

Abstract
R5X4 HIV-1 have impaired utilization of CCR5 on primary CD4+ lymphocytes but the mechanisms responsible are not well defined. Using a panel of diverse R5X4 Envs we identified a spectrum of CCR5 use on CD4+ lymphocytes. Greater lymphocyte CCR5 use correlated with relative resistance to CCR5 mAbs and small molecule antagonists. Increasing CCR5 expression on lymphocytes increased the proportion of entry mediated by CCR5 for all R5X4 isolates except 89.6. In cell lines with regulated CCR5 expression, strains with greater lymphocyte CCR5 use better exploited limiting levels of CCR5. Introduction of an R306S mutation in the 89.6 V3 domain enhanced its utilization of CCR5 at low levels and switched its preference to CCR5 for lymphocyte entry. Thus, the degree to which R5X4 HIV-1 use primary lymphocyte CCR5 is determined by low CCR5 expression coupled with variations in the efficiency of Env-CCR5 interactions, which is in part governed by V3 sequences.
Keywords: R5X4 HIV-1, CD4+ lymphocytes, CCR5, Coreceptor use, Viral entry, HIV-1 tropism
Introduction
HIV-1 coreceptor use is influenced by variation in the interactions between the cellular and viral proteins required for entry into target cells. Viral entry is preceded by a series of structural changes in the viral envelope glycoprotein that begins with binding to CD4, followed by conformational changes that result in coreceptor binding and membrane fusion. CCR5 and CXCR4 are the principal coreceptors used by HIV-1 in vivo. Viruses that use CCR5 (R5 variants) are responsible for nearly all newly transmitted infections whereas strains that use CXCR4 are excluded from transmission and/or establishment of new infections; interestingly, this includes both those that use CXCR4 alone (X4 variants) and those that use CCR5 and CXCR4 (R5X4 variants) (Scarlatti et al., 1997; van’t Wout et al., 1994). R5 strains also predominate during the early and intermediate stages of disease, however, in nearly half of individuals infected with subtype B, CXCR4-using variants arise late in infection and are associated with a precipitous decline in CD4 counts and more rapid disease progression (Connor et al., 1997; Koot et al., 1993; Tersmette et al., 1989). The earliest CXCR4-using viruses typically retain CCR5 use (R5X4 variants), while viruses that are restricted to CXCR4 (X4 variants) may emerge later (Hu et al., 2000; van Rij et al., 2000).
The major targets of HIV-1 infection are CD4+ lymphocytes and macrophages. Previous studies from our lab showed that prototype R5X4 viruses infected monocyte-derived macrophages using CCR5 and CXCR4 (Yi et al., 1999). In CD4+ lymphocytes, however, CCR5-mediated infection by R5X4 HIV-1 was severely impaired despite effective CCR5 use on these cells by R5 viruses and, consequently, R5X4 entry into CD4+ lymphocytes occurred almost exclusively through CXCR4 (Yi, Shaheen, and Collman, 2005). Pathogenesis by R5X4 viruses is also linked to CXCR4 use in several models. Infection and depletion of lymphocytes in human lymphoid tissue infected ex vivo by R5X4 HIV-1 is completely blocked by the CXCR4 antagonist AMD3100, and infection of rhesus macaques with SIV/HIV (SHIV) chimeras carrying R5X4 envelopes induce a disease course similar to infection by X4 SHIVs (Glushakova et al., 1999; Malkevitch et al., 2001; Nishimura et al., 2004). Thus, CXCR4 is the principal entry path used by R5X4 viruses to infect CD4+ lymphocytes whereas CCR5 does not appear to substantially contribute to T cell infection by these viruses.
In contrast to those data, other studies have suggested that lymphocyte CCR5 can be used by some R5X4 strains (Ghezzi et al., 2001; Gray et al., 2006). It is unclear what factors contribute to the variation in lymphocyte CCR5 use by R5X4 viruses, and why many R5X4 viruses are unable to effectively utilize CCR5 as a coreceptor to infect CD4+ lymphocytes. Several studies have suggested that, in general, R5X4 variants tend to differ from R5 strains in a number of factors including their requirements for CCR5 expression levels and the molecular anatomy of CCR5 interactions (Lu et al., 1997; Simmons et al., 2000; Yi, Shaheen, and Collman, 2005; Yi et al., 2003b). Furthermore, the emergence of R5X4 variants in vivo has been associated with increased sensitivity to CCR5 antagonists and reduced ability to infect cells expressing low levels of CCR5 relative to earlier R5 variants (Coetzer et al., 2008). Whether these factors regulate coreceptor utilization on primary lymphocytes, and whether differences among R5X4 strains may exist, is not known.
The goal of this study was to examine the diversity among R5X4 HIV-1 in the efficiency of CCR5 use on lymphocytes, and to identify the mechanisms that regulate its utilization. To do this, we assembled a diverse panel of R5X4 envelope clones, determined the extent to which CCR5 contributed to CD4+ lymphocyte infection by pseudotype reporter viruses, and related CCR5 use on lymphocytes to several measures of envelope-coreceptor interaction. Our findings suggest that the use of lymphocyte CCR5 by R5X4 HIV-1 is associated with the overall efficiency of interaction with CCR5. We also found that increasing CCR5 expression on lymphocytes could overcome the barrier to entry mediated by this coreceptor for most (but not all) R5X4 viruses. Thus, inefficient interactions with CCR5 coupled with low CCR5 expression on CD4+ lymphocytes are responsible for restricting CCR5-mediated infection of T cells by R5X4 HIV-1 isolates.
Results
A spectrum of restricted CCR5-mediated entry into CD4+ lymphocytes exists among R5X4 viruses
In order to examine the diversity in primary cell coreceptor utilization among R5X4 HIV-1, and facilitate the identification of factors that influence primary cell coreceptor use, we assembled a panel of diverse R5X4 env clones derived from divergent sources to study. Luciferase reporter viruses were generated that carried primary isolate R5X4 HIV-1 envelope clones from three different infected individuals (DR, C2, NR), using several individual Envs from within each swarm (Gorry et al., 2002; Gray et al., 2006; Ray et al., 2007). We also studied three widely used prototype R5X4 clones (89.6, DH12 and R3A) which are also primary isolate-derived (Collman et al., 1992; Meissner et al., 2004; Shibata et al., 1995), along with control R5 and X4 Envs Bal and Tybe, respectively (Hwang et al., 1991; Yi et al., 2003a). Of note, 89.6, DH12 and NR envs were derived from late-stage AIDS patients (Collman et al.; 2007; Shibata et al.), which is the most common situation in which these variants are seen. R3A was an R5X4 isolate obtained at the time of seroconversion from an individual infected via intravenous drug use who exhibited rapid disease progression (Meissner et al., 2004; Yu et al., 1998), whereas C2 and DR were isolated from two CCR5-null individuals (Gray et al., 2006). Dual CCR5 and CXCR4 use for each virus was confirmed by infecting U87 cells that expressed CD4 and either CCR5 or CXCR4 (Fig. 1).
Figure 1. Coreceptor use on U87 indicator cells by HIV-1 pseudotype viruses.
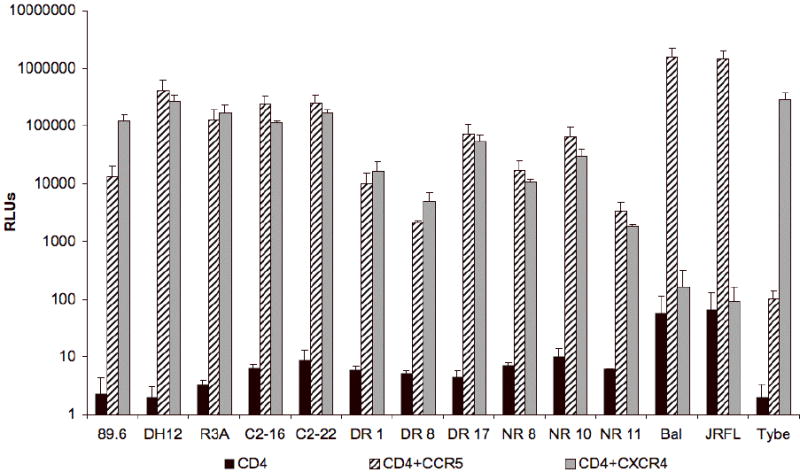
U87 indicator cells expressing only CD4, CD4 and CCR5, or CD4 and CXCR4 were infected with HIV-1 luciferase reporter viruses pseudotyped with primary isolate envelopes (C2, DR, NR), prototype R5X4 envelopes (89.6, DH12 or R3A), R5 (Bal or JRFL) or X4 envelopes (Tybe) as controls. Three days later, infection levels were determined by measuring luciferase activity in cell lysates. The results are expressed in relative light units (RLUs) and represent means +/- standard error (SEM) for two experiments performed in triplicate.
In order to determine coreceptor use by these viruses on primary lymphocytes, CD4+ T cells were infected in the absence or presence of CCR5 or CXCR4 antagonists M657 and AMD3100, respectively (Fig. 2). As expected, Bal was completely inhibited by the CCR5 antagonist M657 and Tybe by the CXCR4 antagonist AMD3100. When CD4+ lymphocytes were infected with R5X4 pseudotypes, a different pattern was evident. All R5X4 strains were markedly inhibited by CXCR4 blockade, whereas CCR5 blocking had no detectable effect. For all R5X4 strains, however, the addition of M657 to AMD3100 further reduced entry levels (Fig. 2a). These results indicate that all R5X4 viruses tested entered CD4+ lymphocytes through both CXCR4 and CCR5, but that infection through CCR5 was markedly less than that through CXCR4, and played little, if any, additional role when both coreceptors were available.
Figure 2. CXCR4 is the principal coreceptor on PHA/IL-2 stimulated CD4+ lymphocytes for R5X4 HIV-1 primary and prototype strains.
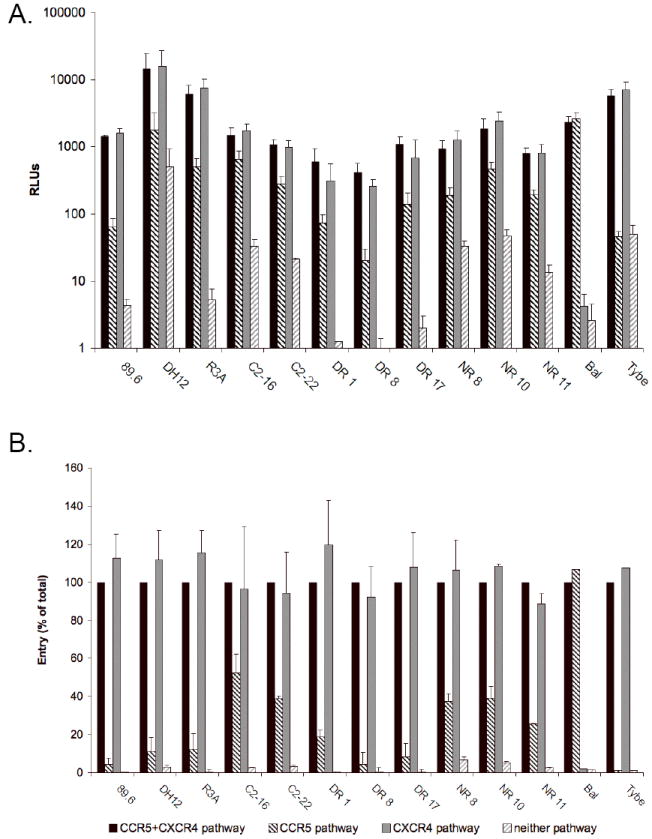
CD4+ T cells were stimulated with PHA for 3 days then treated for 1 hour before infection with or without the CXCR4 blocker AMD3100 (5μg/ml; “CCR5 pathway”), CCR5 blocker M657 (5μM; “CXCR4 pathway”) or both (“neither pathway”). Cells were then infected with HIV-1 luciferase reporter viruses in the continued presence of coreceptor blockers and maintained with IL-2 (10 U/ml). Four days post infection, viral entry was determined by luciferase activity in cell lysates. Results shown for each virus are presented in RLUs (A) and as a percentage of the RLUs seen in the absence of coreceptor blockers (B). Data are means +/- SEM of three experiments using lymphocytes from different donors, each done in duplicate.
