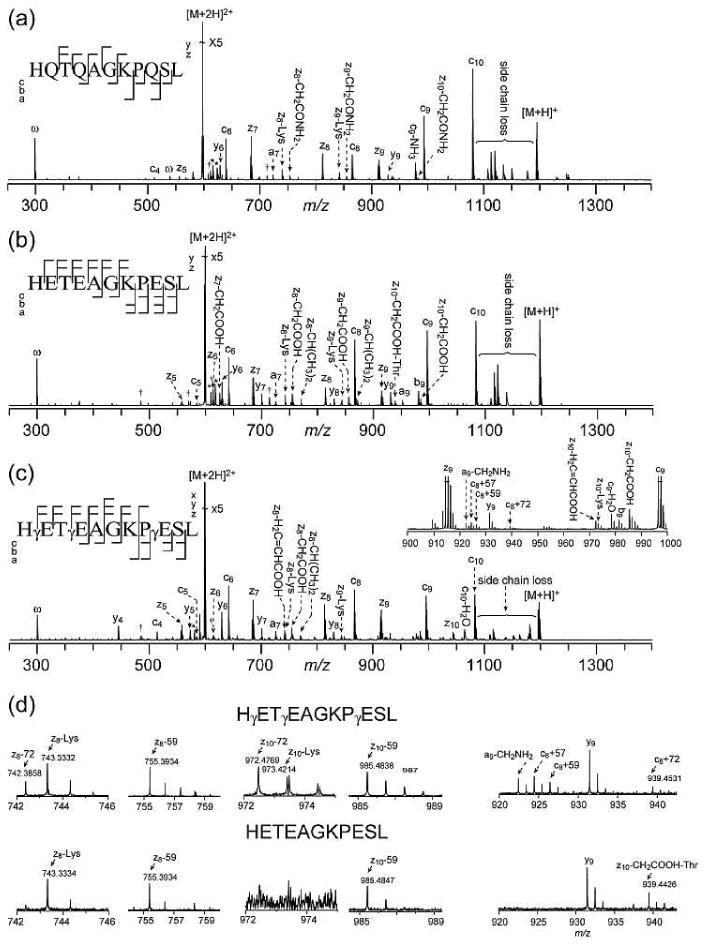
ECD spectra of βB2 crystallin peptide (4-14) in its Gln (a), Glu (b), and γGlu (c) forms. The comparison of the enlarged regions of interest is shown in (d). Peak labeling follows the same convention as in Figure 1. Cleavage patterns are shown as the insets. (Figure 2b modified from reference44 with permission)
Figure 3.
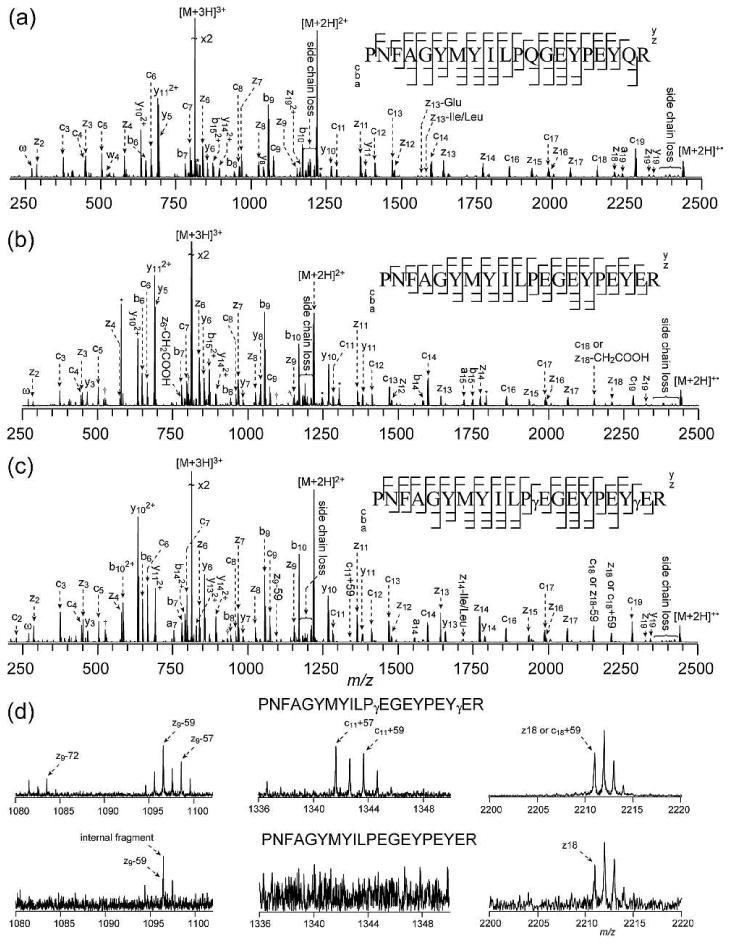
ECD spectra of γS crystallin peptide (52-71) in its Gln (a), Glu (b), and γGlu (c) forms. The comparison of the enlarged regions of interest is shown in (d). Peak labeling follows the same convention as in Figure 1. Cleavage patterns are shown as the insets.
