

Abstract
We introduce several novel visualization and interaction paradigms for visual analysis of published protein-protein interaction networks, canonical signaling pathway models, and quantitative proteomic data. We evaluate them anecdotally with domain scientists to demonstrate their ability to accelerate the proteomic analysis process. Our results suggest that structuring protein interaction networks around canonical signaling pathway models, exploring pathways globally and locally at the same time, and driving the analysis primarily by the experimental data, all accelerate the understanding of protein pathways. Concrete proteomic discoveries within T-cells, mast cells, and the insulin signaling pathway validate the findings. The aim of the paper is to introduce novel protein network visualization paradigms and anecdotally assess the opportunity of incorporating them into established proteomic applications. We also make available a prototype implementation of our methods, to be used and evaluated by the proteomic community.
Index Terms: Biological (genome or protein) databases, Data and knowledge visualization, Graphs and networks, Interactive data exploration and discovery, Visualization techniques and methodologies
1 Introduction
Proteins within a cell interact with one another in order to regulate the cell's activity. The nature of these interactions is diverse. Among others, an external event can be transmitted to the inside of a cell through interactions of signaling molecules; a protein binds to another protein to alter its function; or a protein will carry another protein to a specific cell location.
A cascade of protein interactions peculiar to a specific cell, stimulation, or cellular outcome is called a signaling pathway. An in-depth understanding of these pathways will, among other outcomes, let researchers discover efficient drugs that can influence a cells behavior without causing unwanted side-effects.
Experimental data is an important component that researchers use to understand how signaling pathways function. For instance, researchers can artificially stimulate a cell and measure how the proteins within it respond, possibly over a series of timepoints. To efficiently interpret the results of such experiments, they need to be collated with existing knowledge that can explain some of the observations and provide valuable insights for hypothesis generation. One of the most common such data used in signaling pathway analysis are protein-protein interactions extracted from proteomic publications and stored in online databases.
Advances in proteomic experimental techniques and improved analytical methods have enabled researchers to produce vast quantities of experimental data. Combining it with the sheer complexity of protein interaction networks increases the information space even more. Thus, thinking about the data at its original low level has become impractical. New computational techniques are required that either extract relevant information automatically or let researchers process data faster by looking at condensed visual representations.
This necessity has been acknowledged by the research community and analysis frameworks that build on traditional graph drawing to visualize protein interaction networks have emerged. However, findings presented in this paper, as well as results from more recent work, suggest that additional research is needed to ensure that the visualization methods employed are adequate for proteomic research.
Here we present a design study on several novel visual and interaction paradigms for the analysis of quantitative proteomic data, canonical signaling pathway models, and protein interaction networks along with the proteomic analysis requirements that motivated them. We evaluate our methods anecdotally with domain experts to determine their overall ability to accelerate the proteomic discovery process.
The methods we describe are general and discussed in terms of their benefits as components of established protein networks analysis applications such as Cytoscape. However, for concrete exemplification, we will occasionally frame them in the context of the testbed application used to develop and evaluate them. This prototype is available for download and testing on the projects website at: http://graphics.cs.brown.edu/research/sciviz/proteins/home.htm.
Figure 1 illustrates the main visualization and interaction paradigms presented in the paper: harnessing the researchers existing mental schema and intuition by integrating dynamic interaction data into static but familiar signaling pathway images provided by the user; enabling proteomic specific interaction level analysis of dense networks by integrating a novel Focus+Context technique; and driving exploration by comparative analysis of multiple experimental datasets.
Fig. 1.
