

Abstract
Orthopoxviruses produce two, antigenically distinct, infectious virions, intracellular mature virions and extracellular virions (EV). A33 and B5 are found on EV but not intracellular mature virions. To investigate the function of A33, a recombinant virus that has A33R deleted and expresses B5R-GFP (vB5R-GFP/ΔA33R) was generated. A comparison of vB5R-GFP/ΔA33R to an analogous virus (vΔA33R) revealed an additional defect in infectious EV production that was not apparent when A33R was present. Characterization of these recombinants revealed that EV produced in the absence of A33 had undetectable levels of B5-GFP. Both recombinants released similar amounts of EV but there were differences in their infectivity. Approximately equal numbers of virions produced by these recombinants were able to bind cells even though EV produced by vB5R-GFP/ΔA33R do not contain B5. These results suggest that in the absence of A33, the cytoplasmic tail of B5 contributes to its incorporation into the envelope of progeny virions.
Introduction
Vaccinia virus is the best-studied member of the Orthopoxvirus genus. It has a double-stranded DNA genome of about 200kb that is predicted to encode for approximately 200 functional open reading frames (Moss, 2001). Viral replication occurs entirely in the cytoplasm of infected cells in a specialized area known as the viral factory and results in three commonly recognized, morphologically distinct, forms: intracellular mature virions (IMV), intracellular enveloped virions (IEV), and extracellular virions (EV) (Moss, 2006; Smith, Vanderplasschen, and Law, 2002). IMV is the first infectious progeny virions formed and represents the majority of virions produced by infected cells. IEV are formed by intracellular envelopment of a subset of IMV with an additional double membrane derived from the trans-Golgi network (TGN) or early endosomes (Hiller and Weber, 1985; Schmelz et al., 1994; Tooze et al., 1993). IEV are transported to the cell periphery via microtubules and released from the cytoplasm by fusion of their outermost envelope membrane with the plasma membrane (Geada et al., 2001; Hollinshead et al., 2001; Rietdorf et al., 2001; Ward and Moss, 2001a). These released virions have lost one of their IEV membranes but possess one more membrane than IMV and have been called EV. EV can be further classified as being either cell-associated enveloped virions (CEV) or extracellular enveloped virions (EEV) for those virions that have detached from the cell they were produced in. CEV are believed to be important for efficient cell-to-cell spread via actin tails (Reeves et al., 2005; Ward and Moss, 2001a) while EEV mediate greater dissemination of virus (Appleyard, Hapel, and Boulter, 1971; Payne, 1980).
Presently, six proteins encoded by the virus, A33, A34, A36, B5, F12, and F13, have been found to be both exclusive to either IEV or EV and involved in infectious EV production (Duncan and Smith, 1992; Engelstad, Howard, and Smith, 1992; Hirt, Hiller, and Wittek, 1986; Isaacs et al., 1992; Roper, Payne, and Moss, 1996; van Eijl et al., 2002; van Eijl, Hollinshead, and Smith, 2000). A33 is predicted to play a role in both IEV/EV morphogenesis/egress and subsequent infection. A33R is highly conserved among orthopoxviruses and encodes a 23 kDa type II glycoprotein (Roper, Payne, and Moss, 1996). Deletion of A33R results in a reduction in IEV production and an increase in infectious EEV production (Roper et al., 1998). The cytoplasmic tails of A33 and A36 interact and the interaction is required for the incorporation of A36 into IEV and subsequently, actin tail formation (Ward, Weisberg, and Moss, 2003; Wolffe, Weisberg, and Moss, 2001). It has also been reported that A33 interacts with B5 (Perdiguero and Blasco, 2006). The crystal structure of the ectodomain of A33 suggests that it contains an unusual C-type lectin-like domain that may interact with host cells (Su et al., 2009). Here, we have created a recombinant vaccinia virus that has A33R deleted and expresses B5-GFP in place of the normal B5. This new virus was more defective than the parental virus vΔA33R. Characterization of these viruses revealed that in the absence of A33, B5-GFP was not found on EV. Quantification of progeny EV produced by these recombinants shows that the absence of A33 only slightly reduces the amount of EV produced in addition to making them less infectious. The removal of B5, in addition to A33, from EV further reduces the infectivity of EV but this reduction is not due to a reduction in the ability to bind cells.
Results
Construction and characterization of a recombinant virus that expresses B5R-GFP and has A33R deleted
A recombinant virus expressing B5R fused to the coding sequence of enhanced GFP in place of normal B5R (vB5R-GFP) has been useful for studying virion egress in living cells (Ward, 2009; Ward and Moss, 2001b). We recently reported an important role for A34 in proper targeting and incorporation of B5 into progeny virions utilizing a recombinant virus that has A34R deleted and expresses B5R-GFP in place of B5R (Earley, Chan, and Ward, 2008). In addition to B5 and A34, A33 has been shown to be required for efficient cell-to-cell spread as deletion of A33R results in a reduction in plaque size (Roper et al., 1998). In a similar way, we constructed a recombinant virus that has A33R deleted and expresses B5R-GFP to study IEV/EV morphogenesis in the absence of A33. The new recombinant, vB5R-GFP/ΔA33R, formed plaques that fluoresced green and were noticeably smaller than plaques formed by the parental A33R deletion virus, vΔA33R, (Fig. 1). The two analogous viruses, WR and vB5R-GFP, formed plaques that were similar in size and much larger than those formed by either ΔA33R virus. Plaque size is related to the amount of infectious EV produced and the ability to form actin tails (Blasco and Moss, 1991; Ward and Moss, 2001b). The deletion of A33R abrogates the production of actin tails, therefore, the smaller plaques formed by vB5R-GFP/ΔA33R suggest that the addition of GFP to the cytoplasmic tail of B5 in the absence of A33 is affecting the production of infectious enveloped virions. To test this idea, B5R-GFP was replaced with B5R in vB5R-GFP/ΔA33R to create vB5R/ΔA33R. This replacement restored the plaque size of vB5R/ΔA33R to that of vΔA33R, indicating that the smaller plaque phenotype of vB5R-GFP/ΔA33R is due to the addition of GFP to the cytoplasmic tail of B5.
Fig. 1. Plaque phenotypes.
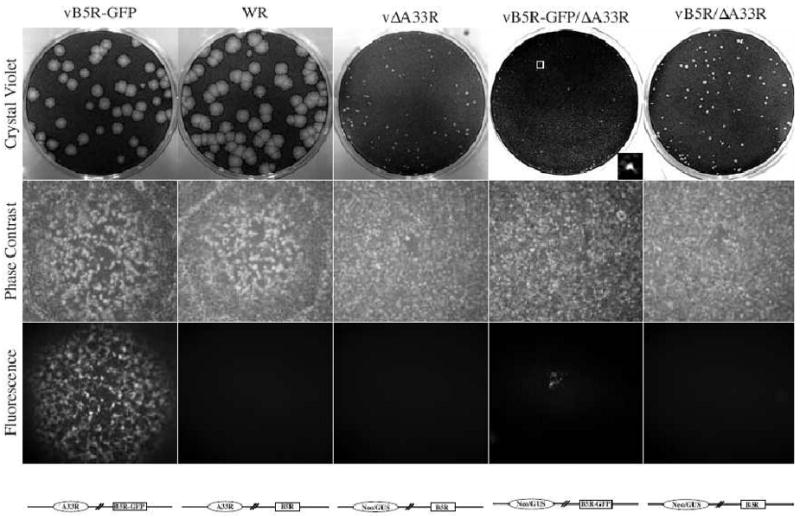
Confluent BS-C-1 cell monolayers were infected with the indicated viruses and overlaid with semi-solid media. Two days PI, phase contrast and fluorescence images were captured using a fluorescent microscope. Cell monolayers were stained with crystal violet three days PI and imaged. For vB5R-GFP/ΔA33R, insert shows an enlarged plaque in the boxed area. Schematic representation of the genome of each recombinant virus is shown below the fluorescence images.