Figure 4.
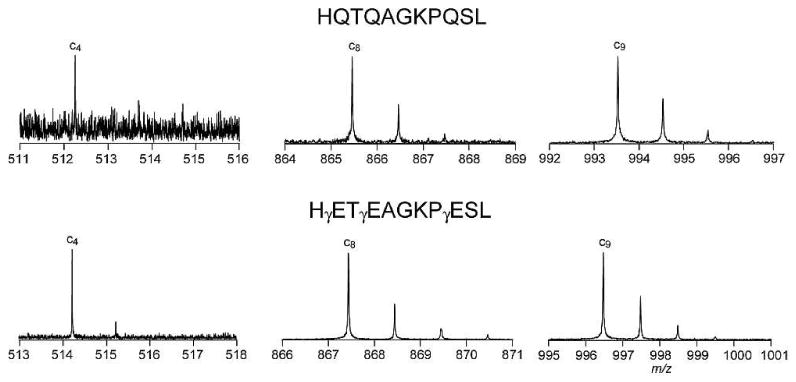
Comparison of several c ions from ECD of βB2 crystallin peptide (4-14) in its Gln and γGlu forms, upper and lower panel, respectively.
Characteristic ions for the differentiation of the α- and γ-Glu containing peptides Alpha A crystallin peptide (1-11)
The αA crystallin peptide (1-11) represents the simplest situation, where there is only one Gln residue in the peptide sequence that can undergo deamidation. The C-terminal lysine residue is the preferred site to retain the charge after the electron capture, and consequently the ECD spectra in Figure 1 are dominated by the C-terminal fragments. In addition to the z• ions, z• fragments with additional Glu side chain losses are also observed in Figure 1b and 1c. The ECD spectra of both Glu and γ-Glu containing αA crystallin peptides have peaks corresponding to z6•-59 ions (Figures 1b and 1c, insets), although they are formed via different mechanisms. In ECD of the Glu containing αA crystallin peptide (Figure 1b), the z6•-59 ion is the w6 ion, generated by the α-cleavage induced by the radical on the z6• ion (Scheme 2a). In addition, a series of peaks corresponding to zn•-59 ions (n>6) are also observed. These ions are generated from the partial loss of the Glu side chain from the corresponding z• ions, following through-space radical rearrangement (Scheme 2b).43,44 Similar radical migration in larger zn• ions (n>6) to the alpha position of the γ-Glu site may also occur in ECD of the γ-Glu containing αA crystallin peptide. However, because the Cβ-Cγ bond is part of the backbone in this γ-Glu peptide, its subsequent cleavage will not result in the formation of a series of zn•-59 ions, but rather z6•-59 ion only (Scheme 2c, Figure 1c). Although the z•-59 ion is observed site specifically in γ-Glu peptides, it can not be used as the diagnostic ion, because of its presence in Glu peptides as well.
Scheme 2.
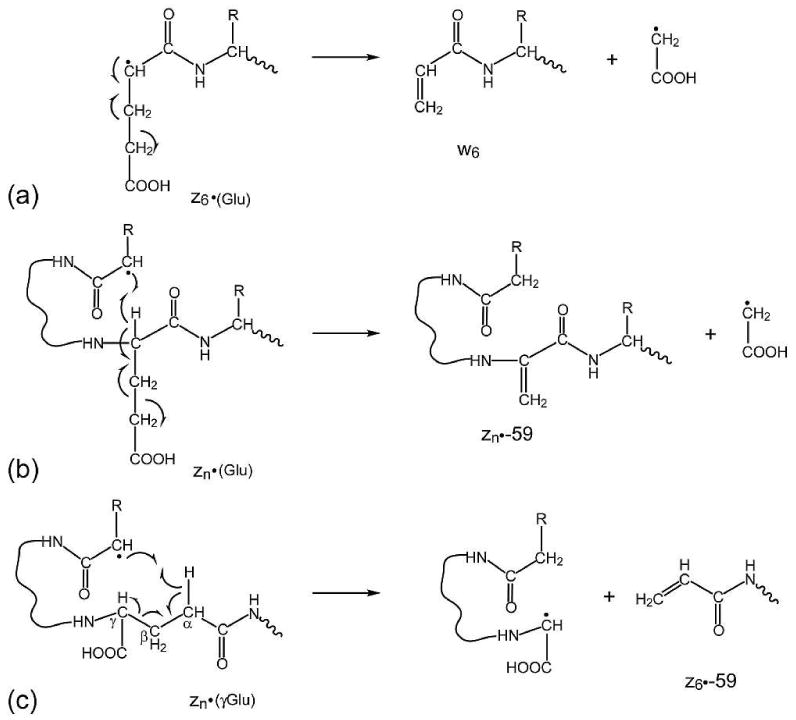
Mechanisms for the formation of z•-59 ions in ECD of the Glu containing αA crystallin peptide (1-11) via (a) direct partial side-chain loss (w6 ion formation) and (b) through-space radical rearrangement. (c) proposed mechanism for the formation of site specific z6•-59 ions in ECD of the γGlu containing αA crystallin peptide (1-11).