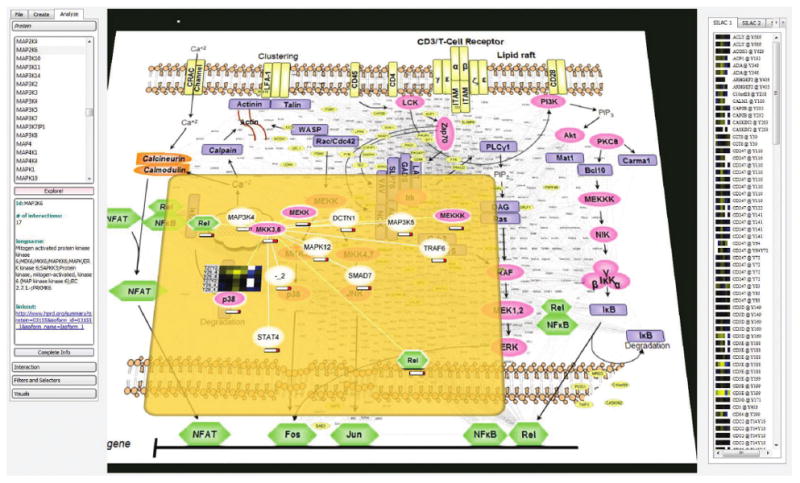
Analysis of a protein interaction network in the context of the T-cell pathway. Proteins and interactions dynamically extracted from the HPRD database (small fonts scattered between the protein icons in the pathway view) are integrated directly into an imported image of a canonical signaling pathway. Heatmaps representing quantitative data from multiple experiments appear on the right and are used to drive analysis. Focus+Context is implemented as a semitransparent plane hovering over the global image and allows researchers to navigate through complex networks in a one-level-at-a-time mode.
Next, we relate our results to previous work in protein interaction network visualization and related techniques. We then introduce our methods by presenting an overview of the visualization workflow and then detailing each of its components. We then present our results as findings and evaluations of how our techniques improve the proteomic workflow. A discussion of our design choices follows. We conclude with a distillation of our findings.
2 Related Work
2.1 Visualizing Signaling Pathways and Protein Interaction Networks
The first representations of protein interaction networks had the form of static, schematic drawings of signaling pathways. Several papers such as [3], [11] discuss guidelines and approaches to drawing such representations. However, the static nature and manual assembly became serious limiting factors when protein-protein interaction databases were first created – researchers needed a way to generate visualizations on the fly based on database queries.
Many popular protein interaction databases – examples include the Human Protein Reference Database (HPRD) [14], Molecular INTeractions Database [24], STRING [22], and the Database of Interacting Proteins [23] – started to provide on their websites visual components that let users navigate the protein interaction space. Most of these visualizations represent protein-protein interactions via a node-link paradigm and produce visual layouts with spring models or other force-directed methods. Recently, more advanced standalone visualization systems have emerged; notable among them, Cytoscape [18] and VisANT [9] offer multiple representation methods, session-saving capabilities, and numerous features for pathway analysis. Moreover, users can add features and customize the software using plugin architectures.
Nevertheless, aspects of these visualization systems can still be improved. For instance, using generic techniques devised by the graph-drawing community sometimes yields visualizations that are far from intuitive to proteomic researchers, since their failure to incorporate protein cellular location and signaling pathway drawing conventions detracts from the visualization's familiarity. This problem is also recognized by [2] and [10].
Another topic not sufficiently investigated is the integration into protein interaction visualizations of quantitative data from large-scale proteomics experiments. Cytoscape uses a flexible plug-in architecture to address this and other functionality needs; other systems simply let one load textual annotations onto a protein network. The visual display, analysis, and comparison of results from multiple quantitative proteomic experiments are still an area of active research. The most recent work identifying and addressing the issues of both layout and experimental data is [2]. It extends Cytoscape with a new protein network layout algorithm that organizes proteins in cellular layers, based on an annotation file supplied by the user. Quantitative data can be loaded and viewed as color mappings on the proteins. Multiple experimental conditions are shown using small multiples (i.e. multiple iconic representations of the protein network for each experimental condition) and a parallel coordinate view. Our work differs in offering an alternate way of drawing the protein network, a different represention of the experimental data and the ability to load multiple experiments, each with several conditions, and in identifying and supporting the need for exploring biological networks at global and local levels simultanously.
2.2 Visualizing and Exploring Networks
There are many techniques or systems for displaying general graphs such as [4], [8], [6], [21]. However they often fail to translate well to biological networks. Protein network layouts require a constraint-based approach in which general aesthetic graph-drawing criteria are met, while satisfying other biological or user-defined constraints. Dwyer and Marriott [5] is the state of the art in constraint-based graph layout but its complexity, while powerful in its adaptability, makes it hard to implement and control. Like [2], we chose to implement our own algorithm that is easier to adapt to our specific problem. The layout algorithm itself is close in several aspects to the one described in [7] for drawing evolving graphs. They place new nodes at the barycenter of existing ones, with subsequent force-directed steps. We use a similar approach to place database-extracted proteins in relation to pathway proteins.