B5-GFP is mis-targeted in the absence of A33
The smaller plaque phenotype of vB5R-GFP/ΔA33R compared to vΔA33R suggests that in the absence of A33, the addition of GFP to the cytoplasmic tail of B5 results in a decrease in the amount of infectious EV produced. This could be due to a decrease in the total amount of EV produced, their infectivity or a combination of both. To determine where the defect was occurring, we looked at the localization of B5 in cells infected with our recombinant viruses by immunofluorescence microscopy. Typically, cells infected with vB5R-GFP display three characteristic hallmarks of GFP fluorescence: an accumulation of GFP fluorescence at the site of wrapping in the juxtanuclear region, at the cell vertices, and GFP-labeled virion-sized particles (VSPs) in the cytoplasm (Fig. 2). Cells infected with WR had a similar B5 localization pattern to that seen in cells infected with vB5R-GFP (Fig. 2). In cells infected with either vΔA33R or vB5R/ΔA33R, B5 localized at the site of wrapping and on VSPs (Fig. 2). In contrast, cells infected with vB5R-GFP/ΔA33R displayed a GFP fluorescence pattern different from that seen in cells infected with vΔA33R (Fig. 2). B5-GFP was found throughout the cytoplasm and accumulated in the juxtanuclear region (Fig. 2). In addition, in the absence of A33, there was a distinct reduction in GFP-labeled VSPs in the cytoplasm, suggesting that B5-GFP was not efficiently incorporated into progeny enveloped virions in the absence of A33 (Fig. 2). The nearly perfect overlap of GFP fluorescence with B5 staining in cells infected with either vB5R-GFP or vB5R-GFP/ΔA33R indicated that the GFP fluorescence was a true representation of the localization of B5 fused to GFP (Fig. 2). In addition, Western blot analysis of the lysates of cells infected with vB5R-GFP or vB5R-GFP/ΔA33R indicated that the B5-GFP chimera was intact in the absence of A33 (data not shown).
Fig. 2. B5-GFP is mis-targeted in the absence of A33.
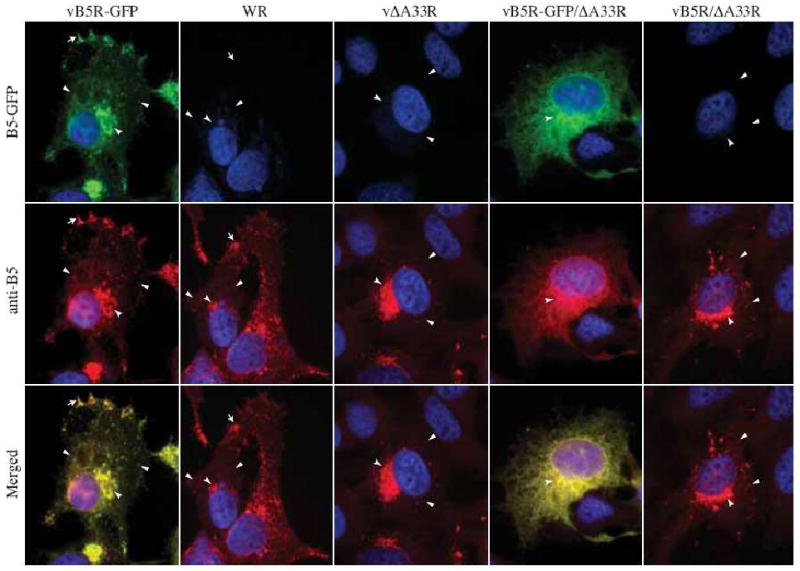
HeLa cells were infected with the indicated viruses at a MOI of 1.0. At 24 h PI, fixed and permeabilized cells were stained with an anti-B5 MAb, followed by Texas Red-conjugated donkey anti-rat antibody (Red), and visualized using a fluorescent microscope. Localization of B5-GFP or B5 at the site of wrapping (concave arrowheads), at the vertices (arrows), and on virion-sized particles (arrowheads) is indicated. The DNA in nuclei and viral factories was stained with DAPI (Blue). B5-GFP is shown in green. The overlap of green and red is shown in yellow.
A33 is required for proper targeting of B5-GFP
We wanted to determine if proper B5-GFP localization could be restored in cells infected with vB5R-GFP/ΔA33R by providing the A33R gene in trans. Therefore, we performed in vivo trans-complementation, followed by immunofluorescence microscopy. A plasmid that expresses A33R with an HA epitope tag under its own promoter was constructed (pA33R-HA-118). Cells infected with vB5R-GFP were transfected with the empty vector pBMW118, (Ward, 2009), while cells infected with vB5R-GFP/ΔA33R were transfected with either pBMW118 or pA33R-HA-118. Both plasmids contain the coding sequence of the HcRed gene under control of the vaccinia virus modified H5 promoter, therefore, cells that are both transfected and infected are easily identified by the expression of HcRed. Cells infected with vB5R-GFP/ΔA33R and transfected with pA33R-HA-118 fluoresced red and displayed a GFP fluorescence pattern identical to that seen in cells infected with vB5R-GFP (Fig. 3). In contrast, cells infected with vB5R-GFP/ΔA33R and transfected with pBMW118 fluoresced red and displayed the mis-targeted B5-GFP fluorescence pattern (Fig. 3). The staining of A33 with anti-HA MAb indicated that A33-HA is recognized by the antibody and suggests that the HA epitope tagged A33R localized properly in these infections (Fig. 3). In addition, the ability of A33-HA to restore proper targeting of B5-GFP in cells infected with vB5R-GFP/ΔA33R suggests that the aberrant localization of B5-GFP is due solely to the absence of A33.
Fig. 3. A33 is required for proper targeting of B5-GFP.
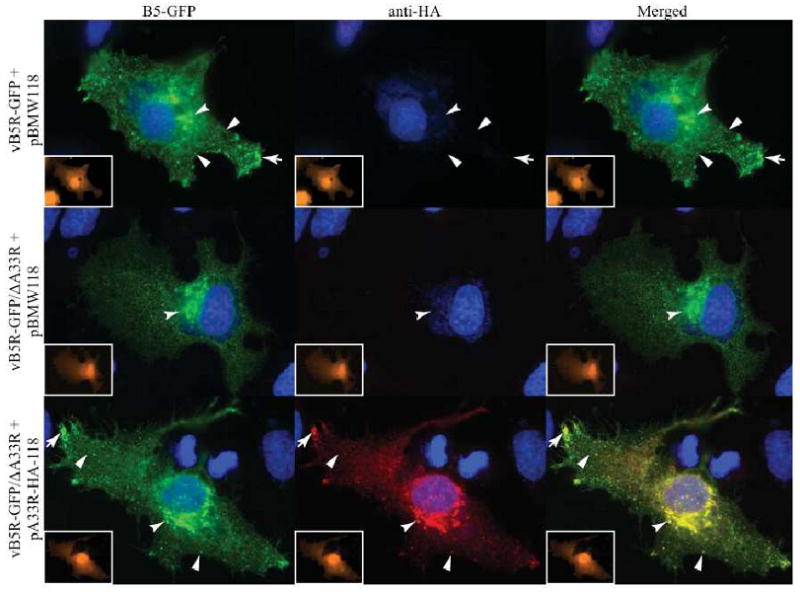
HeLa cells were infected with the indicated viruses at a MOI of 1.0 and transfected with the indicated plasmids. 24 h PI, cells were fixed, permeabilized, and A33-HA was visualized by staining with an anti-HA MAb, followed by Cy-5-labeled donkey-anti-rat antibody (Red). Inserts show HcRed fluorescence (Orange), indicating that cells were transfected and infected. Localization of B5-GFP or A33-HA at the site of wrapping (concave arrowheads), at the vertices (arrows), and on virion-sized particles (arrowheads) is indicated. The DNA in nuclei and viral factories was stained with DAPI (Blue). B5-GFP is shown in green. The overlap of green and red is shown in yellow.