By contrast, the z6•-72 ion is only present in the γ-Glu containing αA crystallin peptide, but not in the Glu peptide (Figures 1b and 1c, insets). It may be formed via a similar mechanism to that of the isoAsp diagnostic ion formation in ECD.22 In addition to the N-Cα bond cleavage, the electron capture at the protonated carbonyl group of the γ-Glu residue could also induce Cα-Cβ cleavage at its N-terminal side, generating the c+71 and z•-71 ions. The primary carbon radical in the c+71 ion is unstable, which may abstract a hydrogen from the z•-71 ion species to form the stable c+72 and z•-72 ions (Scheme 3a). However, because of the unstable radical intermediates involved, this process is not expected to play a significant role. A more plausible mechanism is shown in Scheme 3b, where the radical on the N-terminal α-carbon of the z• ions can initiate α-cleavage along the peptide backbone to generate z•-72 ions. In rare occasions, z•-72 ions corresponding to the entire Glu side chain loss from z• ions may also be present in the ECD spectra of Glu containing peptides. These ions are usually formed via α-cleavage following through-space radical migration to the γ-carbon of the Glu residue,44 and they almost never occur at sites directly N-terminal to the Glu residue. For example, in Figure 1b, the z8•-72 ion is observed, but the z6•-72 ion is absent. Thus, the site specific z•-72 ions may serve as characteristic ions for the differentiation of the Glu and γ-Glu containing peptides. Due to the presence of the C-terminal lysine residue, in the ECD spectrum of the γ-Glu containing αA crystallin peptide, no peaks corresponding to the N-terminal complementary (c+59 or c+72) ions are observed.
Scheme 3.
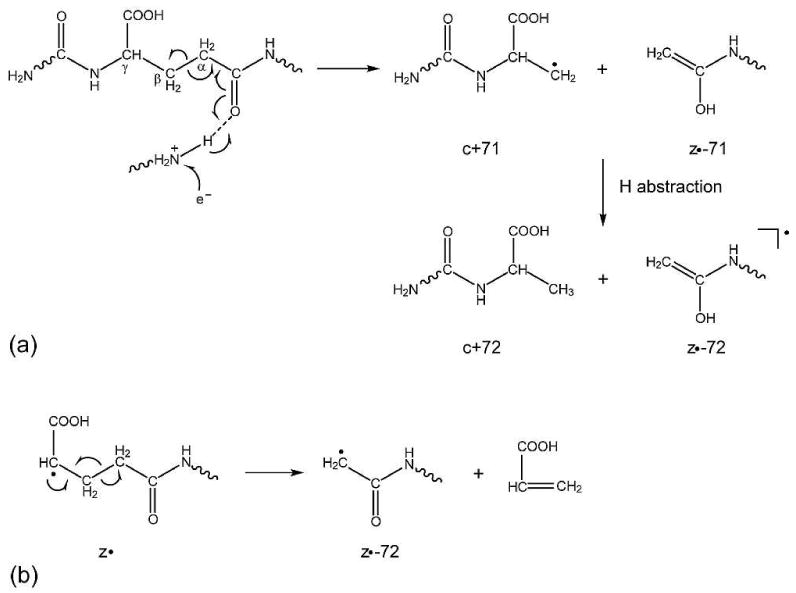
Proposed mechanism for the formation of z•-72 ions in the ECD of the γGlu containing peptide via (a) primary backbone cleavage and (b) secondary fragmentation.
Beta B2 crystallin peptide (4-14)
The βB2 crystallin peptide (4-14) (Figure 2) contains three Gln residues (Gln2, Gln4, and Gln9) in its sequence and one lysine residue in the middle, presenting an opportunity to investigate both N- and C-terminal fragments in ECD. The ECD spectra of the βB2 crystallin peptide and two of its potentially deamidated forms are shown in Figure 2a-c, respectively. Figure 2d shows the comparison of several regions of interest between ECD spectra of the two potentially deamidated forms of βB2 crystallin peptide. ECD of the γ-Glu containing peptide generates site specific zn•-59 ions N-terminal to the γ-Glu residues (n = 8 and 10, Figure 2c), with the exception of the z3•-59 ion, which is not observed due to its lack of a charge carrier. However, these zn•-59 ions are also present in the ECD spectrum of the Glu containing peptide (Figure 2b), as a result of the partial side chain losses from the Glu residues, preventing their use as diagnostic ions for γ-Glu residues.