To quantitate the relative contribution of each coreceptor to infection of CD4+ lymphocytes, the luciferase levels in the presence of coreceptor antagonists were normalized to the total infection capacity for each virus in untreated cells (Fig. 2b). Blocking CCR5 alone had no inhibitory effect on infection for any of the viruses. However, CXCR4 blocking, when compared to dual coreceptor blocking, revealed CCR5-mediated infection that ranged from ≤10% for the three prototype strains and two of the DR primary isolate Envs, to 50% of untreated levels for the C2-16 primary isolate Env. Thus, CXCR4 is the predominant coreceptor used by R5X4 viruses to infect CD4+ lymphocytes. However, some entry can be mediated by CCR5, and that proportion varies between isolates indicating a spectrum of lymphocyte CCR5 use.
Resistance to inhibition by ECL2-specific CCR5 mAbs and small molecule antagonists correlates with the capacity of R5X4 HIV-1 to use CCR5 for lymphocyte infection
Previous studies comparing CCR5 use by R5 and R5X4 Envs have suggested that R5X4 Envs appear to have a lower affinity for CCR5 than do R5 Envs, are generally more sensitive to inhibition by CCR5 antagonists and are more affected by changes in the structure of CCR5 (Cormier et al., 2000; Doranz et al., 1997; Lu et al., 1997; Yi, Shaheen, and Collman, 2005; Yi et al., 2003b). Since R5 strains by definition use CCR5 efficiently for lymphocyte entry, our finding that R5X4 strains vary in their capacity to infect primary lymphocytes through CCR5 raised the possibility that these strains might also vary in their interaction with CCR5.
To examine the relationship between Env-coreceptor interactions and lymphocyte CCR5 use, we took advantage of CCR5-specific mAbs with well-characterized target epitopes (Blanpain et al., 2002; Lee et al., 1999a). U87/CD4/CCR5 cells were infected in the presence or absence of mAbs that target epitopes in the amino terminus (N-terminal), the second extracellular loop (ECL2) or multiple domains (MD) of CCR5, and entry inhibition was determined by normalizing infection levels to those of untreated cells. As shown in Fig. 3a, both mAbs that targeted CCR5 ECL2 (45531 and MC-1) inhibited entry by the R5X4 viruses. Infection levels were reduced 15-70% by mAb 45531 and 30-90% by MC-1. Treatment with 2D7, another ECL2-directed mAb, completely inhibited infection by all viruses, even at four-fold lower concentrations (data not shown). Importantly, R5X4 Envs that were better able to use lymphocyte CCR5 for entry were blocked less efficiently than those viruses that used the pathway poorly, as shown by the significant correlation between lymphocyte CCR5 use and infection in the presence of ECL2-directed mAbs 45531 and MC-1 (Fig. 3a). In contrast to the ECL2-targeted antibodies, multidomain and N-terminal antibodies (45523 and CTC8, respectively) had marginal or no effect on entry (data not shown).
Figure 3. CCR5 use on CD4+ lymphocytes correlates with resistance to inhibition by ECL2-directed CCR5 mAbs and small molecule antagonists.
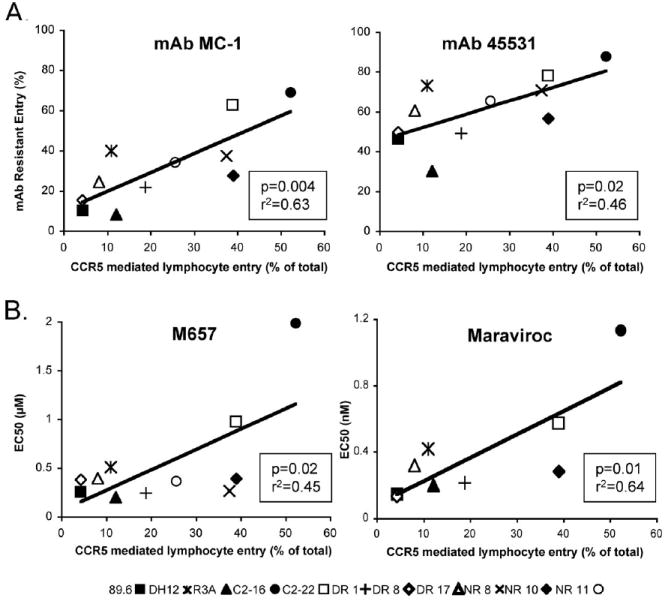
U87/CD4/CCR5 cells were treated for 1 hour with or without 10 μg/ml of CCR5-specific mAbs (A) or increasing concentrations of CCR5 antagonists M657 or Maraviroc (B), then infected with HIV-1 luciferase pseudotype viruses in the continued presence of mAb or blocker. Three days later, infection was determined by luciferase activity in cell lysates and normalized to luciferase activity in untreated cells. Each symbol represents an individual R5X4 virus, and results are presented as the proportion of total lymphocyte entry mediated by CCR5 (from Figure 2b) on the X-axis versus the percentage of U87/CD4/CCR5 infection resistant to inhibition by CCR5 mAbs (A) or the EC50 values for the small molecule CCR5 antagonists (B) on the Y axis. Data are means for three experiments each carried out in duplicate.
We then turned to small molecule CCR5 antagonists, which are frequently used as probes to assess Env-coreceptor interactions. Sensitivity to CCR5 blockers M657 and Maraviroc was determined by infecting U87/CD4/CCR5 cells in the presence of increasing concentrations of inhibitor. Infection by all viruses was extinguished at the highest concentrations of each antagonist (data not shown), but there was variation in sensitivity to both M657 and Maraviroc, exemplified by differences in antagonist EC50 values among the R5X4 Envs (Fig. 3b). M657 and Maraviroc EC50 values were lower for R5X4 Envs with poor lymphocyte CCR5 use, compared to Envs such as C2-16 that are better able to use this coreceptor. Similar to inhibition by ECL2-directed CCR5 mAbs, M657 and Maraviroc EC50 values correlated with lymphocyte CCR5 use. Concordant mAb and antagonist results show a link between lymphocyte CCR5 use and sensitivity to blocking among R5X4 HIV-1, and suggest that the variation in CCR5-mediated infection of lymphocytes results at least in part from differences among these strains in the efficiency of interactions with the coreceptor.
CCR5 upregulation on CD4+ lymphocytes increases the proportion of R5X4 HIV-1 entry mediated by CCR5
These data indicate that CCR5-mediated infection of CD4+ lymphocytes by many R5X4 HIV-1 is restricted (Fig. 2), yet all these viruses can use this coreceptor to readily infect indicator cells such as U87 CD4/CCR5 cells (Fig. 1). CCR5 on indicator cells is typically over-expressed, however, while peripheral blood lymphocytes express CCR5 at low density and on a minority of cells (Lee et al., 1999b; Wu et al., 1997). We therefore sought to test whether low levels of CCR5 expression were an obstacle to efficient CCR5 use on CD4+ lymphocytes by R5X4 viruses, and how CCR5 expression levels regulated pathway-specific R5X4 lymphocyte entry.
To address the role of CCR5 expression, CD4+ lymphocytes were infected with HIV-1 luciferase pseudotype viruses immediately after 3 days of CD3/CD28 costimulation, or after 10 additional days of culture with 300 U/ml of IL-2, which has been shown to upregulate lymphocyte CCR5 (Creson et al., 1999; Yang et al., 2001). Infections were performed in the presence or absence of CCR5 and CXCR4 antagonists to determine proportional use of each entry pathway, and coreceptor expression was measured by FACS at the time of infection. Following CD3/CD28 costimulation, ~1% of CD4+ lymphocytes were CCR5+ as determined by surface staining, whereas 20-40% had detectable CCR5 after prolonged culture. In addition to the increase in percentage of cells within the CCR5+ gate, the mean flourescence intensity (MFI) of cells within the CCR5 gate increased as well (approximately 2-fold). In contrast, the percentage of cells expressing CXCR4 decreased to a modest degree but remained highher after culture with IL-2 (50-70%).
As shown in Fig. 4a, CCR5 use by R5X4 HIV-1 was restricted in day 3 CD3/CD28-stimulated CD4+ lymphocytes, with CXCR4 being the predominant pathway used for entry. However, there was a modest spectrum of CCR5 use similar to that seen in PHA-stimulated lymphocytes (Fig. 2). In contrast, CCR5 upregulation following 10 days of IL-2 stimulation dramatically increased the proportion of infection mediated by CCR5 for all R5X4 viruses tested, except for 89.6 (Fig. 4b). Despite elevated CCR5 expression, CXCR4 remained the predominant coreceptor used for infection of day 13 CD4+ lymphocytes by most R5X4 strains. Exceptions to this coreceptor usage pattern were seen in the two C2 Envs (C2-16 and C2-22) for which CCR5 consistently mediated an equal or greater proportion of entry than did CXCR4 on CCR5-upregulated CD4+ T cells (Fig. 4b). VSV-G pseudotypes, used as a control, showed no change in luciferase expression from day 3 to day 13 cells (data not shown).
Figure 4. CCR5 upregulation on CD4+ lymphocytes increases the proportion of R5X4 infection mediated by CCR5.
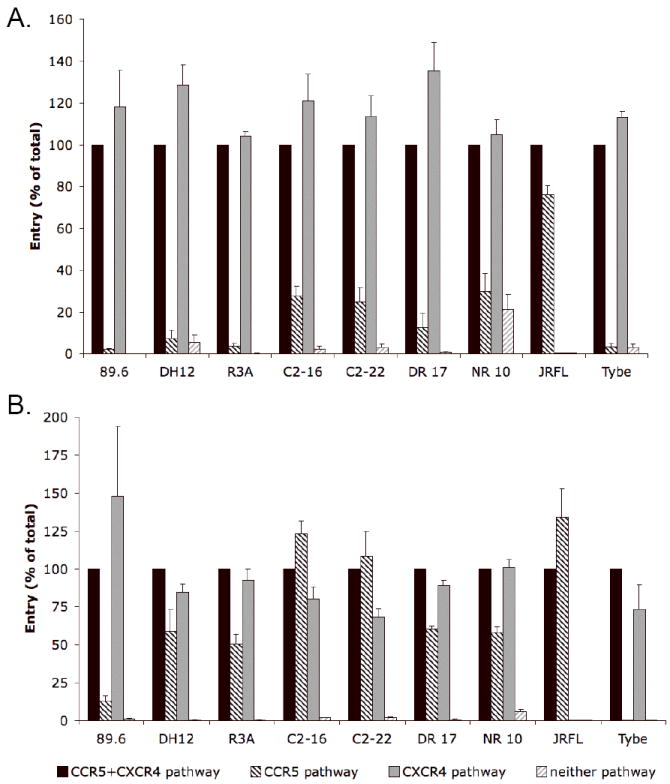
CD4+ lymphocytes were CD3/CD28 costimulated for three days and then infected with HIV-1 luciferase pseudotype viruses (A) or cultured with IL-2 for 10 additional days to upregulate CCR5 expression prior to infection (B). Infections were carried out with or without AMD3100, M657 or both antagonists to block CXCR4 and/or CCR5. Four days later, infection was determined by luciferase activity in cell lysates. Entry was normalized as a percentage of the RLUs seen in the absence of coreceptor blockers for each virus. Data are means +/- SEM of three experiments using lymphocytes from different donors, each done in duplicate
The percentage of CD4+ lymphocytes infected through each coreceptor is modulated following CCR5 upregulation
In addition to total infection levels within the cultures, we also aimed to define the contribution of each pathway to infection of individual CD4+ T lymphocytes following CCR5 upregulation. CD4+ lymphocytes were cultured for 3 or 13 days as described above to establish standard or high CCR5 expression conditions, respectively, and then infected with HIV-1 Env pseudotype virions carrying a GFP reporter gene. Infection-mediated GFP expression therefore enabled per-cell analysis of infection based on GFP expression as determined by FACS. As with luciferase infection, addition of both CCR5 and CXCR4 antagonists completely blocked GFP expression (data not shown).