Targeting of B5-GFP to the site of wrapping is partially defective in the absence of A33
We previously reported that the interaction of A34 and B5-GFP is required for the efficient trafficking of B5-GFP from the ER to the site of wrapping (Earley, Chan, and Ward, 2008). Similarly, we wanted to examine, at the subcellular level, the localization of B5-GFP in the absence of A33. We, therefore, stained cells infected with vB5R-GFP, WR, vΔA33R, or vB5R-GFP/ΔA33R with anti-Calnexin and anti-golgin-97 antibodies to visualize the localization of B5-GFP or B5 relative to the ER and TGN resident proteins, respectively. As shown in Fig. 4, both B5-GFP and B5 localized with golgin-97. However, there appeared to be more B5-GFP localization with Calnexin throughout the cytoplasm in the absence of A33 (Fig. 4).
Fig. 4. More B5-GFP localizes to the ER.
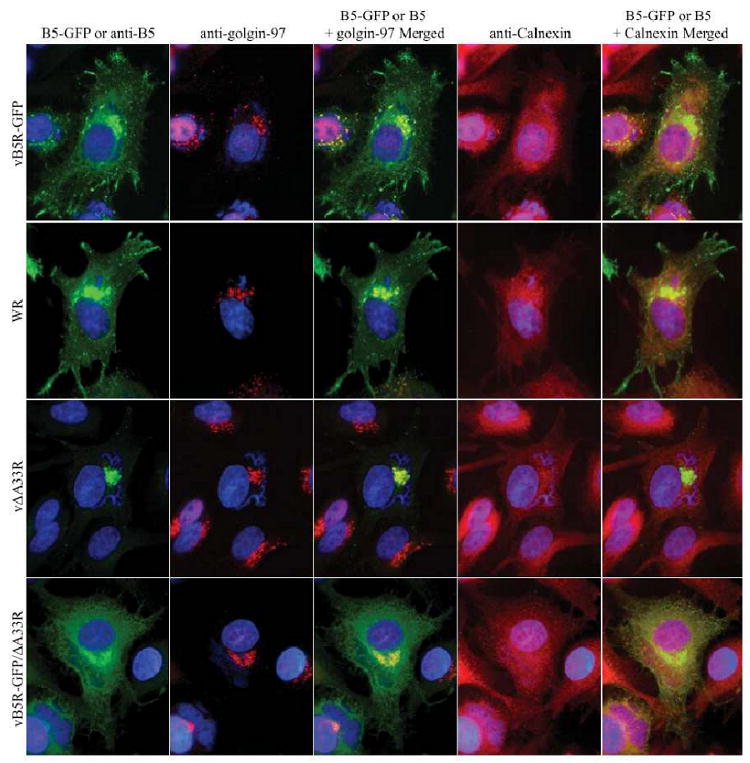
HeLa cells were infected with the indicated viruses at a MOI of 1.0. 12 h PI, cells were fixed, permeabilized, and stained with an anti-golgin-97 MAb and rabbit anti-Calnexin antibody, followed by Cy5-conjugated goat anti-mouse (Red) and Texas Red-conjugated donkey anti-rabbit (Red) antibodies. Cells infected with either WR or vΔA33R were subsequently stained with an anti-B5 MAb, followed by FITC-conjugated donkey anti-rat antibody (Green). Immunostained cells were imaged using a fluorescent microscope to visualize the localization of B5-GFP or B5 relative to the TGN and ER resident proteins, golgin-97 and Calnexin, respectively. The DNA in nuclei and viral factories was stained with DAPI (Blue). The overlap of green (B5-GFP or B5) and red (golgin-97 or Calnexin) is shown in yellow.
B5-labeled VSPs are not present on the surface of cells infected with vB5R-GFP/ΔA33R
On the surface of infected cells, B5 is typically found on the plasma membrane and on virion-sized particles, i.e., CEV. To look for B5 in the absence of A33, we stained unpermeabilized cells infected with vB5R-GFP, WR, vΔA33R, vB5R-GFP/ΔA33R, or vB5R/ΔA33R with an anti-B5 MAb. B5-labeled VSPs were seen on the surface of cells infected with vB5R-GFP, WR, vΔA33R, or vB5R/ΔA33R (Fig. 5). In contrast, there were little to no VSPs stained on the surface of cells infected with vB5R-GFP/ΔA33R although B5-GFP appeared to be efficiently deposited onto the plasma membrane (Fig. 5), suggesting that A33 is required for the incorporation of B5-GFP into progeny enveloped virions while A33 is not required for the incorporation of B5 into progeny enveloped virions.
Fig. 5. A33 is required for efficient incorporation of B5-GFP into enveloped virions on the surface of cells.
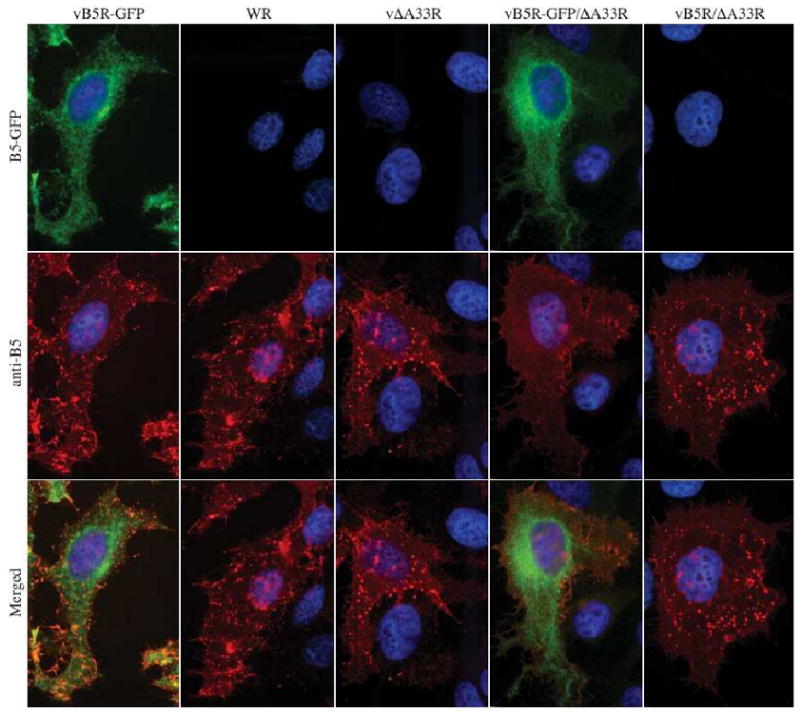
HeLa cells were infected with the indicated viruses at a MOI of 1.0. 24 h PI, cells were fixed and stained with an anti-B5 MAb, followed by Texas Red-conjugated donkey anti-rat antibody (Red). Immunostained cells were imaged using a fluorescent microscope. The DNA in nuclei and viral factories was stained with Hoechst (blue). B5-GFP is shown in green. Merged images of green and red are shown in yellow.
B5-GFP is not efficiently incorporated into progeny virions in the absence of A33
Our previous result suggests that little to no B5-GFP is incorporated into progeny enveloped virions in the absence of A33 (Fig. 6). To test this, radiolabeled EEV released by RK13 cells infected with vB5R-GFP, vB5R-GFP/ΔA33R, WR, or vΔA33R were purified. The resulting viral pellet was lysed and equal amounts of radiation were subjected to immunoprecipitation with either an anti-B5 or anti-F13 MAb. Both B5-GFP or B5 and F13 were precipitated from EEV released by cells infected with vB5R-GFP, WR, or vΔA33R. In contrast, B5-GFP was not precipitated from EEV released by cells infected with vB5R-GFP/ΔA33R while F13 was immunoprecipitated (Fig. 6A), indicating that in the absence of A33, B5-GFP is not efficiently incorporated into EEV. The examination of equilibrated EEV lysates on SDS-PAGE showed that approximately equal amounts of protein were used in the assay (Fig. 6A).