On the other hand, the zn•-72 peaks are present site specifically (n = 8 and 10) in the ECD spectrum of the γ-Glu containing βB2 peptide, but absent in that of the Glu containing βB2 peptide, similar to what was observed in the ECD study of the αA crystallin peptide. In the case of the βB2 crystallin peptide, however, there is an added complexity caused by the interference peaks of the lysine side chain loss from z• ions. For example, the first isotopic peak of the diagnostic z8•-72 ion for the γ-Glu peptide at m/z 743.39 overlaps with the monoisotopic peak of the z8•-Lys peak at m/z 743.33, which is present in the ECD spectra of both the Glu and γ-Glu peptides. However, this interference peak does not present a problem for the identification of the γ-Glu here, as it can be easily distinguished from the diagnostic ion based on the accurate mass measurement afforded by the high performance FT-ICR instrument. A further, and more serious, complication arises from the presence of multiple Glu residues in this peptide. In the ECD spectrum of the Glu peptide, there is a tiny peak at m/z ∼742.38, which likely arises from the loss of Glu9 side chain of the z8• ion, via γ hydrogen abstraction at Glu9. Since this z8•-Glu ion has the same exact mass as that of the z8•-72 diagnostic ion in the ECD spectrum of the γ-Glu peptide, they cannot be differentiated even by an FT-ICR mass analyzer. The result from this peptide illustrates a potential limitation of using z•-72 ion as the diagnostic ion for the identification of γ-Glu residue in peptides with multiple Glu residues. However, since side chain loss occurrence from a remote residue often decreases dramatically as its distance from the backbone cleavage site increases, such interference is likely to be minimal as evident from the very low abundance of the z8•-72 ion and the absence of the z10•-72 ion here. Nonetheless, it would still be advantageous to identify additional characteristic ions for γ-Glu peptides to make the assignment with higher confidence. Of particular interest is the search for N-terminal diagnostic ions, since unlike z• ions, these ions usually do not contain a radical to initiate side chain losses which may produce interference peaks.
The right panel of Figure 2d shows the difference associated with N-terminal fragments between ECD spectra of the two potentially deamidated forms of βB2 crystallin peptide. Of the three γ-Glu residues, only one site (γ-Glu9) produced complementary diagnostic ions that correspond to the c8+57, c8+59, and c8+72 ions. The potential characteristic N-terminal fragments for the other two γ-Glu sites do not contain the charge carrying lysine residue and are hence, unobservable. The peak at m/z 922.43 does not arise from the c8+55 ion, but rather the a9•-CH4N ion. The peak at m/z 939.453 corresponding to the c8+72 ions in the γ-Glu peptide ECD spectrum can be distinguished from the peak at m/z 939.442 in the Glu peptide ECD spectrum, which is the result of multiple side chain losses. The c+72 ions may be formed via the Cα-Cβ cleavage upon electron capture followed by intra-complex hydrogen transfer, although this process is disfavored because of the unstable primary carbon radical involved as discussed earlier (Scheme 3a). An alternative mechanism is shown in Scheme 4, which involves the ring opening process of proline. Dissociations of proline radicals formed by ECD/ETD have been studied in the past, where it was found that although H-atom loss is energetically favored over backbone dissociations, H-atom migrations may also occur,45 sometimes leading to secondary N-C or C-C bond cleavage at the proline side chain.46 In this case, the Cα radical generated by the initial N-Cα bond cleavage at the proline residue can abstract one hydrogen from the α or γ position of the nearby γ-Glu residue to induce α-cleavage and form c+59 and c+72 ions, respectively. All intermediates in Scheme 4 involved are resonantly stabilized by the conjugated carbonyl groups.47 By contrast, in ECD of Glu containing peptides, because there are no additional methylene groups in the Glu peptide backbone, the hydrogen abstraction from the α or γ position of the Glu residue will only lead to partial or entire side chain loss from the molecular ions, as shown in Scheme 5. The amino acid side chain losses from the charge reduced species are usually initiated by electron capture at either charged basic amino acid side chains (e.g. Arg, Lys, and His), or amino acid residues capable of solvating the charge (e.g. Asp and Met). It has been shown that charge neutralization at the solvated Asp side chain can lead to the formation of the diagnostic [M-60]+• ion by losing an even electron species, CH2=C(OH)2.23 The radical is left on the backbone α-carbon, which is stabilized captodatively by the neighboring amine and carbonyl groups. Although the carboxylic acid group of the Glu side chain is also capable of solvating the charge, a similar process is unlikely to occur at Glu residues, as the loss of the CH2=C(OH)2 group will result in the formation of an unstable primary radical on the β-carbon. Nonetheless, partial Glu side chain loss from the charge reduced species is present (peak at m/z 1139.57) in the ECD spectrum of the Glu containing βB2 peptide ECD spectrum here. This 59 Da loss corresponds to the loss of a radical •CH2COOH species, most likely from the Glu side chain following the proline ring opening.