The idea of scaffolding graph drawings on another structure, as we do in this work, is found in [12]. Here, domain knowledge is used to identify spanning trees within graphs, and the simpler tree layouts are used as scaffolds for the general graph structure. Similarly, [1] automatically computes spanning trees as graph scaffolds and demonstrate their methods in the context of biological networks.
2.3 Focus and Context
Revealing global aspects of data while also granting access to details is commonly known as Overview+Detail. A subcategory of Overview+Detail is formed by so-called Focus+Context techniques which show the global and detailed views simultanously. They are often preferred over more traditional Overview+Detail, such as zooming and panning, which can leave the global picture out of view when zoomed in on details. Quantitative evidence that may explain this preference was published by Plumlee and Ware [15] — they show that the cognitive cost is higher when zooming and panning than when viewing local and global aspects of the data simulatanously on side-by-side displays.
Several Focus+Context techniques have been devised. For instance, [16] leverages trained human 3D perception by displaying trees in 3D and using the proximity of objects as a direct focusing mechanism. Another popular Focus+Context approach is to distort the representation space to give more screen real estate to focused regions as opposed to context regions. Other examples of such techniques are [17], [12] or [20].
Our Focus+Context method is closely related to [19], which interposes a separate viewing plane between the viewer and the actual scene. Although similar to a regular lens, this space can be used to display detailed information about the underlying scene.
3 Methods
Here we introduce the design principles and implementation details employed by our visualization methods. We first present an overview of the visualization workflow we propose. We then provide details about each of its components.
3.1 Design Overview
Researchers analyze their data in the following workflow:
import a model of a canonical pathway representation either by loading a signaling pathway image and preprocessing it to help the system infer the structure (Figure 2, lower left) or by specifying the model explicitly by placing proteins and interactions on an empty canvas (Figure 2, lower right);
load one or more quantitative datasets (Figure 3);
automatically extract proteins and interactions from protein interaction databases such as HPRD and build a network around the pathway model specified in step 1 and the quantitative data from step 2;
represent the network graphically using a novel canonical pathway-oriented layout (Figure 4);
explore and analyze the network guided by interesting features noted in the experimental data; investigate the network at interaction level using a Focus+Context technique; analyze how known information blends with the new experimental results using such features as clustering of quantitative proteomic data, filtering, highlighting, and information on demand (Figure 1);
derive insights or generate new hypotheses, design and run new experiments, and restart from step 2.
Fig. 2.
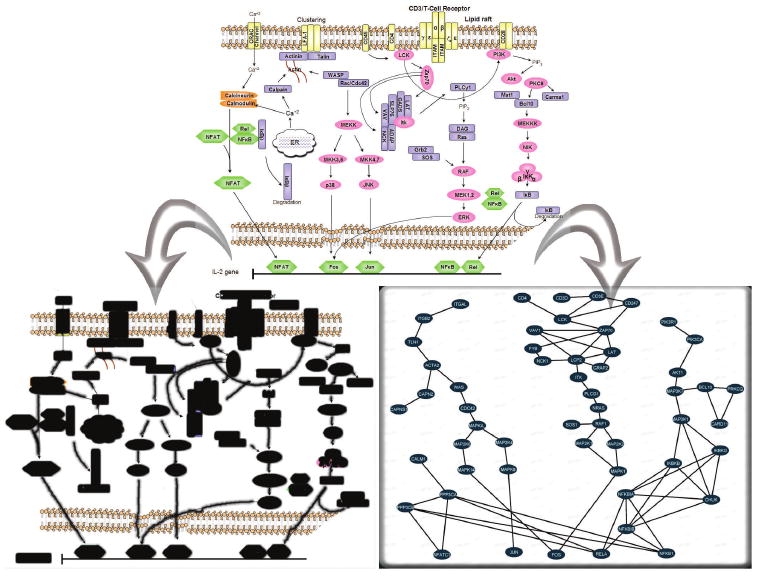
Structuring protein interactions around familiar canonical pathways provides intuitive visualizations. A canonical signaling pathway representation (top) can be imported into the system in two ways: on the lower left the pathway image itself is loaded into the system and preprocessed by circling proteins and drawing over interactions; the pathway features are then inferred from the user strokes and image features and shown here in black; or, on the lower right, protein and interaction icons are placed and dragged on an empty canvas to create a new pathway model. After positional assignment of each protein, the software aids in associating interaction database accession numbers to each of the newly defined canonical pathway proteins.