Fig. 6. A33 is required for efficient incorporation of B5-GFP into EEV.
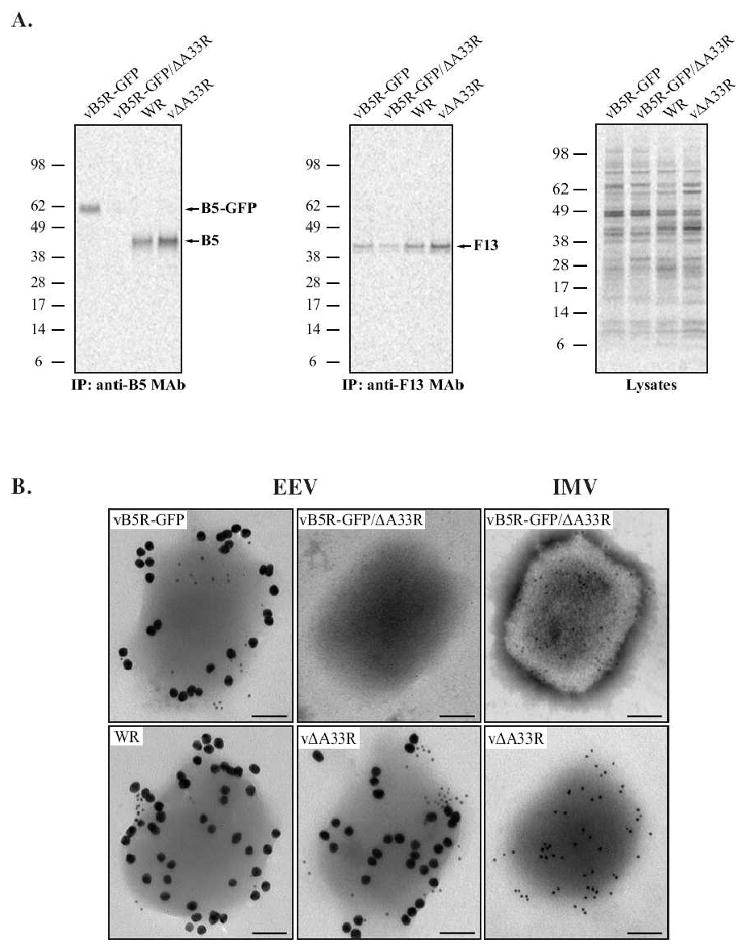
RK13 cells were infected with the indicated viruses at a MOI of 10.0 and incubated with media containing [35S]-Met/Cys. 24 h PI, supernatants were collected, clarified, and loaded onto a 36% sucrose cushion to purify EEV. (A) Immunoprecipitation. Purified EEV were resuspended in RIPA buffer and equal amounts of radiation from each were subjected to immunoprecipitation with either an anti-B5 or anti-F13 MAb. Immune complexes were resolved by SDS-PAGE and radiolabeled proteins were detected by autoradiography. Equilibrated crude EEV lysates were analyzed to show that comparable amounts were used for each immunoprecipitation. The molecular weights in kDa and positions of marker proteins are shown. (B) Immunoelectron microscopy. Purified EEV and IMV were immunolabeled with an anti-B5 MAb and rabbit anti-L1 antibody, followed by 18 nm colloidal gold-conjugated goat anti-rat and 6 nm colloidal gold-conjugated goat anti-rabbit antibodies. Immunogold-labeled virions were negative-stained and visualized using a TEM. A representative virion from each is shown. Scale bars represent 50 nm.
To verify these results, we performed immunoelectron microscopy on purified EEV and IMV, which allows us to directly visualize the incorporation of B5 into enveloped virions. Purified EEV released from cells infected with vB5R-GFP, WR, vΔA33R, or vB5R-GFP/ΔA33R and purified IMV from cells infected with either vΔA33R or vB5R-GFP/ΔA33R were immunolabeled with an anti-B5 MAb and rabbit anti-L1 antibody, which are specific for proteins found in the EEV membrane and IMV membrane, respectively, followed by 18 nm and 6 nm colloidal gold-conjugated secondary antibodies. Anti-B5 MAb readily labeled EEV released from cells infected with vB5R-GFP, WR, or vΔA33R but not vB5R-GFP/ΔA33R (Fig. 6B). With the exception of EEV from cells infected with vB5R-GFP/ΔA33R, L1 was readily detected on all of the virions, including the IMV from cells infected with vB5R-GFP/ΔA33R. The L1 staining of EV is likely due to ruptures in the EV membrane that occurred during processing of the samples, which exposed the IMV antigens. EV that lack B5 were reported to have a more stable outer envelope (Roberts et al., 2009). This stability likely accounts for the lack of L1 staining of the EV particles released from cells infected with vB5R-GFP/ΔA33R as they do not contain B5. Taken together, our data indicate that A33 is required for the incorporation of B5-GFP into progeny enveloped virions while B5 is efficiently incorporated into progeny enveloped virions in the absence of A33.
The absence of either A33 or B5 from EV reduces their infectivity
B5 has been reported to play a role in the dissolution of the outermost membrane of EEV to allow IMV to enter cells (Roberts et al., 2009). Therefore, the smaller plaque phenotype of vB5R-GFP/ΔA33R suggests two possibilities: less enveloped virions are produced by vB5R-GFP/ΔA33R compared to vΔA33R or enveloped virions produced by vB5R-GFP/ΔA33R are less infectious as they lack B5. To test these possibilities, we quantified the amounts of infectious and total EEV and CEV produced by vB5R-GFP, vΔA33R, and vB5R-GFP/ΔA33R after 24 h of infection using plaque assays and real time PCR, respectively. As shown in Fig. 7A & 7B, vΔA33R produced more total EEV, as measured by the copies of genomes, than vB5R-GFP/ΔA33R and more infectious EEV, as measured by plaque forming unit (PFU). In accordance with previously published data that compared WR to vΔA33R, vΔA33R produced 3 fold more infectious EEV than vB5R-GFP (Fig. 7A) (Roper et al., 1998). However, vΔA33R produced 28 fold more EEV genomes compared to vB5R-GFP, indicating that EEV produced by vΔA33R are not fully infectious (Fig. 7). Next, the number of genome copies was compared to PFUs to determine their infectivity. Whereas 1 out of every 1.6 genome copies resulted in a plaque for vB5R-GFP, this number was greatly elevated to 1 out of every 12.9 genome copies in the absence of A33 indicating that the absence of A33 on EEV is detrimental to their infectivity. Surprisingly, the absence of B5 from vΔA33R caused a reduction of this number to 1 out of every 6.1 genome copies resulting in a plaque (Fig. 7C). We next performed a similar analysis on virion released from the cell surface by trypsin treatment. Both vB5R-GFP/ΔA33R and vΔA33R produced fewer infectious CEV than vB5R-GFP while the genome copies were similar between the three viruses (Fig. 7). Similar to EEV, CEV produced by vΔA33R and vB5R-GFP/ΔA33R was less infectious compared to vB5R-GFP. By adding the numbers obtained for EEV and CEV, we quantified the amount of EV produced by all three viruses. Whereas the total number of genomes released by vΔA33R and vB5R-GFP/ΔA33R were very similar, vB5R-GFP/ΔA33R produced less infectious EV than vΔA33R, indicating that EV production may not be the reason for the difference in plaque size (Fig. 7).
Fig. 7. The majority of EEV and CEV produced by A33-deficient viruses are not infectious.