Scheme 4.
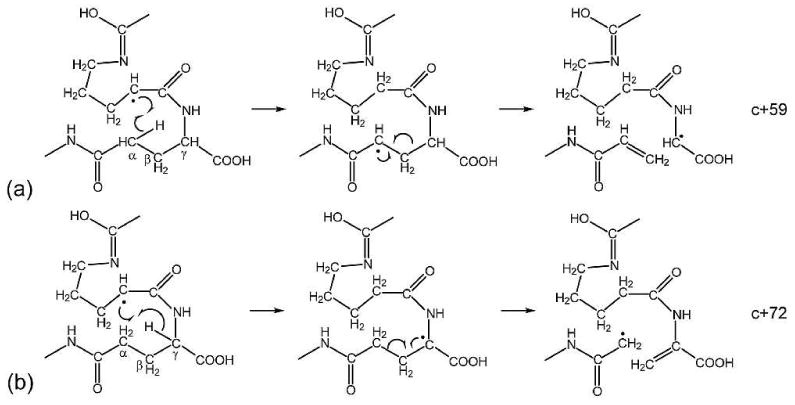
Proposed mechanisms for the formation of (a) c+59 and (b) c+72 ions in the γGlu containing peptide via proline ring opening process.
Scheme 5.
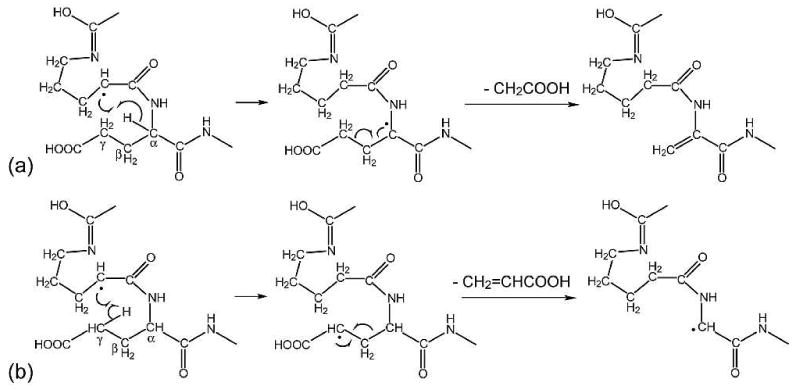
Proposed mechanisms for the loss of (a) -CH2COOH and (b) -CH2=CHCOOH molecules in the Glu containing peptide via proline ring opening process.
The proposed mechanisms for the c+57 ion formation are shown in supplementary Scheme 1. The c8+57 ion may be formed from the a9• ion via loss of an ethylene molecule (supplementary Scheme 1a), although the a9• ion itself contains an unstable primary carbon radical, which makes this mechanism an unattractive one. Alternatively, the c+57 ions could result from the intra-complex hydrogen transfer after the initial N-Cα bond cleavage. For any c ion that contains the γ-Glu residue, the radical on its complementary z• fragment may abstract a hydrogen atom from the α-carbon of the γ-Glu, and the resulting c• radical can undergo α-cleavage to produce the c+57 ion (supplementary Scheme 1b).
The site-specific occurrence of the c+57, c+59, and c+72 ions at the γ-Glu residue observed here suggests that they too can be used as diagnostic ions for the identification of γ-Glu residues.
Gamma S crystallin peptide (52-71)
Both αA crystallin peptide (1-11) and βB2 crystallin peptide (4-14) are short Gln containing peptides, without the interference of other existing Glu residues in their sequences. The γS crystallin peptide (52-71) contains two Gln residues and two Glu residues in its twenty amino acid-long peptide sequence, which makes the differentiation of Glu and γ-Glu residues more complex than in the αA and βB2 crystallin peptides. The two potentially deamidated forms studied here include one containing four Glu residues, and another containing two Glu and two γ-Glu residues.
The comparison of the relevant regions associated with fragmentations near the two γ-Glu sites (γ-Glu12 and γ-Glu19) from Figure 3c and the related Glu sites from Figure 3b is shown in Figure 3d. Peaks corresponding to z9•-59, z9•-57, and z9•-72 ions as well as the complementary c11+57 and c11+59 ions, which are generated by cleavage within the γ-Glu12 residue, are present in the ECD spectrum of the γ-Glu containing γS crystallin peptide. However, the complementary c11+72 ion is not observed here. Incidentally, in the γS crystallin peptide, the γ-Glu12 residue is also preceded by a proline residue, just like the γ-Glu9 residue in the βB2 peptide. Thus, it is not clear whether the c+57 and c+59 ions are sequence specific or they may serve as general diagnostic ions. None of these N- and C-terminal characteristic ions are observed in the ECD spectrum of the Glu containing γS peptide. The tiny peak at m/z 1096 in the Glu peptide spectrum does not correspond to the z9•-59 ion, but rather an internal fragment: MYILPEGEY, based on its accurate mass. Therefore, the presence of these complementary diagnostic ions provides high confidence for the identification and location of the γ-Glu12 residue, even in the presence of multiple Glu residues in the sequence.