As shown in Fig. 5a, CCR5-mediated entry into day 3 CD4+ lymphocytes resulted in a low percentage of GFP-positive infected cells. This value ranged from barely detectable (DH12) to ~5% (C2-16 and the R5 Env JRFL). While this result is concordant with the low percentage of cells positive for CCR5 expression by FACS, it was interesting to note that the fraction of GFP+ cells for C2-16 and JRFL often exceeded the proportion that were CCR5+ by FACS. Entry by these viruses was clearly mediated by CCR5, however, since infection was blocked by M657 or Maraviroc (data not shown). This finding indicates that susceptibility to infection by efficient CCR5-using Envs can be a more sensitive indicator of CCR5 expression than immunostaining, and thus these isolates can use CCR5 at levels that are below the threshold of detection by FACS. Furthermore, the fact that some strains can exploit CCR5 to infect what appear to be CCR5-negative CD4+ T cells by FACS indicates that, following upregulation, the apparent increases in percentage of CCR5+ cells also reflects increased CCR5 expression levels by cells that are initially CCR5-positive but at a level below the threshold detectable by flow cytometry.
Figure 5. Stimulation-dependent changes in coreceptor expression affect the percentage of CD4+ lymphocytes infected via each pathway.
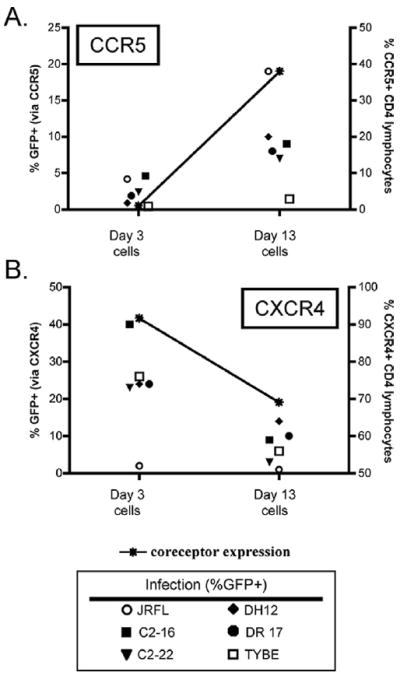
CD4+ lymphocytes were infected with HIV-1 GFP pseudotype viruses after 3 days of CD3/CD28 costimulation or following an additional 10 days of culture with IL-2 to upregulate CCR5. Cells were incubated with coreceptor antagonists for 1 hour prior to and throughout the infection to define the pathway of entry. CCR5 and CXCR4 expression were analyzed by FACS on the day of infection, and four days later, infected cells were determined by FACS analysis for GFP expression. Data indicate the percentage of GFP+ cells infected through CCR5 (A) or CXCR4 (B) at day 3 and day 13 after isolation, and are representative of three independent experiments using cells from different donors. The percentage of cells expressing CCR5 (A) and CXCR4 (B) at the time of infection is indicated by an asterisk (*).
When CCR5 was upregulated on CD4+ lymphocytes by prolonged culture in IL-2 (Fig. 5a, right), the percentage of GFP+ cells infected through CCR5 increased dramatically for the R5 isolate JRFL. Similarly, the percentage of GFP+ cells infected through CCR5 also increased for R5X4 viruses following CCR5 upregulation, although these percentages remained less than that for JRFL. Infection with VSV-G pseudotypes, used as a control for possible effects independent of coreceptor-mediated entry, showed no change in infection (54±6% GFP+ on day 3 cells and 44±8% on day 13 cells; data not shown). Thus, both R5X4 and R5 Envs are highly dependent on CCR5 expression levels, although R5 JRFL is more efficient at exploiting limiting levels than R5X4 Envs (Fig. 5a).
In contrast to CCR5, CXCR4 levels typically decreased somewhat from day 3 to day 13, and this was associated with a modest decrease in the percentage of cells infected via CXCR4 for the R5X4 viruses tested as well as the X4 strain Tybe (Fig. 5b).
Coreceptor-dependent R5X4 entry following lentiviral vector CCR5 over-expression
Although 10 days in culture with IL-2 upregulated CCR5 levels and modulated coreceptor utilization, we considered the possibility that the extended period in vitro could also effect other factors that impact infection despite the lack of changes in VSV-G pseudotype infection. For that reason, we used a second approach to upregulate CCR5. CD4+ lymphocytes were transduced after 3 days of PHA stimulation with a lentiviral vector expressing CCR5 or a mutated version of the coreceptor that is not expressed on the cell surface. CCR5 levels on transduced cells were measured by FACS, and cells were infected with prototype R5X4 Env luciferase viruses in the presence or absence of AMD3100 to block CXCR4. Transduction with the CCR5 vector increased the proportion of CCR5+ cell to 18-70 % of CD4+ lymphocytes compared with 1-19 % of cells transduced with a control vector (Fig. 6), and the MFI of CCR5-positive cells increased as well (fold increase of 2.9±0.9; data not shown). CCR5 over-expression lead to a marked increase in entry mediated by CCR5 for R5X4 pseudotypes R3A and DH12 compared to control vector-transduced cells (Fig. 6). In contrast, entry mediated by CCR5 increased only marginally for strain 89.6. CCR5 transduction had no impact on the MFI of CD4 or CXCR4 expression and did not impact infection by the X4 virus Tybe (data not shown). Results from the lentiviral vector CCR5 over-expression studies are similar to those following stimulation-induced upregulation (Fig. 4), supporting the notion that entry as measured here reflects changes in coreceptor expression and not other features of the cell condition that might modulate infection.
Figure 6. Lentiviral over-expression of CCR5 increases the proportion of R5X4 entry mediated by CCR5 on CD4+ lymphocytes.
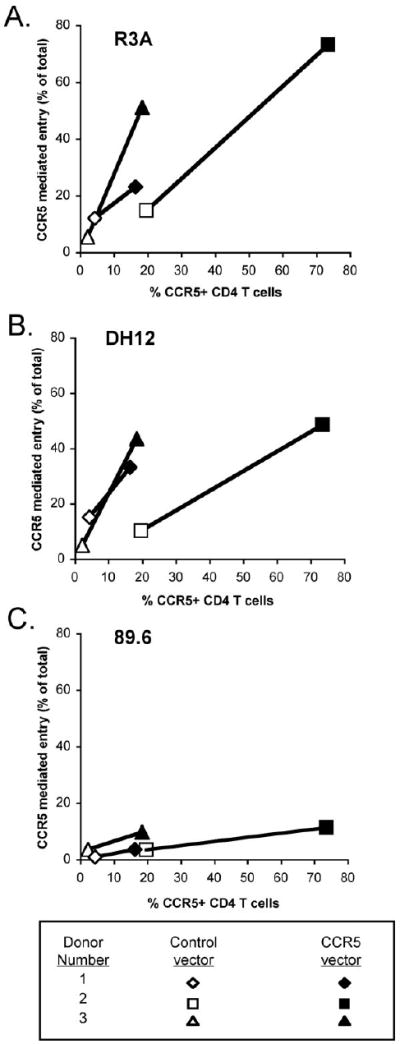
CD4+ lymphocytes were stimulated with PHA for 3 days, transduced with either control or CCR5-expressing lentiviruses, and maintained in the presence of IL-2. Two days post-transduction, cells were pretreated for 1 hour with CXCR4 blocker AMD3100 and infected with HIV-1 luciferase pseudotypes in the continued presence of blocker. Infection was determined by luciferase activity in cell lysates four days later. The results are shown as the proportion of CD4+ lymphocyte infection mediated through CCR5 for R3A (A), DH12 (B) and 89.6 (C) on the Y-axis, plotted against the percentage of CD4+ lymphocytes that express CCR5 after transduction with a control or CCR5 expression vectors. Data are from three independent experiments using cells from different donors.
Spectrum of CCR5 use on CD4+ lymphocytes by R5X4 HIV-1 correlates with the use of low levels of CCR5 on 293 Affinofile cells
Increasing CCR5 expression on CD4+ lymphocytes increased the proportion of infection mediated by CCR5 for R5X4 viruses, suggesting inefficient use of endogenous CCR5 might be linked to sensitivity to coreceptor expression levels. To further address the interplay between CCR5 expression and R5X4 infection, we used 293 Affinofile cells, a cell line in which expression of CD4 and CCR5 can be regulated precisely and independently by minocycline and ponasterone, respectively (Johnston et al., 2009; Lassen et al., 2009). The number of CD4 and CCR5 antibody binding sites (ABS) per cell can be determined for each inducer combination using quantitative FACS analysis. Induction of Affinofile cells resulted in CD4 expression levels that ranged from 3,000 to 200,000 ABS per cell and CCR5 levels from 1,000 to 50,000 ABS per cell. Importantly, these levels encompass the physiologically relevant range of CD4 and CCR5 levels reported on stimulated CD4+ lymphocytes of 65,000-100,000 and 500-7,000 molecules per cell, respectively (Lee et al., 1999b). Therefore, we chose to examine the sensitivity of R5X4 HIV-1 infection to CCR5 expression levels in Affinofile indicator cells by analyzing the effect of varying CCR5 density in the presence of physiologically relevant CD4 expression levels (~83,000 molecules/cell). Cells were maintained with a stable concentration of CD4 inducer and different concentrations of CCR5 inducer, and infected with luciferase pseudotype viruses at the same time that receptor expression levels were confirmed by FACS. Luciferase levels produced at each CCR5 density were normalized to luciferase activity in cells with the highest density of CCR5.
As shown in Fig. 7a, at a stable level of CD4 expression, luciferase virus infection was reduced for all R5X4 viruses as CCR5 density decreased. However, there were marked differences in the response to declining CCR5 levels. R5X4 Envs C2-16 and NR10 were least affected by declining CCR5 expression, showing 60 to 80% of maximal infection even at the lowest CCR5 density. A second group of R5X4 viruses, R3A, DR17 and DH12, were more significantly impaired by low CCR5 expression, achieving 35-45% of maximal infection at the lowest CCR5 expression tested. 89.6 was the R5X4 virus most affected by decreasing CCR5 expression, and at the lowest density of CCR5 expression reached only 25% of its maximal level of infection (Fig. 7a). Thus, R5X4 HIV-1 display a spectrum in their ability to use diminishing levels of CCR5 on Affinofile cells in the presence of CD4 levels approximating those on primary CD4+ T cells.