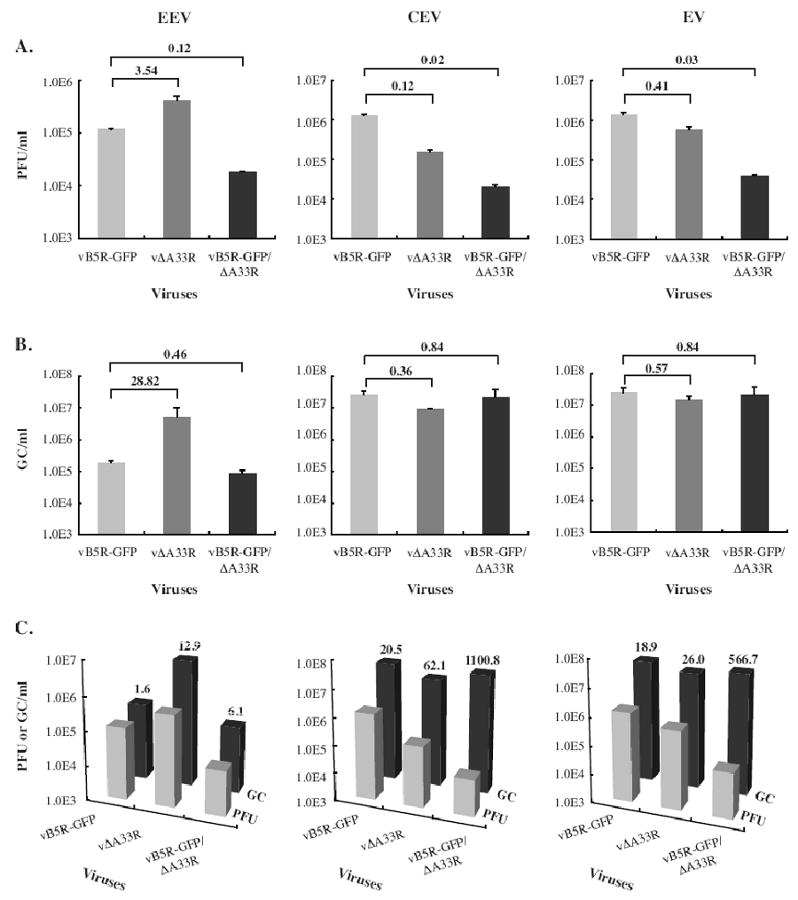
BS-C-1 cells were infected with the viruses indicated on the X-axes at a MOI of 10.0. 24 h PI, EEV containing culture supernatants were collected and a trypsin release assay was performed on cell monolayers to quantify CEV. (A) The amounts of infectious EEV or CEV produced (PFU/ml) were determined by a plaque assay on BS-C-1 monolayers. (B) The total amounts of EEV or CEV produced were determined by absolute quantification of genome copies (GC/ml) using real time PCR. For EV, the amount of EEV and CEV quantified by either PFU (A) or GC (B) were added together. Error bars are plotted for each virus. The fold difference relative to vB5R-GFP is shown. (C) Both PFU and GC of EEV, CEV or EV were plotted and the fold difference between PFU and GC is shown.
EEV produced by A33R-deficient viruses do not efficiently bind BS-C-1 cells
It has been suggested that B5 has a role in cell binding (Smith, Vanderplasschen, and Law, 2002). Therefore, the reduction in the infectivity of EV produced by vB5R-GFP/ΔA33R could be due to a reduction in cell binding. To test this, we equilibrated fresh EEV from cells infected with vB5R-GFP, vB5R-GFP/ΔA33R, or vΔA33R using real-time PCR and quantified their ability to bind BS-C-1 cells using immunofluorescence microscopy. EEV produced by vB5R-GFP, vB5R-GFP/ΔA33R or vΔA33R were labeled with anti-F13 MAb on the surface of BS-C-1 cells indicating that they are capable of binding cells (data not shown). To quantify the amount of binding, 200 nuclei were imaged and the number of labeled VSPs was counted. Fewer EEV produced by either vB5R-GFP/ΔA33R or vΔA33R bound to the cells compared to those produced by vB5R-GFP, indicating EEV produced by A33R-deficient viruses do not efficiently bind BS-C-1 cells (Fig. 8). In addition, our data indicate that the absence of B5 on EEV did not affect their ability to bind BS-C-1 cells as similar number of bound EEVs was detected for both vB5R-GFP/ΔA33R and vΔA33R.
Fig. 8. EEV produced by A33R-deficient viruses bind less efficiently to BS-C-1 cells.
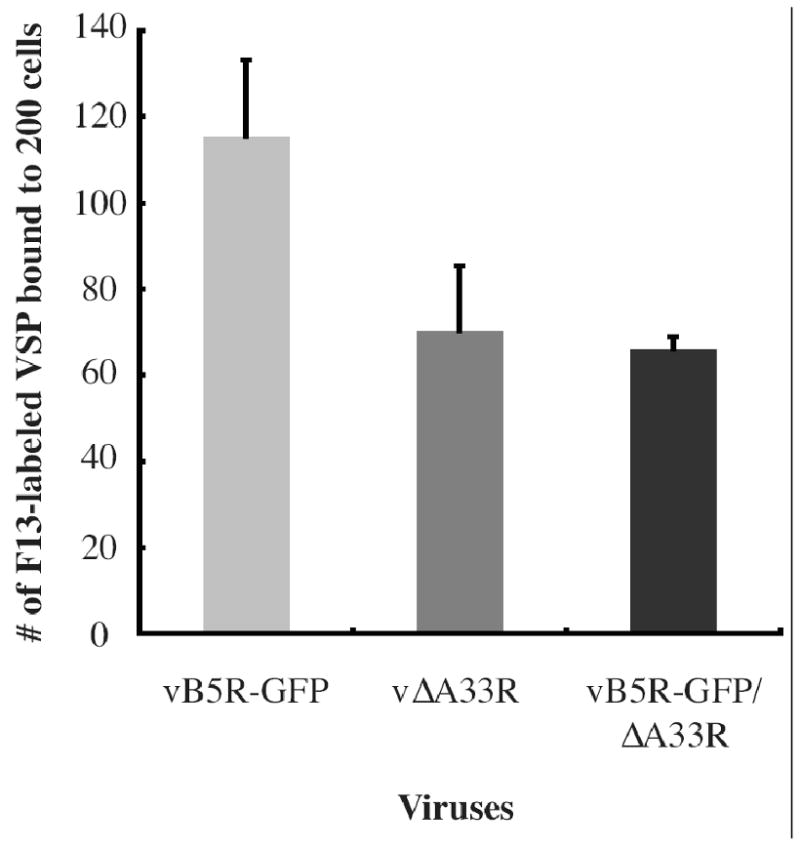
EEV released from cells infected with the indicated viruses were collected and the number of viral genomes released by each virus was determined by real time PCR. The same number of genome copies for each virus was bound to BS-C-1 cells at 4°C for 1 h. Unbound virions were removed by washing. Cells were fixed, permeabilized, and stained with an anti-F13 MAb, followed by Texas Red-conjugated donkey anti-mouse antibody. DNA in the nuclei was stained with DAPI and 200 nuclei were imaged. The number of F13-labeled VSPs was counted. The average of two independent experiments is shown and error bars are plotted for each virus.
Discussion
Initially, we wanted to use B5-GFP to track IEV production in the absence of A33 but found that a virus that has A33R deleted and expresses B5-GFP in place of B5 formed plaques that were smaller than the parental virus that only had A33R deleted. This was surprising because a recombinant virus that has B5R replaced with B5R-GFP appears to be similar to its parental virus, WR (Fig. 1) (Ward 2001). We have determined that the addition of GFP to the cytoplasmic tail prevents the incorporation of B5 into EV in the absence of A33. This finding points to the cytoplasmic tail of B5 as an additional determinant for its targeting to the EV membrane. While this may seem at odds with previous reports that the cytoplasmic tail of B5 was dispensable for EV formation and incorporation (Lorenzo et al., 1998; Mathew et al., 1998), both of these studies were carried out in the presence of A33. One explanation is that GFP is preventing the interaction of the cytoplasmic tail of B5 with some other protein, either viral or cellular, that targets B5 to the EV membrane in the absence of A33. A likely viral protein candidate would be F13, which is found exclusively on the cytoplasmic side of the membrane and was recently shown to coimmune precipitate B5 (Chen et al., 2009). In addition, the cytoplasmic tail of B5 was shown to have two cellular targeting motifs, a membrane-proximal tyrosine-based endocytosis signal and a C-terminal dileucine signal (Ward and Moss, 2000). These signals interact with various adapter proteins (AP) for incorporation into clatherin-coated vesicles and intracellular transport between the compartments of the endomembrane system including bi-directional transport between the trans-Golgi network and the plasma membrane (Robinson, 2004). It is not difficult to imagine that this trafficking could target B5-GFP to the proper location for EV incorporation. Although it should be pointed out that these signals were shown to be functional for plasma membrane retrieval of B5-GFP in the absence of infection (Ward and Moss, 2000).