For the γ-Glu19 residue in the γ-Glu containing γS crystallin peptide, the diagnostic z2•-59 and c18+59 ions have the same elemental composition as the c2 and z18• ions, respectively. This ambiguity has a similar origin as the homeometric peptides, which are different peptides with similar theoretical tandem mass spectra.48,49 This does not constitute a systematic limitation to the method developed here, since its occurrence is expected to be rare, although it does emphasize the importance of multiple diagnostic ions to increase the confidence in γ-Glu identification. Unfortunately, in this particular case, the z2•-72 and c18+72 ions are not observed in the γ-Glu peptide ECD spectrum either. Thus, the γ-Glu19 residue could not be identified by the N- or C-terminal characteristic ions established here.
There are, in general, no equivalent Glu diagnostic ions to the abundant [M-60]+• ion present in the ECD spectra of Asp containing peptides.22 The [M-60]+• ion is produced via the charge neutralization at the solvated Asp side chain, a process that is inhibited at the Glu side chain, due to the instability of the resulting radical. Direct Glu side chain loss from the charge reduced species either partially or completely, is rarely observed in ECD. Of the three Glu containing peptides studied here, only one, the βB2 crystallin peptide, produced the partial Glu side chain loss peak at [M-59]+•, and it is likely an unusual case due to the presence of the proline residue. On the other hand, a series of zn•-59 ions in the ECD spectra of the Glu containing peptides may be used to infer the existence of Glu residue(s), but they can not be generally used to locate the position or to determine the number of Glu residues. Finally, while it is possible to quantify the relative content of γ-Glu in a mixture of deamidation products, the presence of zn•-59 and zn•-72 ions due to remote Glu side chain losses makes the quantification of α- and γ-Glu residues a much more difficult task, particularly when there are multiple Glu/γ-Glu residues present in the sequence.
Conclusion
Three synthetic human crystallin peptides and their potentially deamidated forms were analyzed by ECD in this study. The C- and N-terminal diagnostic ions associated with the γ-Glu residue were established and the fragmentation mechanisms were discussed. In the ECD spectra of these peptides, peaks corresponding to the z•-72 fragment ions were observed at multiple γ-Glu sites in the γ-Glu containing peptides, but were generally absent in the Glu containing peptides. Further, these z•-72 fragment ions happened only at sites N-terminal to the γ-Glu residues. Thus, the z•-72 fragment ions can be used as diagnostic ions for the differentiation of the α- and γ-Glu residues, and for the determination of the γ-Glu sites. This C-terminal diagnostic ion is especially useful in the bottom-up approach, which typically involves the analysis of tryptic peptides containing a C-terminal charge carrier that facilitates the detection of C-terminal fragments in ECD.
The z•-59 fragment ions, although detected site specifically in ECD of γ-Glu containing peptides, were also present in the ECD spectra of Glu containing peptides. This 59 Da loss from Glu containing z• ions arose from the partial loss of the Glu side chain, either directly at the cleavage site (w ion) or following through-space radical rearrangement within the corresponding z• ions, which led to the formation of a series of zn•-59 ions. The presence of a series of zn•-59 ions is suggestive of the existence of Glu residue(s), but cannot be used to determine their exact locations.
In the ECD spectra of the three crystallin peptides studied, the N-terminal diagnostic ions (c+57, c+59 and/or c+72 ions) were only observed at two γ-Glu sites, both of which contain proline residue located directly before γ-Glu residue. Because of the relative location of the γ-Glu residues and the charge carriers, the only γ-Glu residue in a non-Pro-γGlu sequence capable of generating detectable N-terminal diagnostic ions here is the γ-Glu19 in the γS crystallin peptide. However, it is impossible to either confirm or rule out the formation of the diagnostic c+59 ion related to this γ-Glu19 residue, due to the presence of an interference ion. Thus, it remains unclear whether the N-terminal diagnostic ions are Pro-γ-Glu sequence specific or they may serve as general diagnostic ions. Even if the proline effect is generally applicable, the c+59 and c+72 ions will still be useful in the study of Gln deamidations in the Pro-Gln rich regions of crystallin, gluten and other biological proteins.50,51
In conclusion, multiple diagnostic ions including both N- and C-terminal fragments for the identification of γ-Glu residue were established using ECD, which provided confident differentiation of the Glu and γ-Glu residues at the peptide level. This knowledge will facilitate the bottom-up analysis of Gln deamidations in aged or diseased proteins. The high mass accuracy afforded by the FT-ICR instrument is crucial for confident identification, particularly in larger peptides, where interference peaks abound.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the financial support from the NIH/NCRR P41RR10888, NIH/NHLBI N01HV28178, NIH/NIGMS R01GM078293, MDS SCIEX, and the ACS Petroleum Research Fund. They thank Dr. Chunxiang Yao and Dr. Weidong Cui for helpful discussions.