Fig. 3.
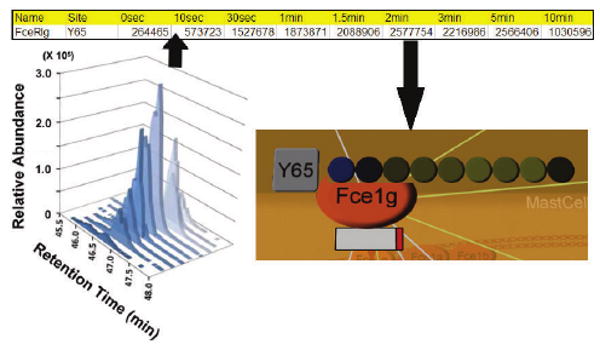
Loading and visualizing quantitative proteomic data. (a) Selected-ion chromatogram of a peptide belonging to the FceRI gamma protein. (b) Line in a file containing experimental data ready to be loaded. (c) Software display of a heatmap representing the same peptides relative abundance across the nine time points, attached to the FceRI-gamma protein. Black represents the average value for a certain peptide across all conditions, blue corresponds to a below-average abundance and yellow to an above-average abundance. The intensity of the color corresponds to the magnitude of the fold change across all peptides: the most intense yellow and blue represent the single peptide that changes the most across all peptides. Missing values would be indicated in white.
Fig. 4.
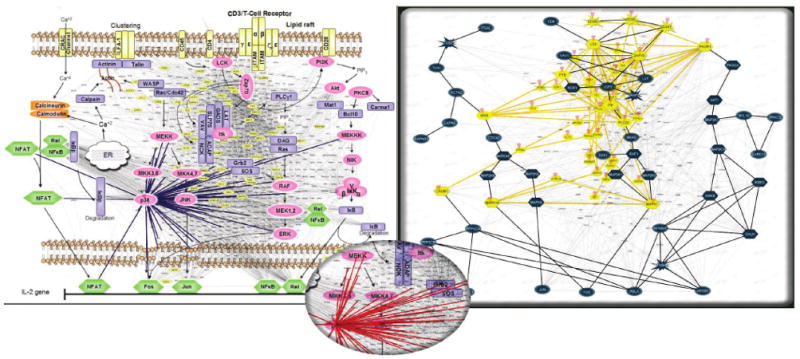
Proteins and interactions from HPRD (small fonts) that are connected to the canonical pathway model are: (left) integrated directly into the signaling pathway image with one protein selected and its interactions highlighted; (right) structured around a user-constructed model; different classes of proteins have different appearances: experimental proteins are colored yellow, kinases are drawn as hexagons and receptors as irregular stars; several experimental proteins are not known to be connected to the pathway and are therefore located in the lower right corner. HPRD proteins are placed in a structured manner between the pathway proteins based on their separation from the pathway proteins. (cutout) Disadvantage of simply drawing the network on top of the pathway image: HPRD interactions obscure elements of the canonical pathway; compare to the improved method (left) in which important pathway elements remain in the foreground.
3.2 Pathway Model Specification
Our solution requires the user to specify, using a simple interface, the canonical pathway representation of the signaling pathway under investigation. This can be done either by putting proteins and interactions on an empty canvas or by using a pathway image that is preprocessed to help the system extract the pathway structure; the preprocessing entails drawing single, continuous strokes over or around each pathway element – proteins, interactions and other entities. These strokes aid the software in identifying image features (Figure 2) as detailed below.
If the stroke endpoints are far apart compared to the stroke length, the image feature is probably an interaction and the endpoints are matched against protein positions to find which proteins are involved. The interaction strokes snap to image features in a manner similar to a lasso tool. This is done in order to obtain the correct image region that the interaction is covering, for reasons described in Section 3.7.
If the stroke endpoints are close relative to the total length of the stroke, the feature-detection algorithm decides to classify the feature as a protein. It then computes an average color for the area enclosed by the stroke and removes all points dissimilar to it. In most cases this leaves only the image shape selected. The protein position on the canvas can be inferred through this computation.