Figure 7. Efficiency of CCR5 use on Affinofile cells correlates with CCR5 use on CD4+ lymphocytes by R5X4 HIV-1.
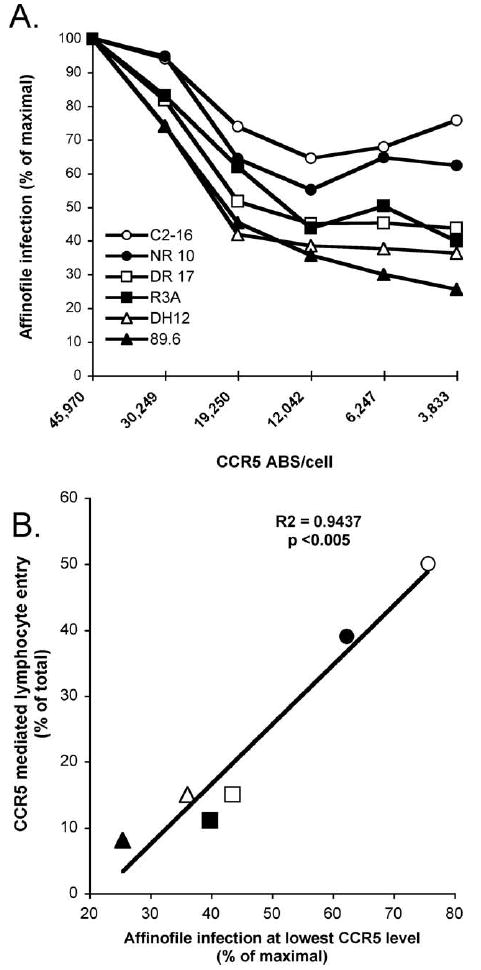
Affinofile cells were induced to express a fixed level of CD4 (83,000 antibody binding sites (ABS)/cell) and varying levels of CCR5, then infected with HIV-1 luciferase pseudotypes in the presence of AMD3100 to block endogenous CXCR4. Infection was quantified 4 days later by luciferase activity in cell lysates. The results are shown as the luciferase levels at each CCR5 density normalized to infection levels on cells expressing the maximum density of CCR5 (A). The relationship between normalized infection of Affinofile cells expressing the lowest density of CCR5 and the proportion of total CD4+ lymphocyte infection that is mediated by CCR5 for each R5X4 Env pseudotype is also shown (B).
We then asked if there was an association between R5X4 use of lymphocyte CCR5 and entry into Affinofile cells expressing CCR5 at the lowest density (Fig. 7a). Overall, there was a strong correlation for R5X4 HIV-1 between the proportion of CD4+ lymphocyte entry mediated by CCR5 and the ability to infect Affinofile cells expressing diminishing levels of CCR5 (Fig. 7b). C2-16 and NR-10 used CCR5 on lymphocytes more efficiently than other R5X4 viruses and were least sensitive to decreasing levels of CCR5 on the Affinofile cells. Lymphocyte CCR5 use was lowest for 89.6 and infection of Affinofile cells was most impaired by decreasing CCR5 levels for this virus. DR17, R3A and DH12 showed an intermediate phenotype in both lymphocyte CCR5 use and infection of Affinofile cells expressing low levels of CCR5. These results confirm that there is biological heterogeneity among R5X4 HIV-1, and suggest that strains with marginal CCR5 use on lymphocytes are unable to effectively scavenge for CCR5 when it is expressed at low density.
Relationship between V3 sequences and CD4+ lymphocyte coreceptor preference
The V3 region is a major determinant of coreceptor use, and position-specific scoring matrix (PSSM) algorithms based on variations in this domain are frequently used to predict viral phenotype. Two widely used algorithims are based on viruses phenotyped by syncytia induction in CXCR4/CD4+ cell lines (SI/NSI PSSM) or on coreceptor use in indicator cells (X4/R5 PSSM), which are highly related although not completely concordant features (Jensen et al., 2003). However, such algorithms are typically less able to identify dual-tropic R5X4 than single coreceptor R5 and X4 variants, nor have they been applied to selective use of primary cell coreceptors. Therefore, we applied the PSSM algorithms to V3 sequences from Envs in our panel to determine whether primary CD4+ T cell coreceptor usage among R5X4 HIV-1 might be associated with sequences in V3 (Table 1). Strikingly, we found that, for these R5X4 variants, prediction of an NSI phenotype based on V3 sequence was associated with relatively more efficient use lymphocyte CCR5. Conversely, R5X4 viruses with more restricted entry through lymphocyte CCR5 were predicted to be SI. Thus, while unable to identify these R5X4 strains as a group, the SI/NSI algorithm appears to discriminate among R5X4 strains and predict relative efficiency of lymphocyte CCR5 use based on V3 determinants. In contrast, there was no link between primary lymphocyte coreceptor usage and predicted coreceptor phenotype based on V3 sequences using the X4/R5 coreceptor prediction algorithm. Interestingly, most of these R5X4 Envs were predicted to be R5 by the X4/R5 PSSM matrix, which differed from the SI/NSI algorithm results. Thus, while the V3-based PSSM is an incomplete predictor of coreceptor use for R5X4 strains, it does appear to predict the relative coreceptor preference on lymphocytes seen in this study. This result also suggests that elements within V3 contribute to regulation of coreceptor use by R5X4 viruses on lymphocytes.
Table 1.
V3 sequence alignment and PSSM viral phenotype predictiona
| Strain | V3 sequence | CD4+ PBL %CCR5 useb | SI/NSIc | X4R5d |
|---|---|---|---|---|
| Bal | CTRPNNNTRKSIHIGPGRALYTTGEIIGDIRQAHC | 100 | 0 | 0 |
| C2-16 | ............TM....VY.............. | 52 | 0 | 0 |
| C2-22 | ............TM....VY............... | 39 | 0 | 0 |
| NR10 | ............A.....SF.A..R.......... | 39 | 0 | 0 |
| NR8 | ............A.....SF.A..R.......... | 35 | 0 | 0 |
| NR11 | ............A.....SF.A..R.......... | 26 | 0 | 0 |
| DH12 | ..........G.TL....VF......V....K... | 12 | 1 | 0 |
| R3A | ....G.....RVTL....VY....Q......K... | 11 | 1 | 1 |
| DR1 | .I.........MTL...KVF....-VT....K... | 15 | 1 | 0 |
| DR17 | .I.........MTL...KVF....-VT....K... | 8 | 1 | 0 |
| DR8 | .I.........MTL...KVF....-VT....K... | 4 | 1 | 0 |
| 89.6 | .........RRLS......F.ARRN.......... | 4 | 1 | 1 |
Position specific scoring matrix (Jensen et al., 2003)
Percentage of total primary lymphocyte entry in the presence of CXCR4 blocking
Predicted SI phenotype based on SI/NSI PSSM algorithm; 0=NSI, 1=SI
Predicted coreceptor phenotype based on X4R5 PSSM algorithm; 0=R5, 1=X4 or R5X4
Impaired CCR5 use on CD4+ lymphocytes by 89.6 is reversed by an R306S mutation within the V3 region that enhances CCR5 interactions
While CCR5 use on CD4+ lymphocytes by R5X4 envelopes was bolstered by increasing CCR5 expression, this measure had little effect on lymphocyte CCR5 use by 89.6. Therefore, for this virus we wished to address the Env side of the Env-CCR5 interaction, by determining if specific changes within V3 might alter the efficiency of CCR5 use and affect lymphocyte entry. The presence of positively charged amino acids at positions 11 and 24 or 25 within the V3 loop is associated with CXCR4 use (Cardozo et al., 2007; Xiao et al., 1998), and 89.6 exhibits a positively charged arginine in both the 11 and 24 positions. In a recent study of brain derived R5X4 envelopes, the efficiency of CCR5 and CXCR4 use on primary cells was linked to the presence of a serine or arginine, respectively, at position 11 of V3, corresponding to residue 306 of the Env glycoprotein (Gray et al., 2009). Another study attempted to derive variants of 89.6 resistant to CXCR4 inhibitors on cell lines expressing both coreceptors and identified a change from arginine to serine at the same residue (Maeda, Yusa, and Harada, 2008).
We therefore speculated that the amino acid at position 306 within Env might be involved in determining the efficiency of 89.6 CCR5 interactions, and that it might thus regulate the ability or inability of 89.6 to use CCR5 on lymphocytes. To test this notion, R306 in 89.6 was mutated to serine (89.6 R306S; Fig. 8a). In contrast to 89.6, the R5X4 Env with greatest lymphocyte CCR5 use, C2-16, has a serine at position 11 in V3. Therefore, we also mutated the serine at this position in C2-16 to arginine in order to assess the converse effect (C2-16 S306R) (Fig. 8a). Both Envs remained R5X4 in U87/CD4/coreceptor cells (data not shown).
Figure 8. Arginine at position 11 within the V3 region influences the efficiency of CCR5 use by 89.6 and coreceptor preference on CD4+ lymphocytes.
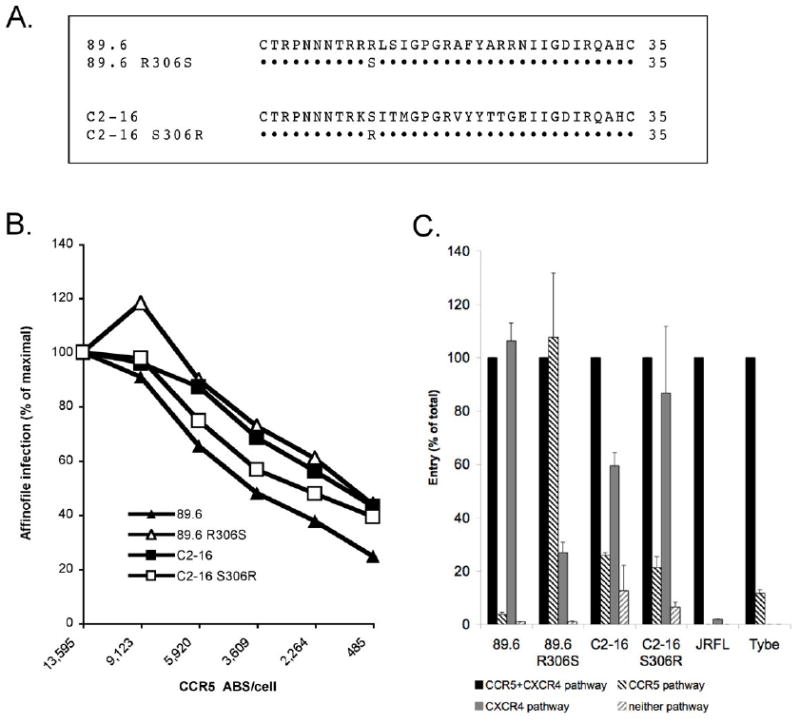
The amino acids of envelope V3 regions of 89.6 and C2-16 are shown, along with the 89.6 R306S and C2-16 S306R mutations, which correspond to position 11 of the V3 domain (A). Affinofile cells expressing a fixed level of CD4 and differing levels of CCR5 were infected with wild-type and mutant 89.6 and C2-16 luciferase pseudotype viruses. The results are shown as the luciferase levels at each CCR5 density normalized to infection levels on cells expressing the maximum density of CCR5 (B). PHA stimulated CD4+ lymphocytes were infected with wild-type and mutant 89.6 and C2-16 luciferase pseudotype viruses in the presence or absence of AMD3100, Maraviroc or a combination of both inhibitors. The results are presented as the RLUs measured from cell lysates 4 days after infection (C).