Our results suggest that inclusion into the IEV/EV membrane is an active process and therefore, only select viral membrane proteins get incorporated. Alternatively, incorporation into EV may be passive and that the addition of GFP to B5 actively inhibits its incorporation. This seems unlikely when considering that unmodified A36 requires an interaction with A33 to be targeted to the IEV membrane (Ward, Weisberg, and Moss, 2003; Wolffe, Weisberg, and Moss, 2001). Furthermore, similar to A36, B5 and B5-GFP have been shown to interact with A33 (Perdiguero and Blasco, 2006). It is tempting to speculate that B5-GFP requires an interaction with A33 for inclusion into IEV/EV. However, it was reported, using fluorescence microscopy, that DNA staining particles in infected cells colocalized with B5 in the absence of A33 but not with A33 in the absence of B5 (Perdiguero and Blasco, 2006). It was concluded that A33 requires B5 for incorporation into IEV/EV membranes but B5 does not require A33. This result is in contrast to a similar study in which particles in the cytoplasm of infected cells were stained, albeit infrequently, with both A27 and A33 in the absence of B5 (Rottger et al., 1999). Furthermore, Rottger et al. reported that actin tails were formed, a process that requires A33 for the incorporation of A36, in the absence of B5. Using both immunoprecipitation and immunoelectron microscopy, we confirmed that B5 was incorporated into EV in the absence of A33 but B5-GFP was not. Similarly, we were able to detect F13 on purified EV made in the absence of A33, albeit at reduced levels which supports the conclusion that F13 does not require A33 for incorporation into EV. Indeed, F13 was previously found in EV by immuno-EM of infected cells and by Western blot of purified virions (Roberts et al., 2009; Roper et al., 1998).
For orthopoxviruses, plaque formation is a multistep-process that utilizes no less than six IEV/EV specific proteins. With the exception of A56R, deletion of any individual vaccinia virus IEV/EV specific protein leads to a reduction in plaque size on cell monolayers, but not a complete abrogation. A priori, one could predict that plaque size is directly related to the amount of infectious EV produced. A small plaque phenotype could be due to a decrease in EV production, and/or a decrease in their infectivity. We have quantified the amount of EV produced by vB5R-GFP, vΔA33R, and vB5R-GFP/ΔA33R and their infectivity. The deletion of A33 seems to have only a small effect on the amount of EV produced as measured by genome copy. In contrast, the absence of A33 decreases the infectivity of EEV almost 10 fold. This could account for the reduction in plaque size seen between WR/vB5R-GFP and vΔA33R except that the total amount of infectious EEV released by vΔA33R is 3.5× greater (Fig. 7 and (Roper et al., 1998)). A more likely explanation is the greater reduction of infectious CEV and loss of actin tail formation by vΔA33R. Indeed, the abrogation of actin tail formation has been shown to result in a 3-fold reduction in plaque size (Ward and Moss, 2001a).
After our initial observation that vB5R-GFP/ΔA33R forms plaques that are smaller than the parental virus vΔA33R (Fig. 1), we sought to determine the underlying mechanism. Although a fraction of B5-GFP appears to be mistargeted in the absence of A33 (Figs. 4), this does not appear to have a large effect on EV production. Subsequently, we determined that in the absence of A33, B5-GFP was not incorporated into EV while B5 was. Our data suggest that the difference in plaque size between these two viruses is due to the reduced amounts of infectious EV produced by vB5R-GFP/ΔA33R. Our data also suggest that the presence of B5 does not enhance EEV binding. Therefore, B5 must have a role other than cell binding during infection. One possibility is EV membrane dissolution, which is required to liberate IMV from the EV membrane in subsequent infection (Law et al., 2006; Roberts et al., 2009).
Surprisingly, vB5R-GFP, vΔA33R and vB5R-GFP/ΔA33R released similar amounts of EV. B5 is thought to have a function in IEV formation and egress and therefore B5 either does not require EV incorporation to perform this function, or other viral proteins can compensate for the lost function. In a similar fashion, the small reduction in EV production seen in the absence of A33 could be due to either a small function in EV production/egress, or compensation by other proteins for the lost function. The results presented here support the latter idea. Given the importance of EV for the spread of orthopoxviruses, it seems highly likely that other proteins are, to some extent, able to compensate for missing functions. This idea is supported by the fact that IEV/EV production cannot be completely abrogated by deleting any individual IEV/EV specific gene.
Our results show that the absence of A33 causes a shift of CEV to EEV. Previous reports have attributed CEV retention in part to the putative C-type lectin domain of A34 (Blasco, Sisler, and Moss, 1993). It is possible that the absence of A33 has an indirect effect on A34 and the retention of CEV on the cell surface. Alternatively, a recent paper describing the crystal structure of A33 produced in bacteria indicates that it also has a lectin-like domain that could potentially bind ligands (Su et al., 2009), raising the possibility that A33 is directly involved in the retention of CEV. This would indicate that both A33 and A34 are independently involved in CEV retention. In the absence of A33, there was a substantial number of CEV released by trypsin suggesting that other EV proteins are capable of retaining CEV in the absence of A33, and that A33 and A34 may have a cumulative effect on CEV retention.
Materials and Methods
Cells and viruses
Monolayers of HeLa, BS-C-1, and RK13 cells were maintained as previously described (Ward, 2005). Construction of vB5R-GFP and vΔA33R has been described previously (Roper et al., 1998; Ward and Moss, 2001b). To construct vB5R-GFP/ΔA33R, cells infected with vΔA33R were transfected with pB5R-GFP (Ward and Moss, 2001b). A recombinant virus that has A33R deleted and expresses B5R-GFP was screened and amplified as previously described (Earley, Chan, and Ward, 2008). To construct vB5R/ΔA33R, cells infected with vB5R-GFP/ΔA33R were transfected with a plasmid containing the coding sequence of B5R and 500 bp upstream and downstream regions. Plaques that did not fluoresce green were selected and amplified. The coding sequence of B5-GFP or B5 in each virus was verified by sequencing.
Plaque Assay
The procedure for imaging plaque assays has been described previously (Ward, 2005). Briefly, confluent BS-C-1 monolayers were infected with vB5R-GFP, WR, vΔA33R, vB5R-GFP/ΔA33R, or vB5R/ΔA33R and overlaid with semi-solid media. Two days after infection, phase contrast and fluorescence images of plaques were captured using a fluorescent microscope. Cell monolayers were stained with crystal violet three days after infection and imaged.
Plasmid constructs
Construction of pBMW118 and pB5R-GFP has been described previously (Ward, 2009; Ward and Moss, 2001b). The 500 bp upstream region and the coding sequence of A33R followed by the hemagglutinin (HA) epitope sequence was inserted into pBMW118 to make pA33R-HA-118. The construct was verified by sequencing.