If a selection is unsatisfactory the user can cancel it and try again – depending on the previous selection, the algorithm will attempt to correct the image-processing parameters for the second try. For instance, if the area selected by the user is much larger than that returned by the algorithm, the color-similarity threshold is increased.
Once the graphical model is complete, either by pathway processing or by pathway drawing, the placed proteins need to be linked to protein identifiers in the protein interaction database. The user chooses the correct protein by searching the database for keywords using a dedicated dialog box. In our test cases this process took between 15 and 30 minutes for medium pathways such as those in the figures, but these times vary with image complexity and user training.
3.3 Interaction Data
In our experimental prototype we use the HPRD protein interaction database. HPRD is a protein interaction and metadata source based on manual literature search. The database information is stored and loaded as flat files.
We have also experimented with the STRING interaction database, version 7.0. STRING searches multiple sources for evidence of protein-pair interactions: database occurrence (HPRD, KEGG, REACTOME), genomic context, coexpression, high-throughput experiments, and the literature. A score is computed for each source and aggregated into a number that quantifies the likelihood that a protein pair interacts. Due to STRING's unsupervized automatic parsing and computation, it has greater naming redundancy.
The network exploration paradigms defined here could be used with any protein interaction database. One of the main challenges in supporting a protein interaction database is providing access to useful metadata from other databases. This is due to the inherent difficulty of translating protein identifiers across independent protein databases.
3.4 Experimental Data
The quantitative proteomic data is loaded as XML or flat files upon pathway creation and can contain multiple quantitative data points as well as protein identifiers and other metadata. For graphical representation, the quantitative proteomic data are transformed into a colored heatmap representation (Figure 3) indicating fold changes of a given peptide across different experimental conditions (time course of receptor activation or comparison between wild type and mutant cells). The following color-coding is used: blue – decrease of proteomic quantity, yellow – increase of proteomic quantity, black – no change.
If multiple experimental files are loaded, as in a comparison between wild type and mutant cells, special types of heatmaps are computed for each pair of experiments to reflect changes between experiments: yellow then indicates a major change between the two experiments, while black corresponds to no change. A single protein can have multiple heatmaps, one for each assigned peptide. The heatmap icon appears in two places: displayed in the expanded network exploration upper plane, attached to proteins revealed in the experiment (Figure 1), and in a dedicated panel on the right (Figure 1) containing all peptides discovered in an experiment.
For multiple quantitative data sets, the heatmap experimental data panel on the right (Figure 1) is configured to contain tabs not only for each separate experimental data-set but also for changes observed between pairs of data-sets. For instance, in a phosphoproteomic receptor activation timecourse experiment involving wild type and cells lacking critical signaling proteins, the heatmap tab contains one tab dedicated to timecourse phosphopeptide heatmaps in the wild type cell, another tab for the mutated cell, and a third tab displaying the fold change of individual phosphopeptides observed between the two cell types through the receptor activation timecourse. This feature can be particularly useful in knockout-type experiments since the differences in behavior between a normal and a mutated cell become evident immediately.
The experimental data panel is kept visible at all times so that researchers can use it to explore the new quantitative data systematically. The items in the experimental data panel can be used to start the exploration by linking directly to Focus+Context representation.
Using experimental data to guide exploration was also discussed in [2]. Our work differs both in the way we present the information to the user and in the emphasis we put on comparative analysis of multiple experiments. Such analysis can also be performed with their system, but we believe the small multiple approach would overload the display if used with dense networks and large quantities of experimental data. Their parallel coordinates view was also not extended for both multiple time-points and multiple experiments.
3.5 Network Generation
From the user-provided pathway skeleton, the software constructs a protein-protein interaction network by loading proteins and interactions from the HPRD database. The network is grown iteratively in a breadth-first manner: first, proteins interacting directly with the canonical signaling pathway model are imported, and then in subsequent steps, proteins interacting with those added in the previous iteration are extracted from HPRD and included. Finally, interactions among all proteins are loaded.