We infected 293 Affinofiles expressing a fixed level of CD4 and varying levels of CCR5 to test the effect of the Env mutations on the efficiency of CCR5 use in that system. As noted earlier, infection by 89.6 decreased markedly as CCR5 density on Affinofiles dropped from maximal to minimal levels. Replacing the arginine at position 306 with serine resulted in a two-fold enhancement of entry at the lowest CCR5 levels, reaching a relative efficiency of entry at low compared to high CCR5 approximately that for C2-16 (Fig. 8b). In contrast, the C2-16 S306R mutation had some effect on entry efficiency at intermediate CCR5 levels, but not at the lowest CCR5 density; which most closely mimics those on primary CD4+ lymphocytes (Fig. 8b).
Next, coreceptor use on primary CD4+ T cells by wild type and mutant 89.6 and C2-16 was determined by infecting CD4+ lymphocytes with luciferase viruses bearing each envelope in the absence or presence of coreceptor antagonists. As shown in Fig. 8c, converting arginine to serine at position 306 in Env dramatically enhanced use of lymphocyte CCR5 for 89.6. In fact, 89.6 R306S showed a marked preference for infecting CD4+ lymphocytes using CCR5, in contrast to wild type 89.6, which was essentially restricted to CXCR4 for entry into these cells. Unlike 89.6, CCR5 use on CD4+ lymphocytes by strain C2-16 was not affected by the converse S306R mutation (Fig. 8c).
Thus, replacing the positively charged arginine with serine at residue 306 in the 89.6 Env increased the efficiency of CCR5 use, and resulted in preferential infection of CD4+ lymphocytes through CCR5, reversing its otherwise strict dependence on CXCR4 in those cells. However, the amino acid at this position is not a critical determinant for all R5X4 viruses since converting serine to arginine at that residue did not notably impact the efficiency of CCR5 use or coreceptor preference on primary lymphocytes by the C2-16 Env, indicating that other context-dependent factors are involved as well.
Discussion
Coreceptor use and target cell tropism are important determinants of HIV-1 transmission and pathogenesis. In previous studies of primary cell tropism and coreceptor use by prototype R5X4 HIV-1 strains, we found that these viruses could use CCR5 and CXCR4 to infect macrophages, but in CD4+ lymphocytes, CXCR4 was the predominant entry pathway while CCR5 was used poorly, if at all. Here we sought to determine, employing a variety of dual-tropic isolates from disparate sources that would represent a broad spectrum of phenotypes, whether and how R5X4 strains might vary in their ability to use CCR5 on lymphocytes, and what factors were responsible for determining use of this pathway. We found limited CCR5-mediated infection of CD4+ T cells by the R5X4 strains tested, which did not substantially contribute to overall infection levels when CXCR4 was available. We did find, however, that a spectrum existed among the R5X4 variants in the extent to which lymphocyte CCR5 could be used if CXCR4 was not available. We also found that lymphocyte CCR5 use by R5X4 HIV-1 was restricted primarily by the low CCR5 expression levels on CD4+ T cells combined with inefficient interactions between the R5X4 envelopes and CCR5. Thus, we conclude that the spectrum of lymphocyte CCR5 use by R5X4 viruses is determined by the efficiency of Env-CCR5 interactions.
Even for R5X4 viruses with relatively greater lymphocyte CCR5 use, blocking this pathway had no impact on CD4+ T cell infection, as entry through CXCR4 alone was equivalent to infection when both coreceptors were available. CXCR4 is expressed on the majority of peripheral blood CD4+ lymphocytes, whereas CCR5 is expressed on a smaller subset of cells (Lee et al., 1999b; Ostrowski et al., 1998; van Rij et al., 2000; Wu et al., 1997). Most cells that are CCR5+ also express CXCR4; so blocking CCR5 may have little net effect on infection because entry can still occur through CXCR4. Conversely, since many CXCR4+ lymphocytes do not express CCR5, entry in the presence of CXCR4 blockers should be representative of the subset of cells that express CCR5. Nevertheless, the level of CCR5-mediated lymphocyte infection by R5X4 viruses varied and was determined by the efficiency of CCR5 use, as indicated by lower sensitivity to CCR5 blocking agents and a better ability to use low levels of CCR5 on indicator cells. The density of CCR5 expressed on individual CD4+ lymphocytes varies within the CCR5+ population (Reynes et al., 2000) and, thus, while potential CCR5-mediated infection is determined by the proportion of cells that express CCR5, actual CCR5 use by a specific R5X4 virus is also regulated by the threshold of CCR5 it requires for infection. Thus, the differences in CCR5/Env interactions identified among the R5X4 strains here, which regulate the threshold of CCR5 required for entry, is the principal factor underlying differences in CCR5-mediated target cell availability for each isolate.
Our studies suggest that the basis for differential lymphocyte CCR5 use among R5X4 variants is due to the efficiency with which Env engages CCR5. Multiple steps are involved in coreceptor-dependent entry and infection, including Env-chemokine receptor binding, which is influenced by the amino acids within the V3 loop (Cormier et al., 2001; Platt et al., 2001; Sakaida et al., 1998; Wu et al., 1996). While the presence of arginine at position 11 within the V3 loop of 89.6 was associated with inefficient CCR5 use and poor CCR5-mediated primary lymphocyte infection, this amino acid in the context of the C2-16 region had no impact on CCR5 use. Characterization of R5X4 clones from subtype D strains on dual coreceptor expressing cell lines revealed that more efficient CCR5 use was associated with V3 sequences that were more “R5 like”, while “X4 like” V3 sequences were linked to less efficient CCR5 use (Huang et al., 2007). The 89.6 V3 region has hallmarks of CXCR4 use such as a high net positive charge and basic amino acids at both positions 11 and 24, determinants that are lacking in C2-16, and CCR5 use by 89.6 and C2-16 are similar to the subtype D Envs with more “X4 like” or “R5 like” V3 sequences, respectively. This result is consistent with recent studies indicating a critical role for this region within the V3 loop in determining coreceptor utilization by other R5X4 isolates (Gray et al., 2009; Nolan, Jordan, and Hoxie, 2008), although our data indicate that amino acid 11 is not a critical determinant for all R5X4 isolates. Thus, our findings suggest that different R5X4 viruses interact with this coreceptor in distinct ways. Of note, this residue was also shown to regulate the relative dependence of R5X4 HIV-1 Envs on specific regions within CCR5 and CXCR4, particularly the N-terminal domain (Gray et al., 2009), suggesting that in addition to overall efficiency, it can also affect the manner in which Env-coreceptor interactions occur.
Extending the role of V3, we also evaluated the relationship to coreceptor use predicted by PSSM algorithms, which have been developed to take into account multiple V3 sequence factors in order to predict viral phenotype (Jensen et al., 2003). PSSM predictive algorithms are not highly accurate for distinguishing R5X4 from single coreceptor-tropic viruses, but we found that among R5X4 variants the SI/NSI matrix showed a strong correlation between “NSI-like” characteristics and greater lymphocyte CCR5 use, and “SI-like” and poorer lymphocyte CCR5 use. In contrast, the X4/R5 predictive matrix, based on coreceptor use in indicator cell lines, did not distinguish between R5X4 strains with greater or lesser lymphocyte CCR5 use. These results further support the role of V3 in determining coreceptor preference in the context of primary cells and, in addition, further emphasize the limitations of indicator cells in predicting primary cell coreceptor use. Future studies will be needed to characterize the strains for which phenotype predictions differ between the scoring matrices, and determine whether these Envs may exhibit other distinguishing features in their interactions with primary cells.
The R5X4 strains in this report exhibited a spectrum in their efficiency of lymphocyte CCR5 use, but none preferentially infected CD4+ T cells using this coreceptor. On one hand, this is not surprising given the difference in number of cells positive for each coreceptor. On the other hand, R5X4 strains emerge in vivo from R5 strains in memory lymphocytes that express CCR5 and CXCR4, while later in infection, R5X4 along with X4 viruses are found mostly in the CCR5-/CXCR4+ naïve subset (van Rij et al., 2000). Thus, it seems likely that some R5X4 strains would use CCR5 as the predominant coreceptor to infect CD4+ T cells. The fact that the R306S mutation in 89.6 changes coreceptor preference on lymphocytes from CXCR4 to CCR5 indicates that this type of R5X4 Env can exist and that only minor changes may be required to shift between these two phenotypes. Perhaps R5X4 viruses that predominately use CCR5 to infect lymphocytes emerge but then rapidly disappear from the viral quasispecies. Alternatively, evolution of R5X4 strains might never transit through a phase were CCR5 use is dominant. In a study of in vitro-derived R5X4 switch variants, the earliest CXCR4-using Envs were more sensitive to CCR5 antagonists and less efficient at using CCR5 for entry when compared to the parental R5 virus (Pastore, Ramos, and Mosier, 2004), and the mutations conferring CXCR4 use were detrimental except when introduced with compensatory changes elsewhere in the envelope (Pastore et al., 2006). In vivo, R5 strains showed reduced ability to infect CCR5-low cells prior to the emergence of R5X4 viruses (Coetzer et al., 2008). Thus, CXCR4 use may only emerge after compensatory mutations occur that have the effect of decreasing CCR5 use, suggesting that R5X4 evolution may not involve a phase of predominate lymphocyte CCR5 use.
R5 viruses are responsible for the majority of new HIV-1 infections, but it is still not clear why only this type of strain is able to transmit or successfully establish new infections. R5 strains are typically more macrophage-tropic than X4 variants, and CCR5-dependent macrophage tropism was classically proposed as a possible reason (Liu et al., 1996; van’t Wout et al., 1994; Zhu et al.). However, R5X4 strains also use CCR5 to infect primary macrophages, yet this strain is rarely transmitted (Liu et al., 1996; van’t Wout et al., 1994; Yi, Shaheen, and Collman, 2005). Furthermore, recent studies using transmission pair isolates obtained near the time of transmission enabling donor/recipient comparison, or PCR amplification and phylogenetic reconstruction of the successfully transmitted virus, reveal that these founder viruses are restricted to CCR5 and readily infect lymphocytes but typically infect macrophages inefficiently and/or no better than the donor variants (Isaacman-Beck et al., 2009; Keele et al., 2008; Salazar-Gonzalez et al., 2009). The fact that R5X4 variants generally use lymphocyte CCR5 poorly relative to R5 viruses raises the possibility that these strains do not establish infections in recipients because use of CCR5 on lymphocytes is the critical determinant of transmission. Alternatively, it is possible that neither R5X4 nor X4 strains efficiently transmit because CXCR4 use is selected against during transmission or establishment of new infections in recipients (Cornelissen et al., 1995).