Immunofluorescence microscopy
For in vivo trans-complementation, HeLa cells grown on glass coverslips and infected with either vB5R-GFP or vB5R-GFP/ΔA33R at a multiplicity of infection (MOI) of 1.0 were transfected with either pBMW118 or pA33R-HA-118 at 2 h post infection (PI) using Lipofectamine (Invitrogen) according to the manufacturer's instructions. The following day, cells were fixed, permeabilized, and incubated with an anti-HA MAb (Roche), followed by Cy5-conjugated donkey anti-rat antibody (Jackson ImmunoResearch Laboratories). Stained cells were mounted in Mowiol containing 1 μg per ml of 4′,6-diamidino-2-penylindole dihydrochloride (DAPI) (EM sciences). For subcellular localization, HeLa cells were infected at a MOI of 1.0. The following day, cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100, both in phosphate buffered saline (PBS). The following antibodies were used for staining: anti-B5 MAb (19C2), (Hooper et al., 2000; Schmelz et al., 1994), anti-Calnexin antibody (Stressgen), anti-golgin-97 MAb (Invitrogen, Molecular Probes). For surface staining, infected cells were fixed and incubated with an anti-B5 MAb on ice, followed by incubation with Texas Red-conjugated donkey anti-rat antibody (Jackson ImmunoResearch Laboratories). Stained cells were incubated with 0.5 mg per ml of Hoechst. Cells were imaged using a Leica DMIRB inverted fluorescent microscope connected to a cooled charged-coupled device (Cooke) controlled by Image-Pro Plus software (MeidaCybernetics). Images were colored, minimally processed, and overlaid using Adobe Photoshop (Adobe).
EEV isolation and immunoprecipitation
RK13 cells were infected with vB5R-GFP, WR, vΔA33R, or vB5R-GFP/ΔA33R at a MOI of 10.0. 2 h later, the inoculum was removed and replaced with fresh media. 4 h PI, media was replaced with media containing 25 μCi per ml of [35S]-Met/Cys (Perkin Elmer). 24 h PI, supernatants were collected and spun at 225 × g to remove cell debris. To purify EEV, clarified supernatants were centrifuged through a 36% sucrose cushion at 140,000 × g for 20 min. Purified EEV were lysed in radioimmunoprecipitation assay (RIPA) buffer (0.5× phosphate buffered saline, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1% NP-40, and 0.5% sodium deoxycholate) containing protease inhibitors on ice for 20 min. B5 or F13 was immunoprecipitated from equilibrated lysates with either an anti-B5 or anti-F13 MAb (kindly provided by Jay Hooper) as previously described (Earley, Chan, and Ward, 2008). Bound immune complexes were washed three times in RIPA buffer, resolved on 4-12% Bis-Tris gels (Invitrogen), and detected by autoradiography. Crude equilibrated EEV lysates were analyzed to show that equal amounts of lysates were used in each immunoprecipitation.
Immunoelectron microscopy
RK13 cells were infected with vB5R-GFP, WR, vΔA33R, or vB5R-GFP/ΔA33R at a MOI of 5.0. 2 h PI, inoculum was removed and cells were washed three times with fresh media to remove unbound virus. 48 h PI, EEV were purified through a 36% sucrose cushion as described above. The procedure for purification of IMV has been described previously (Earl and Moss, 1991). To visualize virions by immunoelectron microscopy, purified IMV or EEV were adsorbed to Formvar-coated nickel grids (EM Sciences). Grids were washed in 0.2% glycine in PBS for 5 min, blocked with 1% BSA in PBS for 45 min. Grids were incubated with an anti-B5 MAb and rabbit anti-L1 antiserum (kindly provided by Gary H. Cohen and Roselyn J. Eisenberg) followed by 18 nm colloidal gold-conjugated goat anti-rat and 6 nm colloidal gold-conjugated goat anti-rabbit antibodies (Jackson ImmunoResearch Laboratories). Immunogold-labeled EEV and IMV were negative-stained and visualized using a Hitachi 7650 TEM with a Gatan 11 Megapixel digital camera.
Quantification of EEV and CEV
To determine the amount of EEV and CEV produced by A33-deficient viruses, BS-C-1 cells were infected with vB5R-GFP, vΔA33R, or vB5R-GFP/ΔA33R at a MOI of 10.0 in duplicate. 2 h PI, the inoculum was removed and cell monolayers were washed with phosphate buffered saline to remove unbound virus. 1.0 ml of EMEM containing no serum (EMEM-) was added to each well. 24 h PI, supernatants were collected and centrifuged to remove detached cells. Clarified supernatants were transferred to fresh tubes and the amount of EEV in the culture supernatant was determined by a plaque assay and real time PCR as described below. After EEV containing supernatants were collected, a trypsin release assay was performed on cell monolayers as previously described (Blasco and Moss, 1992). Briefly, cell monolayers were washed with EMEM- twice. 1.0 ml of EMEM- containing 1 μg of trypsin per ml was added to each well and cells were incubated at 37°C for 1 h. After incubation, supernatants were collected and centrifuged to remove detached cells. Clarified supernatants were transferred to fresh tubes and the amount of CEV in the media was determined by a plaque assay and real time PCR as described below. The amounts of infectious EEV and CEV were determined by a plaque assay on BS-C-1 cells. For absolute quantification of EEV and CEV, viral genomes were isolated using Wizard plus minipreps DNA purification system (Promega) following manufacturer's instructions. TaqMan probes and primers specific for E9L were generated. Real time PCR was performed in quadruplicate for each sample using TaqMan Universal PCR Master Mix (Applied Biosystems) following manufacturer's instructions.
Binding assay
BS-C-1 cells were infected with vB5R-GFP, WR, vΔA33R, or vB5R-GFP/ΔA33R at a MOI of 10.0. 24 h PI, supernatants were collected and clarified as described above. The number of viral genomes released by each virus was determined by real time PCR as described above. An equal number of genomes for each virus was bound to BS-C-1 cells grown on poly-D-lysine treated glass coverslips on ice for 1 h. Unbound virions were removed by washing. Cells were fixed with 4% paraformaldehyde overnight, and permeabilized with 0.1% Triton X-100. Cells were stained with an anti-F13 MAb, followed by Texas Red-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch Laboratories). Stained cells were mounted in Mowiol containing DAPI as described above. For quantification, 200 nuclei were imaged and the number of F13-labeled VSPs counted.