The number of levels to grow the network and optional filters used to exclude proteins from the build process are specified by the user. However, growing the pathway from the user-specified proteins alone may leave experimental proteins outside the network. To ensure inclusion of all experimental proteins in the final visualization, we also grow the network from the experimental proteins themselves. This solution increases the chances of linking the experimental proteins to the pathway since two networks are grown simultaneously toward each other.
3.6 Computing Protein Positions
While the canonical pathway proteins have user-provided predefined positions, our prototype must compute where to put the proteins extracted from the interaction database. These proteins are placed depending on their distance, in terms of number of interactions, from each of the pathway proteins. If protein P is interacting directly with protein A and is three interactions away from protein B, it is placed on the line segment between A and B, closer to A. The distances are not necessarily directly proportional to the path lengths: they can be weighted so that direct connections are much shorter then longer interaction paths.
Essentially the nodes are placed at a path-length weighted barycenter of the pathway nodes. Barycenter positioning was also used in [7] to place new nodes in relation to already existing ones in the context of evolving graph drawings. This algorithm produces positions close to those computed by a traditional spring layout algorithm, since a node is dragged by the edge springs to a similar location.
This methodology leads to identical positions for some proteins, however, and a force-directed approach based on [8] is used to perturb the layout and remove overlaps; a simple linear grid approach is used to improve the performance of the layout algorithm by using vicinities to reduce the number of comparisons needed to compute forces on protein-nodes. We also apply a force to keep the nodes close to their initial position computed by barycentric placement.
The sizes of nodes are taken into consideration when computing repulsive forces. The aspect ratio of nodes in relation to the force vectors can also be taken into account so that forces are applied anisotropically. This leads to slightly longer run times but minimizes overlap, especially in augmented pathway images where some nodes can be much larger in one direction.
As a special case, positions cannot be computed for proteins linked only to the experimental data and not to the known pathway. These are placed in the lower right side of the display, yielding a cluster of proteins that are not known to be connected to the pathway (Figure 2, lower right).
This algorithm is relatively fast, interactive, and achieves the desired results without the complexities of more powerful constraint-based techniques such as [5]. The layouts in Figure 4 took around 2 minutes to compute. We also experimented with simulated annealing methods. These, however, were much slower and did not improve the layouts significantly due to the high network density. Some parameters inherent to force-directed methods still require user adjustment.
3.7 Augmenting a Pathway Image with Dynamic Data
The case of specifying a pathway image and integrating dynamic information seamlessly into the already existing representation is more complicated than assembling a completely new visualization. Simply drawing the database extracted elements on top of the pathway image has several disadvantages, as shown in the cutout of Figure 4. In contrast, our method creates the illusion that the proteins and interactions drawn dynamically are part of the pathway image (Figure 4, left).
The following specialized operations are used to create the illusion that the HPRD proteins and interactions are part of the pathway image. The shapes and locations of proteins and interactions in the image are computed in the image preprocessing step. They are then used in the layout stage to minimize overlap (dynamically loaded proteins tend to move to empty image areas). Finally, they are copied from the image and redrawn as masks on top of the final network. This technique ensures that the pathway model stays on top of the dynamic network and gives the illusion that the canonical pathway representation and the dynamic network coexist and interact (Figure 4, left).
3.8 Exploring the network
In our design the interaction network can be explored at two levels simultaneously: at a global level, where the signaling pathway and other high-level structures are evident, and at a local level, where only one protein and its neighbors appear in detail as the researcher jumps from protein to protein in the network. The two types of visualization coexist as two parallel planes, the local one gliding above the global one (Figure 1). With these complementary views of the pathway space, the user explores the network in the detailed space that is rich in focused protein information while maintaining an overview of the explored area and orienting the expanded exploration to his or her location within the global view.
Exploration is done in a plane that hovers above the global view and shows in detail only one protein and its interactors. Initial access to the exploration plane can be obtained by double-clicking proteins in the global-view, in the experimental lists, or in a list of all proteins present in the visualization. While in exploration view, clicking one of the interactors shifts the center of the view to this selected protein, a change performed through smooth animation to maintain context understanding. Standard zooming and panning using mouse controls are also available, but testing has found them less favored by users. Proteins in the exploration plane are arranged so as to mimic their placement in the global layer while satisfying aesthetic criteria such as minimum distances between proteins or interaction overlap (Figure 5, left). The effect is achieved by applying a simulated annealing [4] algorithm that attempts to maximize layout similarities while ensuring a pleasing drawing. The area allocated to the exploration view is computed dynamically on the basis of the number of proteins to be displayed. A view that places the main protein in the center and its interactors circularly around it is also provided.