The limited CCR5-mediated infection of lymphocytes by R5X4 HIV-1 raise questions of whether CCR5 use by R5X4 viruses has any impact on viral pathogenesis. Although rare, R5X4 strains have been transmitted and these viruses were able to establish infection in recipients (Meissner et al., 2004; Yu et al., 1998). Given the importance of CCR5 use in transmission, these viruses might be expected to utilize lymphocyte CCR5 more efficiently than R5X4 viruses that arise later. Strain R3A was obtained from an unusual R5X4 acute infection (Meissner et al., 2004), but R3A was not among the R5X4 Envs with more efficient lymphocyte CCR5 use. It is interesting to note, and somewhat unexpected, that the two variants with greatest relative efficiency in lymphocyte CCR5 use were NR10 from a late stage patient and C2-16 from a CCR5-null subject, neither of which are situations in which one might expect more efficient lymphocyte CCR5 use. Macrophage infection is thought to be the major source of virus in tissue, a critical component of neuropathogenesis, and responsible for sustaining virus replication at very late stages of disease (Igarashi et al., 2001; Koenig et al., 1986; Schuitemaker et al., 1992). Prototype and some primary R5X4 strains readily infect macrophages using CCR5, which may be explained by the higher density of CCR5 expressed on these cells compared with primary CD4+ lymphocytes (Lee et al., 1999b; Yi, Shaheen, and Collman, 2005). However, use of this coreceptor may be dispensable since CXCR4 also serves as a viable coreceptor for R5X4 entry into these cells (Yi, Shaheen, and Collman, 2005). Thus, it remains to be determined how the efficiency of lymphocyte CCR5 use among R5X4 variants is linked to specific aspects of pathogenesis. In addition, while X4 use is common in subtype B HIV-1, it is less frequent among subtype C strains and, furthermore, the factors that regulate coreceptor interactions may differ for different subtypes (Lynch et al., 2009), so further studies will be needed to determine the extent to which these findings apply to other subtypes.
In summary, this study confirms with a panel of primary and prototype R5X4 HIV-1 variants that these strains are highly skewed towards CXCR4 use for entry into CD4+ T cells. However, there does exist a range among the isolates in the relative ability to use CCR5 on primary lymphocytes, although we did not identify an evident relationship between relative lymphocyte coreceptor use and the clinical context in which the R5X4 Envs were derived. The ability to use lymphocyte CCR5 by R5X4 variants was linked to greater efficiency in CCR5 interactions, based on antibody and small molecule antagonist blocking studies. Furthermore, the barrier to CCR5 use could be overcome by CCR5 over-expression in lymphocytes for most of the R5X4 isolates or, if not, by mutating Env to enhance coreceptor interaction efficiency. These results indicate that efficiency of the Env-CCR5 interactions combined with low lymphocyte CCR5 expression together determine the extent of restriction to R5X4 entry through this pathway, although additional obstacles to primary cell coreceptor use may still exist. Better understanding of the viral and cellular factors that control coreceptor use by HIV-1 strains is critical to understanding the impact of viral evolution on target cell tropism.
Materials and Methods
Cells
CD4+ lymphocytes were isolated by negative selection from whole blood (Stem Cell Technologies, Vancouver, BC, Canada). Purified lymphocytes were maintained at 106 cells/ml and stimulated for 3 days with 5 μg/ml of phytohemagglutinin (PHA; MP Biomedical, Solon, OH) or in 24 well plates coated with 5 μg/ml of anti-CD3 (OKT3; a gift of M. Betts, U. of Pa.) and anti-CD28 (clone 28.2; Beckman Coulter, Fullerton, CA) antibodies. Cells were then either infected and maintained thereafter in IL-2, or cultured for 10 days in IL-2 (300 U/ml; Proleukin, Novartis, Basel, Switzerland) to upregulate CCR5 prior to infection. 293 Affinofile cells that express CCR5 and CD4 under independent regulation were maintained in blasticidin, G418, hygromycin and zeocin (Invitrogen, Carlsbad, CA) (Johnston et al., 2009; Lassen et al., 2009). U87/CD4, U87/CD4/CXCR4 and U87/CD4/CCR5 cells were obtained from the NIH AIDS Research and Reference Reagent Program (Björndal et al., 1997) and were maintained in selective media containing 1 μg/ml of puromycin (MP Biomedical, Solon, OH) or 300 μg/ml of G418 (Invitrogen, Carlsbad, CA) for selection of CD4 or CCR5 and CXCR4 respectively.
HIV-1 Env clones
HIV-1 R5X4 prototypes 89.6 and DH12 and primary isolate NR Env clones were derived from patients with advanced disease and have been previously described (Collman et al.; Ray et al.; Shibata et al.). Env clone R3A was derived from an acute seroconverter found to harbor R5X4 variants and has been described previously (Meissner et al.; Yu et al.). R5X4 variants C2 and DR were obtained from two HIV-infected CCR5-null individuals homozygous for the Δ32 allele (Gray et al.). The X4 isolate Tybe and R5 strains JRFL and Bal have also been described previously (Gartner et al., 1986; Koyanagi et al.; Yi et al.). Prototype 89.6, R3A, JRFL and Tybe Envs along with NR Envs were subcloned into the expression vector pCAGGS (Niwa, Yamamura, and Miyazaki, 1991) using standard methods. The expression vectors used to subclone the remaining vectors have been previously described (Helseth et al., 1990; Kim et al., 2001). 89.6 and C2-16 envelope clones with mutations at position 11 within the V3 loop were generated using a Quick Change XL mutagenesis kit (Stratagene, La Jolla, CA). NR Env clones were a gift from R. Doms (U. of Pa.), R3A was obtained from J. Hoxie (U. of Pa.) and kindly provided by L. Su (UNC-Chapel Hill), and JRFL, DH12 and Bal Env clones were generously provided by M. Cho (Case Western Reserve).
Pseudotype Virus Production
HIV-1 Env luciferase pseudotype viruses were generated by cotransfecting a plasmid that expressed HIV-1 structural proteins (pCMVΔP1ΔenvpA), a plasmid that expressed the packaged luciferase reporter (pHIV-1 luc), and an HIV-1 or VSV-G Env plasmid. Plasmids were cotransfected into 293 cells using Fugene transfection reagent (Roche, Palo Alto, CA) at ratios of 1:3:1 as previously described (Sterjovski et al., 2007; Yang et al., 2004). GFP reporter viruses were created by cotransfection of structural gene plasmid pHp1, GFP reporter plasmid pHRET-GFP, the tat expression plasmid pCep4-tat, and a plasmid expressing an HIV-1 or VSV-G Env. Plasmids were cotransfected into 293 cells at a ratio of 10:10:1:10 as previously described (Chang et al., 1999). The CCR5 lentiviral expression vector pNL-CCR5 was created from pNL-CD4 by digestion with NotI and XhoI to remove CD4, and the CCR5 gene was amplified (sense primer 5’-TAG TGC TGT TAA CTT GCT CAA TGC-3’ and antisense 5’-GAT CAA GGA TAT CTT GTC TTC-3’) and subcloned into the NotI and XhoI sites. CCR5 transduction vectors were produced by cotransfecting pRev, pNL-CCR5 and pVSV-G into 293 cells at a ratio of 1:2:1.
HIV-1 Env pseudotype viruses and lentiviral expression vectors were harvested 48 hours after transfection, clarified by centrifugation at 250xg, then stored in 5% sucrose at -80°C until use. Plasmids pCMVΔP1ΔenvpA (Parolin et al., 1996) and pHIV-1-luc (Yang et al., 2004) were generously provided by J. Sodroski (Harvard University), and pNL-CD4 (Tokunaga et al., 2001) was kindly provided by B. Cullen (Duke University). Plasmid pHRET-GFP (Lin et al., 2002) was provided by P. Corbeau (Hôpital Saint Eloi) and pHp1 and pCep4-tat (Chang et al., 1999) were a gift from J. Zucali (University of Florida) obtained through the NIH AIDS Research and Reference Reagent Program.
CCR5 over-expression in CD4+ lymphocytes
PHA-stimulated CD4+ lymphocytes were pelleted (250×g at 25°C for 5 minutes) and resuspended at 2×106 cells per ml in media containing 8 μg/ml of Polybrene. Cells were then transduced using 200 μl of CCR5 expression vector pNL-CCR5, spin inoculated for 2 hours as described (O’Doherty, Swiggard, and Malim, 2000), then incubated in the presence of 10 U/ml of IL-2 at 37°C and 5% CO2 for 72 hours. After this period, coreceptor expression was analyzed by FACS and cells were used for infection.
Infection of cell lines and CD4+ lymphocytes
U87 cells were plated in 96 well plates at 1.5×104 cells per well one day prior to infection, infected by spin inoculation at 1200×g for 2 hours with HIV-1 pseudotype viruses using 5 ng of p24 antigen per virus, and cultured at 37°C and 5% CO2 for three days. For blocking studies, U87/CD4/CCR5 cells were pretreated for 1 hour and then infected in the presence of 10 μg/ml of CCR5 mAb or serial dilutions of the CCR5 small molecule antagonists. Luciferase activity was measured by lysing cells in PBS containing 0.1% Triton X-100, combining cell lysate 1:1 with luciferase assay substrate (Luciferase Assay System; Promega, Madison, WI) and measuring luciferase relative light units (RLUs) using a Dynex technologies microtiter plate luminometer. Monoclonal antibodies 2D7, 45531 and CTC8 were obtained from the NIH AIDS Research and Reference Reagent Program, and mAb MC-1 (Blanpain et al., 2002) was a gift from M. Mack (University of Munich). The CCR5 blocker M657 (Finke, 2000) was a gift from M. Miller (Merck, W. Point, P.A.), and Maraviroc (Dorr et al., 2005) (Pfizer Inc., New York City, NY) was obtained through the NIH AIDS Research and Reference Reagent Program.
For infection of primary cells, PHA or CD3/CD28-stimulated CD4+ lymphocytes were added to 96 well plates at 2×105 cells per well. Cells were pretreated for 1 hour with saturating concentrations of CCR5 antagonists M657 (5 μg/ml) or Maraviroc (2 μM), the CXCR4 antagonist AMD3100 (5μg/ml; Sigma-Aldrich, St. Louis, MO) or a combination of CCR5 and CXCR4 blockers. Following pretreatment, CD4+lymphocytes were infected in the presence of the inhibitors using an equal volume or 10 ng of p24 antigen of each virus for PHA or CD3/CD28 stimulated cells, respectively. CD4+ lymphocytes were infected by spin inoculation at 1200×g for 2 hours, and then cultured at 37°C and 5% CO2 in the presence of IL-2 for 3 days. Infection was measured by luciferase activity in the cell lysate as previously described, or by FACS analysis of GFP expression.
Infection of 293 Affinofiles has been described in detail previously (Johnston et al., 2009; Lassen et al., 2009). Briefly, cells were plated at 1.5×104 cells per well in 96 well plates in media containing 2% dialyzed FBS. Cell were allowed to adhere for two days, then media was removed and replaced with media containing a fixed concentration of the CD4-inducing reagent Minocycline (0.625 ng/ml) and varying concentrations of the CCR5 inducer Ponasterone (0-2μM). After an overnight incubation, cells were pre-treated for at least 1 hour with 1 μg/ml of AMD3100 to block CXCR4 in media containing 10% dialyzed FBS, then infected by spin inoculation using 1 ng of p24 antigen of each luciferase pseudotype virus. Infected cells were cultured for 4 days at 37°C and 5% CO2, then luciferase activity was measured as described. FACS analysis of CCR5 and CD4 expression was carried out the day of infection on cells that were plated in 6 well plates and maintained in an identical manner.
FACS analysis of CD4, CCR5, CXCR4 and GFP expression
CD4+ lymphocytes were pelleted (250×g for 5 minutes), washed with FACS buffer (PBS containing 1% fetal bovine serum and 0.1% NaN3), resuspended in 50 μl of FACS buffer and stained with 1 μl of mAbs to CD4 (clone RPAT-4- Fluorescein isothiocyanate [FITC]-conjugated), CCR5 (clone 2D7-phycoerythrin [PE]-conjugated), and CXCR4 (clone 12G-PE-Cy5-conjugated) (all from BD Biosciences, San Jose, CA). Fluorescence minus one controls as well as cells stained with single antibodies were carried out in parallel. Cells were incubated at room temperature in the dark for 30 minutes then washed and resuspended in FACS buffer at 106 cells per ml. Cells were analyzed by a FACS Calibur flow cytometer (BD) using Cell Quest (BD) and FloJo software (Tree Star Inc, Ashland, OR).