Acknowledgments
We thank Bernard Moss for recombinant viruses and plasmids. We thank Jay Hooper for anti-F13 MAb, Gary H. Cohen and Roselyn J. Eisenberg for anti-L1 antisera. We also thank the Electron Microscope Research Core at the University of Rochester Medical Center and Karen Bentley for her help in developing the protocol for the EM study. Parts of this work were funded by National Institute of Allergy and Infectious Diseases grant AI067391 and contract N01-AI-50020. WMC is supported by National Institute of Allergy and Infectious Diseases Molecular Pathogenesis of Bacteria and Viruses Training Grant T32 AI007362.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appleyard G, Hapel AJ, Boulter EA. An antigenic difference between intracellular and extracellular rabbitpox virus. J Gen Virol. 1971;13:9–17. doi: 10.1099/0022-1317-13-1-9. [DOI] [PubMed] [Google Scholar]
- Blasco R, Moss B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000- Dalton outer envelope protein. J Virol. 1991;65(11):5910–5920. doi: 10.1128/jvi.65.11.5910-5920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R, Moss B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol. 1992;66(7):4170–4179. doi: 10.1128/jvi.66.7.4170-4179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R, Sisler JR, Moss B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol. 1993;67(6):3319–3325. doi: 10.1128/jvi.67.6.3319-3325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Honeychurch KM, Yang G, Byrd CM, Harver C, Hruby DE, Jordan R. Vaccinia virus p37 interacts with host proteins associated with LE-derived transport vesicle biogenesis. Virol J. 2009;6:44. doi: 10.1186/1743-422X-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SA, Smith GL. Identification and characterization of an extracellular envelope glycoprotein affecting vaccinia virus egress. J Virol. 1992;66:1610–1621. doi: 10.1128/jvi.66.3.1610-1621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl PL, Moss B. Generation of recombinant vaccinia viruses. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Vol. 2. Greene Publishing Associates & Wiley Interscience; New York: 1991. pp. 16.17.1–16.17.16. [Google Scholar]
- Earley AK, Chan WM, Ward BM. The vaccinia virus B5 protein requires A34 for efficient intracellular trafficking from the endoplasmic reticulum to the site of wrapping and incorporation into progeny virions. J Virol. 2008;82(5):2161–9. doi: 10.1128/JVI.01971-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstad M, Howard ST, Smith GL. A constitutively expressed vaccinia gene encodes a 42-kDa glycoprotein related to complement control factors that forms part of the extracellular virus envelope. Virology. 1992;188:801–810. doi: 10.1016/0042-6822(92)90535-w. [DOI] [PubMed] [Google Scholar]
- Geada MM, Galindo I, Lorenzo MM, Perdiguero B, Blasco R. Movements of vaccinia virus intracellular enveloped virions with GFP tagged to the F13L envelope protein. J Gen Virol. 2001;82(Pt 11):2747–60. doi: 10.1099/0022-1317-82-11-2747. [DOI] [PubMed] [Google Scholar]
- Hiller G, Weber K. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J Virol. 1985;55(3):651–659. doi: 10.1128/jvi.55.3.651-659.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt P, Hiller G, Wittek R. Localization and fine structure of a vaccinia virus gene encoding an envelope antigen. J Virol. 1986;58:757–764. doi: 10.1128/jvi.58.3.757-764.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead M, Rodger G, Van Eijl H, Law M, Hollinshead R, Vaux DJ, Smith GL. Vaccinia virus utilizes microtubules for movement to the cell surface. J Cell Biol. 2001;154(2):389–402. doi: 10.1083/jcb.200104124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JW, Custer DM, Schmaljohn CS, Schmaljohn AL. DNA vaccination with vaccinia virus L1R and A33R genes protects mice against a lethal poxvirus challenge. Virology. 2000;266(2):329–339. doi: 10.1006/viro.1999.0096. [DOI] [PubMed] [Google Scholar]
- Isaacs SN, Wolffe EJ, Payne LG, Moss B. Characterization of a vaccinia virus-encoded 42-kilodalton class I membrane glycoprotein component of the extracellular virus envelope. J Virol. 1992;66(12):7217–7224. doi: 10.1128/jvi.66.12.7217-7224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Carter GC, Roberts KL, Hollinshead M, Smith GL. Ligand-induced and nonfusogenic dissolution of a viral membrane. Proc Natl Acad Sci USA. 2006;103(15):5989–94. doi: 10.1073/pnas.0601025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo MD, Herrera E, Blasco R, Isaacs SN. Functional analysis of vaccinia virus B5R protein: Role of the cytoplasmic tail. Virology. 1998;252(2):450–457. doi: 10.1006/viro.1998.9483. [DOI] [PubMed] [Google Scholar]
- Mathew E, Sanderson CM, Hollinshead M, Smith GL. The extracellular domain of vaccinia virus protein B5R affects plaque phenotype, extracellular enveloped virus release, and intracellular actin tail formation. J Virol. 1998;72:2429–2438. doi: 10.1128/jvi.72.3.2429-2438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Fourth. Vol. 2. Lippincott-Raven Publishers; Philadelphia: 2001. pp. 2849–2883. 2 vols. [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344(1):48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Payne LG. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J Gen Virol. 1980;50(1):89–100. doi: 10.1099/0022-1317-50-1-89. [DOI] [PubMed] [Google Scholar]
- Perdiguero B, Blasco R. Interaction between vaccinia virus extracellular virus envelope A33 and B5 glycoproteins. J Virol. 2006;80(17):8763–77. doi: 10.1128/JVI.00598-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PM, Bommarius B, Lebeis S, McNulty S, Christensen J, Swimm A, Chahroudi A, Chavan R, Feinberg MB, Veach D, Bornmann W, Sherman M, Kalman D. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med. 2005;11(7):731–9. doi: 10.1038/nm1265. [DOI] [PubMed] [Google Scholar]
- Rietdorf J, Ploubidou A, Reckmann I, Holmstrom A, Frischknecht F, Zettl M, Zimmermann T, Way M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat Cell Biol. 2001;3(11):992–1000. doi: 10.1038/ncb1101-992. [DOI] [PubMed] [Google Scholar]
- Roberts KL, Breiman A, Carter GC, Ewles HA, Hollinshead M, Law M, Smith GL. Acidic residues in the membrane-proximal stalk region of vaccinia virus protein B5 are required for glycosaminoglycan-mediated disruption of the extracellular enveloped virus outer membrane. J Gen Virol. 2009;90(Pt 7):1582–91. doi: 10.1099/vir.0.009092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14(4):167–74. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Roper RL, Payne LG, Moss B. Extracellular vaccinia virus envelope glycoprotein encoded by the A33R gene. J Virol. 1996;70(6):3753–62. doi: 10.1128/jvi.70.6.3753-3762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RL, Wolffe EJ, Weisberg A, Moss B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J Virol. 1998;72(5):4192–4204. doi: 10.1128/jvi.72.5.4192-4204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottger S, Frischknecht F, Reckmann I, Smith GL, Way M. Interactions between vaccinia virus IEV membrane proteins and their roles in IEV assembly and actin tail formation. J Virol. 1999;73(4):2863–2875. doi: 10.1128/jvi.73.4.2863-2875.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz M, Sodeik B, Ericsson M, Wolffe EJ, Shida H, Hiller G, Griffiths G. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J Virol. 1994;68(1):130–147. doi: 10.1128/jvi.68.1.130-147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GL, Vanderplasschen A, Law M. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol. 2002;83(Pt 12):2915–31. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- Su HP, Singh K, Gittis AG, Garboczi DN. The structure of the poxvirus A33 protein reveals a dimer of unique C-type lectin-like domains. J Virol. 2009 doi: 10.1128/JVI.02247-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J, Hollinshead M, Reis B, Radsak K, Kern H. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur J Cell Biol. 1993;60(1):163–178. [PubMed] [Google Scholar]
- van Eijl H, Hollinshead M, Rodger G, Zhang WH, Smith GL. The vaccinia virus F12L protein is associated with intracellular enveloped virus particles and is required for their egress to the cell surface. J Gen Virol. 2002;83(Pt 1):195–207. doi: 10.1099/0022-1317-83-1-195. [DOI] [PubMed] [Google Scholar]
- van Eijl H, Hollinshead M, Smith GL. The vaccinia virus A36R protein is a type Ib membrane protein present on intracellular but not extracellular enveloped virus particles. Virology. 2000;271(1):26–36. doi: 10.1006/viro.2000.0260. [DOI] [PubMed] [Google Scholar]
- Ward BM. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J Virol. 2005;79:4755–63. doi: 10.1128/JVI.79.8.4755-4763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM. Using fluorescent proteins to study poxvirus morphogenesis. Methods Mol Biol. 2009;515:1–11. doi: 10.1007/978-1-59745-559-6_1. [DOI] [PubMed] [Google Scholar]
- Ward BM, Moss B. Golgi network targeting and plasma membrane internalization signals in vaccinia virus B5R envelope protein. J Virol. 2000;74(8):3771–3780. doi: 10.1128/jvi.74.8.3771-3780.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Moss B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J Virol. 2001a;75(23):11651–63. doi: 10.1128/JVI.75.23.11651-11663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol. 2001b;75(10):4802–4813. doi: 10.1128/JVI.75.10.4802-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Weisberg AS, Moss B. Mapping and functional analysis of interaction sites within the cytoplasmic domains of the vaccinia virus A33R and A36R envelope proteins. J Virol. 2003;77(7):4113–26. doi: 10.1128/JVI.77.7.4113-4126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe EJ, Weisberg AS, Moss B. The vaccinia virus A33R protein provides a chaperone function for viral membrane localization and tyrosine phosphorylation of the A36R protein. J Virol. 2001;75(1):303–310. doi: 10.1128/JVI.75.1.303-310.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]