Footnotes
Supplementary data: The detailed peak list tables are shown in the supplementary tables 1-9.
References
- 1.Robinson NE, Robinson AB. Molecular clocks: Deamidation of asparaginyl and glutaminyl residues in peptides and proteins. Althouse Press; Cave Junction, OR: 2004. [Google Scholar]
- 2.Wright HT. Crit Rev Biochem Mol Biol. 1991;26:1–52. doi: 10.3109/10409239109081719. [DOI] [PubMed] [Google Scholar]
- 3.Reissner KJ, Aswad DW. Cell Mol Life Sci. 2003;60:1281–1295. doi: 10.1007/s00018-003-2287-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benaroudj N, Tarcsa E, Cascio P, Goldberg AL. Biochimie. 2001;83:311–318. doi: 10.1016/s0300-9084(01)01244-5. [DOI] [PubMed] [Google Scholar]
- 5.Carrard G, Bulteau AL, Petropoulos I, Friguet B. Int J Biochem Cell Biol. 2002;34:1461–1474. doi: 10.1016/s1357-2725(02)00085-7. [DOI] [PubMed] [Google Scholar]
- 6.Halliwell B. Antioxid Redox Signal. 2006;8:2007–2019. doi: 10.1089/ars.2006.8.2007. [DOI] [PubMed] [Google Scholar]
- 7.Capasso S, Mazzarella L, Sica F, Zagari A. J Chem Soc-Chem Commun. 1991:1667–1668. [Google Scholar]
- 8.Robinson NE, Robinson ZW, Robinson BR, Robinson AL, Robinson JA, Robinson ML, Robinson AB. J Pept Res. 2004;63:426–436. doi: 10.1111/j.1399-3011.2004.00151.x. [DOI] [PubMed] [Google Scholar]
- 9.Hayes C, Setlow P. J Bacteriol. 1997;179:6020–6027. doi: 10.1128/jb.179.19.6020-6027.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A. Prog Biophys Mol Biol. 2004;86:407–485. doi: 10.1016/j.pbiomolbio.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Wistow GJ, Piatigorsky J. Annu Rev Biochem. 1988;57:479–504. doi: 10.1146/annurev.bi.57.070188.002403. [DOI] [PubMed] [Google Scholar]
- 12.Wilmarth PA, Tanner S, Dasari S, Nagalla SR, Riviere MA, Bafna V, Pevzner PA, David LL. J Proteome Res. 2006;5:2554–2566. doi: 10.1021/pr050473a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lampi KJ, Amyx KK, Ahmann P, Steel EA. Biochemistry. 2006;45:3146–3153. doi: 10.1021/bi052051k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miesbauer LR, Zhou XJ, Yang ZC, Yang ZY, Sun YP, Smith DL, Smith JB. J Biol Chem. 1994;269:12494–12502. [PubMed] [Google Scholar]
- 15.Lund AL, Smith JB, Smith DL. Exp Eye Res. 1996;63:661–672. doi: 10.1006/exer.1996.0160. [DOI] [PubMed] [Google Scholar]
- 16.Hoehenwarter W, Klose J, Jungblut PR. Amino Acids. 2006;30:369–389. doi: 10.1007/s00726-005-0283-9. [DOI] [PubMed] [Google Scholar]
- 17.Hanson SRA, Hasan A, Smith DL, Smith JB. Exp Eye Res. 2000;71:195–207. doi: 10.1006/exer.2000.0868. [DOI] [PubMed] [Google Scholar]
- 18.Zhang ZL, Smith DL, Smith JB. Exp Eye Res. 2003;77:259–272. doi: 10.1016/s0014-4835(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 19.Zhang ZL, David LL, Smith DL, Smith JB. Exp Eye Res. 2001;73:203–211. doi: 10.1006/exer.2001.1023. [DOI] [PubMed] [Google Scholar]
- 20.Feng JH, Smith DL, Smith JB. J Biol Chem. 2000;275:11585–11590. doi: 10.1074/jbc.275.16.11585. [DOI] [PubMed] [Google Scholar]
- 21.Hanson SRA, Smith DL, Smith JB. Exp Eye Res. 1998;67:301–312. doi: 10.1006/exer.1998.0530. [DOI] [PubMed] [Google Scholar]
- 22.Cournoyer JJ, Lin C, O'Connor PB. Anal Chem. 2006;78:1264–1271. doi: 10.1021/ac051691q. [DOI] [PubMed] [Google Scholar]