For CD4 and CCR5 quantification on 293 Affinofiles, cells were detached by incubation for 10 minutes in PBS containing 2mM EDTA, pelleted, washed in FACS buffer then stained with the CCR5 PE mAb or with CD4 PE mAb (clone S3.5-Invitrogen, Carlsbad, CA) as described above. CD4 and CCR5 binding sites were quantified using FloJo Software by comparing the average mean fluorescence intensity of stained cells to a standard curve created using PE-labeled beads with four distinct, defined quantities of fluorophores (QuantiBrite Beads, BD) as described (Davis et al., 1998). For detection of GFP expression after CD4+ T cell infection by HIV-1 pseudotypes, cells were washed and resuspended in FACS buffer as described 3 days post-infection and analyzed for GFP fluorescence using the FACS Calibur flow cytometer and FloJo software.
Statistical analysis and PSSM matrix scoring
Statistical analysis was performed using GraphPad Prism 4 software. The EC50 values for M657 and Maraviroc were determined using a sigmoidal dose response equation. Correlations were derived by linear regression analysis, and correlation coefficients and p-values were determined using a 2-tailed Pearson test.
The predicted viral phenotype of R5X4 Envs used in this study was determined using the position specific scoring matrix (PSSM) program (http://indra.mullins.microbiol.washington.edu/webpssm/). The PSSM algorithm compares amino acids at each position within the V3 of submitted envelopes to the corresponding residues from a database of Envs experimentally analyzed for coreceptor use (R5/X4 matrix) or viral isolates tested for T cell line synctium inducing capacity (SI/NSI matrix) as previously described (Jensen et al., 2003).
Acknowledgments
We thank M. Mack for mAb MC-1 and M. Miller for M657. We also thank R. Doms, J. Hoxie, L. Su and M. Cho for HIV-1 Envs. We are grateful to J. Sodroski, J. Zucali and B. Cullen for various constructs and vectors. We also acknowledge valuable support for this work from the Immunology and the Viral/Molecular Cores of the Penn Center for AIDS Research. This work was supported by NIH grant R01 AI035502 (R.G.C.). L.L. was supported in part by NIH T32 AI007632 and P.R.G. is the recipient of an Australian National Health and Medical Research Council Level 2 Biomedical Career Development Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Björndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman DR, Fenyö EM. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol. 1997;71(10):7478–87. doi: 10.1128/jvi.71.10.7478-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Vanderwinden JM, Cihak J, Wittamer V, Le Poul E, Issafras H, Stangassinger M, Vassart G, Marullo S, Schlndorff D, Parmentier M, Mack M. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol Biol Cell. 2002;13(2):723–37. doi: 10.1091/mbc.01-03-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, Kimura T, Philpott S, Weiser B, Burger H, Zolla-Pazner S. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res Hum Retroviruses. 2007;23(3):415–26. doi: 10.1089/aid.2006.0130. [DOI] [PubMed] [Google Scholar]
- Chang LJ, Urlacher V, Iwakuma T, Cui Y, Zucali J. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 1999;6(5):715–28. doi: 10.1038/sj.gt.3300895. [DOI] [PubMed] [Google Scholar]
- Coetzer M, Nedellec R, Salkowitz J, McLaughlin S, Liu Y, Heath L, Mullins JI, Mosier DE. Evolution of CCR5 use before and during coreceptor switching. J Virol. 2008;82(23):11758–66. doi: 10.1128/JVI.01141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman R, Balliet JW, Gregory SA, Friedman H, Kolson DL, Nathanson N, Srinivasan A. An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. J Virol. 1992;66(12):7517–21. doi: 10.1128/jvi.66.12.7517-7521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med. 1997;185(4):621–8. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier EG, Persuh M, Thompson DA, Lin SW, Sakmar TP, Olson WC, Dragic T. Specific interaction of CCR5 amino-terminal domain peptides containing sulfotyrosines with HIV-1 envelope glycoprotein gp120. Proc Natl Acad Sci USA. 2000;97(11):5762–7. doi: 10.1073/pnas.97.11.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier EG, Tran DN, Yukhayeva L, Olson WC, Dragic T. Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J Virol. 2001;75(12):5541–9. doi: 10.1128/JVI.75.12.5541-5549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen M, Mulder-Kampinga GA, Veenstra J, Zorgdrager F, Kuiken CL, Hartman S, Dekker J, van der Hoek L, Sol C, Coutinho RA. Syncytium-inducing (SI) phenotype suppression at seroconversion after intramuscular inoculation of a non-syncytium-inducing/SI phenotypically mixed human immunodeficiency virus population. J Virol. 1995;69(3):1810–8. doi: 10.1128/jvi.69.3.1810-1818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creson JR, Lin AA, Li Q, Broad DF, Roberts MR, Anderson SJ. The mode and duration of anti-CD28 costimulation determine resistance to infection by macrophage-tropic strains of human immunodeficiency virus type 1 in vitro. J Virol. 1999;73(11):9337–47. doi: 10.1128/jvi.73.11.9337-9347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KA, Abrams B, Iyer SB, Hoffman RA, Bishop JE. Determination of CD4 antigen density on cells: role of antibody valency, avidity, clones, and conjugation. Cytometry. 1998;33(2):197–205. doi: 10.1002/(sici)1097-0320(19981001)33:2<197::aid-cyto14>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Doranz BJ, Lu ZH, Rucker J, Zhang TY, Sharron M, Cen YH, Wang ZX, Guo HH, Du JG, Accavitti MA, Doms RW, Peiper SC. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J Virol. 1997;71(9):6305–14. doi: 10.1128/jvi.71.9.6305-6314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721–32. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke PE, Caldwell C, Dorn C, Meurer L, Oates B, Chen P, MacCoss M, Mills S, Malkowitz S, Sicilano S, Springer M, DeMartino J, Gould S, Chen Q, Braun J, Hajdu R, Kwei G, Carella A, Carver G, Holmes K, Schleif W, Danzeisen R, Hazuda D, Kessler J, Lineberger J, Miller M, Emini E. 10th National Conference of the Inflammation Research Association; Hot Springs, Va. 2000. [Google Scholar]
- Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986;233(4760):215–9. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- Ghezzi S, Menzo S, Brambilla A, Bordignon PP, Lorini AL, Clementi M, Poli G, Vicenzi E. Inhibition of R5X4 Dualtropic HIV-1 Primary Isolates by Single Chemokine Co-receptor Ligands. Virology. 2001;280(2):253–261. doi: 10.1006/viro.2000.0753. [DOI] [PubMed] [Google Scholar]
- Glushakova S, Yi Y, Grivel JC, Singh A, Schols D. Preferential coreceptor utilization and cytopathicity by dual-tropic HIV-1 in human lymphoid tissue …. Journal of Clinical Investigation. 1999 doi: 10.1172/JCI7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry PR, Zhang C, Wu S, Kunstman K, Trachtenberg E, Phair J, Wolinsky S, Gabuzda D. Persistence of dual-tropic HIV-1 in an individual homozygous for the CCR5Δ32 allele. The Lancet. 2002 doi: 10.1016/S0140-6736(02)08681-6. [DOI] [PubMed] [Google Scholar]
- Gray L, Churchill MJ, Keane N, Sterjovski J, Ellett AM, Purcell DF, Poumbourios P, Kol C, Wang B, Saksena NK, Wesselingh SL, Price P, French M, Gabuzda D, Gorry PR. Genetic and functional analysis of R5X4 human immunodeficiency virus type 1 envelope glycoproteins derived from two individuals homozygous for the CCR5delta32 allele. J Virol. 2006;80(7):3684–91. doi: 10.1128/JVI.80.7.3684-3691.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray L, Roche M, Churchill MJ, Sterjovski J, Ellett A, Poumbourios P, Sheffief S, Wang B, Saksena N, Purcell DF, Wesselingh S, Cunningham AL, Brew BJ, Gabuzda D, Gorry PR. Tissue-specific sequence alterations in the human immunodeficiency virus type 1 envelope favoring CCR5 usage contribute to persistence of dual-tropic virus in the brain. J Virol. 2009;83(11):5430–41. doi: 10.1128/JVI.02648-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helseth E, Kowalski M, Gabuzda D, Olshevsky U, Haseltine W, Sodroski J. Rapid complementation assays measuring replicative potential of human immunodeficiency virus type 1 envelope glycoprotein mutants. J Virol. 1990;64(5):2416–20. doi: 10.1128/jvi.64.5.2416-2420.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu QX, Barry AP, Wang ZX, Connolly SM, Peiper SC, Greenberg ML. Evolution of the human immunodeficiency virus type 1 envelope during infection reveals molecular corollaries of specificity for coreceptor utilization and AIDS pathogenesis. J Virol. 2000;74(24):11858–72. doi: 10.1128/jvi.74.24.11858-11872.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Eshleman SH, Toma J, Fransen S, Stawiski E, Paxinos EE, Whitcomb JM, Young AM, Donnell D, Mmiro F, Musoke P, Guay LA, Jackson JB, Parkin NT, Petropoulos CJ. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J Virol. 2007;81(15):7885–93. doi: 10.1128/JVI.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253(5015):71–4. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, Hirsch V, Martin MA. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc Natl Acad Sci USA. 2001;98(2):658–63. doi: 10.1073/pnas.021551798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacman-Beck J, Hermann EA, Yi Y, Ratcliffe SJ, Mulenga J, Allen S, Hunter E, Derdeyn CA, Collman RG. Heterosexual transmission of human immunodeficiency virus type 1 subtype C: Macrophage tropism, alternative coreceptor use, and the molecular anatomy of CCR5 utilization. J Virol. 2009;83(16):8208–20. doi: 10.1128/JVI.00296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MA, Li FS, van ’t Wout AB, Nickle DC, Shriner D, He HX, McLaughlin S, Shankarappa R, Margolick JB, Mullins JI. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J Virol. 2003;77(24):13376–88. doi: 10.1128/JVI.77.24.13376-13388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SH, Lobritz MA, Nguyen S, Lassen K, Delair S, Posta F, Bryson YJ, Arts EJ, Chou T, Lee B. A quantitative affinity-profiling system that reveals distinct CD4/CCR5 usage patterns amongst HIV-1 and SIV strains. J Virol. 2009 doi: 10.1128/JVI.01242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105(21):7552–7. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YB, Lee MK, Han DP, Cho MW. Development of a safe and rapid neutralization assay using murine leukemia virus pseudotyped with HIV type 1 envelope glycoprotein lacking the cytoplasmic domain. AIDS Res Hum Retroviruses. 2001;17(18):1715–24. doi: 10.1089/08892220152741414. [DOI] [PubMed] [Google Scholar]
- Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–93. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- Koot M, Keet IP, Vos AH, de Goede RE, Roos MT, Coutinho RA, Miedema F, Schellekens PT, Tersmette M. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann Intern Med. 1993;118(9):681–8. doi: 10.7326/0003-4819-118-9-199305010-00004. [DOI] [PubMed] [Google Scholar]
- Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science. 1987;236(4803):819–22. doi: 10.1126/science.3646751. [DOI] [PubMed] [Google Scholar]
- Lassen K, Lobritz M, Bailey J, Johnston S, Nguyen S, Lee B, Chou T, Siliciano R, Markowitz M, Arts E. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog. 2009;5(4):e1000377. doi: 10.1371/journal.ppat.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Sharron M, Blanpain C, Doranz BJ, Vakili J, Setoh P, Berg E, Liu G, Guy HR, Durell SR, Parmentier M, Chang CN, Price K, Tsang M, Doms RW. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J Biol Chem. 1999a;274(14):9617–26. doi: 10.1074/jbc.274.14.9617. [DOI] [PubMed] [Google Scholar]
- Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci USA. 1999b;96(9):5215–20. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YL, Mettling C, Portales P, Reynes J, Clot J, Corbeau P. Cell surface CCR5 density determines the postentry efficiency of R5 HIV-1 infection. Proc Natl Acad Sci U S A. 2002;99(24):15590–5. doi: 10.1073/pnas.242134499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86(3):367–77. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Lu Z, Berson JF, Chen Y, Turner JD, Zhang T, Sharron M, Jenks MH, Wang ZX, Kim J, Rucker J, Hoxie JA, Peiper SC, Doms RW. Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc Natl Acad Sci USA. 1997;94(12):6426–31. doi: 10.1073/pnas.94.12.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RM, Shen T, Gnanakaran S, Derdeyn CA. Appreciating HIV type 1 diversity: subtype differences in Env. AIDS Res Hum Retroviruses. 2009;25(3):237–48. doi: 10.1089/aid.2008.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Yusa K, Harada S. Altered sensitivity of an R5X4 HIV-1 strain 89.6 to coreceptor inhibitors by a single amino acid substitution in the V3 region of gp120. Antiviral Research. 2008;77(2):128–35. doi: 10.1016/j.antiviral.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Malkevitch N, McDermott DH, Yi Y, Grivel JC, Schols D, De Clercq E, Murphy PM, Glushakova S, Collman RG, Margolis L. Coreceptor choice and T cell depletion by R5, X4, and R5X4 HIV-1 variants in CCR5-deficient (CCR5delta32) and normal human lymphoid tissue. Virology. 2001;281(2):239–47. doi: 10.1006/viro.2000.0807. [DOI] [PubMed] [Google Scholar]
- Meissner EG, Duus KM, Gao F, Yu XF, Su L. Characterization of a thymus-tropic HIV-1 isolate from a rapid progressor: role of the envelope. Virology. 2004;328(1):74–88. doi: 10.1016/j.virol.2004.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Igarashi T, Donau OK, Buckler-White A, Buckler C, Lafont BA, Goeken RM, Goldstein S, Hirsch VM, Martin MA. Highly pathogenic SHIVs and SIVs target different CD4+ T cell subsets in rhesus monkeys, explaining their divergent clinical courses. Proc Natl Acad Sci U S A. 2004;101(33):12324–9. doi: 10.1073/pnas.0404620101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Nolan KM, Jordan AP, Hoxie JA. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor tropism and sensitivity to entry inhibitors. J Virol. 2008;82(2):664–73. doi: 10.1128/JVI.01793-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74(21):10074–80. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski MA, Justement SJ, Catanzaro A, Hallahan CA, Ehler LA, Mizell SB, Kumar PN, Mican JA, Chun TW, Fauci AS. Expression of chemokine receptors CXCR4 and CCR5 in HIV-1-infected and uninfected individuals. J Immunol. 1998;161(6):3195–201. [PubMed] [Google Scholar]
- Parolin C, Taddeo B, Palu G, Sodroski J. Use of cis- and trans-acting viral regulatory sequences to improve expression of human immunodeficiency virus vectors in human lymphocytes. Virology. 1996;222(2):415–22. doi: 10.1006/viro.1996.0438. [DOI] [PubMed] [Google Scholar]
- Pastore C, Nedellec R, Ramos A, Pontow S, Ratner L, Mosier DE. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J Virol. 2006;80(2):750–8. doi: 10.1128/JVI.80.2.750-758.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore C, Ramos A, Mosier DE. Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J Virol. 2004;78(14):7565–74. doi: 10.1128/JVI.78.14.7565-7574.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Kuhmann SE, Rose PP, Kabat D. Adaptive mutations in the V3 loop of gp120 enhance fusogenicity of human immunodeficiency virus type 1 and enable use of a CCR5 coreceptor that lacks the amino-terminal sulfated region. J Virol. 2001;75(24):12266–78. doi: 10.1128/JVI.75.24.12266-12278.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray N, Harrison JE, Blackburn LA, Martin JN, Deeks SG, Doms RW. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J Virol. 2007;81(7):3240–50. doi: 10.1128/JVI.02413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynes J, Portales P, Segondy M, Baillat V, Andre P, Reant B, Avinens O, Couderc G, Benkirane M, Clot J, Eliaou JF, Corbeau P. CD4+ T cell surface CCR5 density as a determining factor of virus load in persons infected with human immunodeficiency virus type 1. J Infect Dis. 2000;181(3):927–32. doi: 10.1086/315315. [DOI] [PubMed] [Google Scholar]
- Sakaida H, Hori T, Yonezawa A, Sato A, Isaka Y, Yoshie O, Hattori T, Uchiyama T. T-tropic human immunodeficiency virus type 1 (HIV-1)-derived V3 loop peptides directly bind to CXCR-4 and inhibit T-tropic HIV-1 infection. J Virol. 1998;72(12):9763–70. doi: 10.1128/jvi.72.12.9763-9770.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. 2009;206(6):1273–89. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlatti G, Tresoldi E, Björndal A, Fredriksson R, Colognesi C, Deng HK, Malnati MS, Plebani A, Siccardi AG, Littman DR, Fenyö EM, Lusso P. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nature Medicine. 1997;3(11):1259–65. doi: 10.1038/nm1197-1259. [DOI] [PubMed] [Google Scholar]
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, Lange JM, Schattenkerk JK, Miedema F, Tersmette M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol. 1992;66(3):1354–60. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Hoggan MD, Broscius C, Englund G, Theodore TS, Buckler-White A, Arthur LO, Israel Z, Schultz A, Lane HC. Isolation and characterization of a syncytium-inducing, macrophage/T-cell line-tropic human immunodeficiency virus type 1 isolate that readily infects chimpanzee cells in vitro and in vivo. J Virol. 1995;69(7):4453–62. doi: 10.1128/jvi.69.7.4453-4462.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Reeves JD, Hibbitts S, Stine JT, Gray PW, Proudfoot AE, Clapham PR. Co-receptor use by HIV and inhibition of HIV infection by chemokine receptor ligands. Immunol Rev. 2000;177:112–26. doi: 10.1034/j.1600-065x.2000.17719.x. [DOI] [PubMed] [Google Scholar]
- Sterjovski J, Churchill MJ, Ellett A, Gray LR, Roche MJ, Dunfee RL, Purcell DF, Saksena N, Wang B, Sonza S, Wesselingh SL, Karlsson I, Fenyo EM, Gabuzda D, Cunningham AL, Gorry PR. Asn 362 in gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with AIDS. Retrovirology. 2007;4:89. doi: 10.1186/1742-4690-4-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersmette M, Gruters RA, de Wolf F, de Goede RE, Lange JM, Schellekens PT, Goudsmit J, Huisman HG, Miedema F. Evidence for a role of virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J Virol. 1989;63(5):2118–25. doi: 10.1128/jvi.63.5.2118-2125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga K, Greenberg ML, Morse MA, Cumming RI, Lyerly HK, Cullen BR. Molecular basis for cell tropism of CXCR4-dependent human immunodeficiency virus type 1 isolates. J Virol. 2001;75(15):6776–85. doi: 10.1128/JVI.75.15.6776-6785.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rij RP, Blaak H, Visser JA, Brouwer M, Rientsma R, Broersen S, de Roda Husman AM, Schuitemaker H. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and syncytium-inducing HIV-1. J Clin Invest. 2000;106(8):1039–52. doi: 10.1172/JCI7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van’t Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-van Lent N, Scherpbier HJ, Veenstra J, Boer K, Coutinho RA, Miedema F, Schuitemaker H. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J Clin Invest. 1994;94(5):2060–7. doi: 10.1172/JCI117560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384(6605):179–83. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- Wu L, Paxton WA, Kassam N, Ruffing N, Rottman JB, Sullivan N, Choe H, Sodroski JG, Newman W, Koup RA, Mackay CR. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185(9):1681–91. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Owen SM, Goldman I, Lal AA, deJong JJ, Goudsmit J, Lal RB. CCR5 coreceptor usage of non-syncytium-inducing primary HIV-1 is independent of phylogenetically distinct global HIV-1 isolates: delineation of consensus motif in the V3 domain that predicts CCR-5 usage. Virology. 1998;240(1):83–92. doi: 10.1006/viro.1997.8924. [DOI] [PubMed] [Google Scholar]
- Yang X, Tomov V, Kurteva S, Wang L, Ren X, Gorny MK, Zolla-Pazner S, Sodroski JG. Characterization of the outer domain of the gp120 glycoprotein from human immunodeficiency virus type 1. J Virol. 2004;78(23):12975–86. doi: 10.1128/JVI.78.23.12975-12986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YF, Tomura M, Iwasaki M, Mukai T, Gao P, Ono S, Zou JP, Shearer GM, Fujiwara H, Hamaoka T. IL-12 as well as IL-2 upregulates CCR5 expression on T cell receptor-triggered human CD4+ and CD8+ T cells. J Clin Immunol. 2001;21(2):116–25. doi: 10.1023/a:1011059906777. [DOI] [PubMed] [Google Scholar]
- Yi Y, Chen W, Frank I, Cutilli J, Singh A, Starr-Spires L, Sulcove J, Kolson DL, Collman RG. An unusual syncytia-inducing human immunodeficiency virus type 1 primary isolate from the central nervous system that is restricted to CXCR4, replicates efficiently in macrophages, and induces neuronal apoptosis. J Neurovirol. 2003a;9(4):432–41. doi: 10.1080/13550280390218706. [DOI] [PubMed] [Google Scholar]
- Yi Y, Isaacs SN, Williams DA, Frank I, Schols D, De Clercq E, Kolson DL, Collman RG. Role of CXCR4 in cell-cell fusion and infection of monocyte-derived macrophages by primary human immunodeficiency virus type 1 (HIV-1) strains: two distinct mechanisms of HIV-1 dual tropism. J Virol. 1999;73(9):7117–25. doi: 10.1128/jvi.73.9.7117-7125.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y, Shaheen F, Collman RG. Preferential use of CXCR4 by R5X4 human immunodeficiency virus type 1 isolates for infection of primary lymphocytes. J Virol. 2005;79(3):1480–6. doi: 10.1128/JVI.79.3.1480-1486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y, Singh A, Shaheen F, Louden A, Lee C, Collman RG. Contrasting use of CCR5 structural determinants by R5 and R5X4 variants within a human immunodeficiency virus type 1 primary isolate quasispecies. J Virol. 2003b;77(22):12057–66. doi: 10.1128/JVI.77.22.12057-12066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XF, Wang Z, Vlahov D, Markham RB, Farzadegan H, Margolick JB. Infection with dual-tropic human immunodeficiency virus type 1 variants associated with rapid total T cell decline and disease progression in injection drug users. J Infect Dis. 1998;178(2):388–96. doi: 10.1086/515646. [DOI] [PubMed] [Google Scholar]
- Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science. 1993;261(5125):1179–81. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]