- 23.Cournoyer JJ, Pittman JL, Ivleva VB, Fallows E, Waskell L, Costello CE, O'Connor PB. Protein Sci. 2005;14:452–463. doi: 10.1110/ps.041062905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cournoyer JJ, Lin C, Bowman MJ, O'Connor PB. J Am Soc Mass Spectrom. 2007;18:48–56. doi: 10.1016/j.jasms.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Cournoyer JJ, Lin C, O'Cormora PB. J Am Soc Mass Spectrom. 2008;19:855–864. doi: 10.1016/j.jasms.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Connor PB, Cournoyer JJ, Pitteri SJ, Chrisman PA, McLuckey SA. J Am Soc Mass Spectrom. 2006;17:15–19. doi: 10.1016/j.jasms.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 27.Zubarev RA, Kelleher NL, McLafferty FW. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 28.Zubarev RA, Haselmann KF, Budnik B, Kjeldsen F, Jensen F. Eur J Mass Spectrom. 2002;8:337–349. [Google Scholar]
- 29.Syrstad EA, Turecek F. J Am Soc Mass Spectrom. 2005;16:208–224. doi: 10.1016/j.jasms.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coon JJ, Shabanowitz J, Hunt DF, Syka JEP. J Am Soc Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JEP, Shabanowitz J, Hunt DF. BBA-Proteins Proteomics. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novak P, Man P, Tuckova L, Hogenova HT, Bezouska K, Havlicek V. J Mass Spectrom. 2002;37:507–511. doi: 10.1002/jms.305. [DOI] [PubMed] [Google Scholar]
- 34.Joshi AB, Kirsch LE. J Pharm Sci. 2002;91:2332–2345. doi: 10.1002/jps.10213. [DOI] [PubMed] [Google Scholar]
- 35.Liu HC, Gaza-Bulseco G, Chumsae C. Rapid Commun Mass Spectrom. 2008;22:4081–4088. doi: 10.1002/rcm.3831. [DOI] [PubMed] [Google Scholar]
- 36.Nagasawa HT, Magnan SDJ, Foltz RL. Biomed Mass Spectrom. 1982;9:252–256. [Google Scholar]
- 37.Lloyd JR, Cotter ML, Ohori D, Doyle DL. Biomed Environ Mass Spectrom. 1988;15:399–402. doi: 10.1002/bms.1200150707. [DOI] [PubMed] [Google Scholar]
- 38.Okada K, Kawase M. Chem Pharm Bull. 1977;25:1497–1508. [Google Scholar]
- 39.Schucker SC, Scriba GKE. J Chromatogr A. 2000;888:275–279. doi: 10.1016/s0021-9673(00)00566-5. [DOI] [PubMed] [Google Scholar]
- 40.Harrison AG. J Mass Spectrom. 2004;39:136–144. doi: 10.1002/jms.515. [DOI] [PubMed] [Google Scholar]
- 41.Jebanathirajah JA, Pittman JL, Thomson BA, Budnik BA, Kaur P, Rape M, Kirschner M, Costello CE, O'Connor PB. J Am Soc Mass Spectrom. 2005;16:1985–1999. doi: 10.1016/j.jasms.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 42.O'Connor PB, Pittman JL, Thomson BA, Budnik BA, Cournoyer JC, Jebanathirajah J, Lin C, Moyer S, Zhao C. Rapid Commun Mass Spectrom. 2006;20:259–266. doi: 10.1002/rcm.2307. [DOI] [PubMed] [Google Scholar]
- 43.Lin C, Cournoyer JJ, O'Connor PB. J Am Soc Mass Spectrom. 2008;19:780–789. doi: 10.1016/j.jasms.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Lin C, Han L, Costello CE, O'Connor PB. J Am Soc Mass Spectrom. 2010 doi: 10.1016/j.jasms.2010.01.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayakawa S, Hashimoto M, Matsubara H, Turecek F. J Am Chem Soc. 2007;129:7936–7949. doi: 10.1021/ja0712571. [DOI] [PubMed] [Google Scholar]
- 46.Cooper HJ, Hudgins RR, Hakansson K, Marshall AG. Int J Mass Spectrom. 2003;228:723–728. [Google Scholar]
- 47.Tsybin YO, Hamidane HB, Vorobyev A, Wodrich M, Corminboeuf C. 57th ASMS conference on mass spectrometry; Philadelphia, PA. 2009. [Google Scholar]
- 48.Zubarev RA, Zubarev AR, Savitski MM. J Am Soc Mass Spectrom. 2008;19:753–761. doi: 10.1016/j.jasms.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. J Proteome Res. 2006;6:114–123. doi: 10.1021/pr060271u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiao SW, Bergseng E, Molberg O, Jung G, Fleckenstein B, Sollid LM. J Immunol. 2005;175:254–261. doi: 10.4049/jimmunol.175.1.254. [DOI] [PubMed] [Google Scholar]
- 51.Arentz-Hansen H, McAdam SN, Molberg O, Fleckenstein B, Lundin KEA, Jorgensen TJD, Jung G, Roepstorff P, Sollid LM. Gastroenterology. 2002;123:803–809. doi: 10.1053/gast.2002.35381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.