Fig. 5.
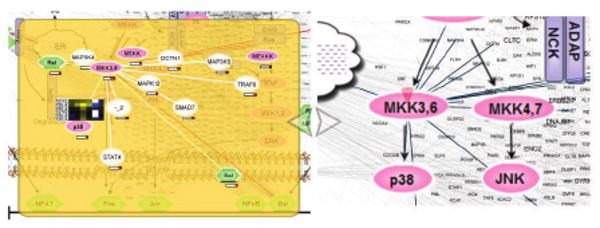
Exploration plane versus zoom-and-pan. (left) The network is explored in a separate plane showing only one protein and its interactors. Selecting an interactor changes the view of that particular protein via a smooth animation. This interaction network crawling method allows systematic discovery of connections among proteomic data and existing protein knowledge. Transparency keeps the global view visible and the same protein is highlighted within both planes. The protein layout in the exploration plane mimics the layout in the global plane, but is slightly distorted to achieve a more attractive representation. Changes in peptide abundance are represented as linear heatmaps. (right) Zooming and panning, while also available to explore the network, have several drawbacks: the view is cluttered, some interactors reach outside the viewing area, there is no space for additional details, and the global perspective is lost.
Clicking a protein in the exploration view highlights it and its neighbors in the lower plane, making it easier for the user to establish a correspondence between the two.
3.9 Visualization prototype
The visualization prototype we used to develop and evaluate our methods can be downloaded and tested at http://graphics.cs.brown.edu/research/sciviz/proteins/home.htm.
A compact set of features were added to allow our researchers to operate on the network data and pose visual queries. For instance, selectors and the ability to adjust appearance allow the researcher to highlight interesting aspects of the visualization. In the right panel of Figure 4, a user has selected various groups or classes of proteins and attached to them special visual attributes such as shape and color a technique often used in stylized signaling pathway representations. The method described in [13] is used to highlight interactions of one or more selected proteins; interaction highlighting can also be restricted to interactions occurring only between selected proteins (Figure 6, right).
Fig. 6.
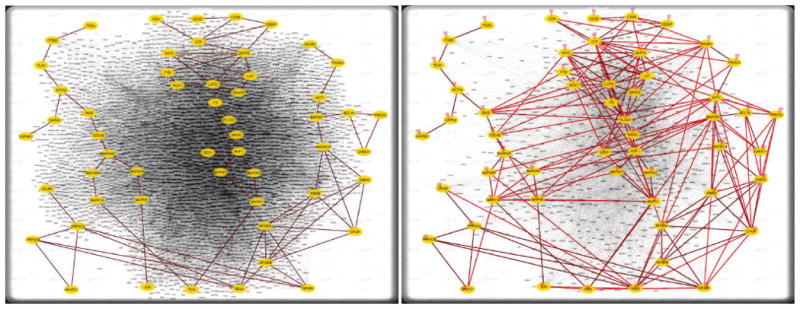
Improving network readability through filtering. (left) unfiltered and un-highlighted, (right) filtered by keeping only the pathway proteins and those proteins that connect the pathway proteins (proteins that are not on interaction-paths linking any two pathway proteins are removed); the pathway proteins and direct interactions between them are highlighted.
Easily extensible filters allow a researcher to remove proteins deemed uninteresting. One potentially useful filter with significant effects keeps only proteins that connect a set of user selected proteins. As an example, Figure 6 shows how a heavily cluttered network was filtered to keep only pathway proteins and those proteins known to connect them through interactions. These filters are crucial since protein interaction networks often contain thousands of proteins and interactions, making comprehension and interaction tedious.
3.10 Implementation Details
The prototype application was written in C++. The G3D 6.7 graphics library was used for 3D graphics and rendering and the Qt 4.3 library for user interface elements. The HPRD database can be downloaded as flat files together with the application.
4 Results
The results of this work are findings about ways to improve analysis of protein interaction networks and quantitative proteomic data, and novel visualization and exploration paradigms motivated by these findings.
The research presented in this paper was driven and validated by insights obtained during our collaborative development process and by an anecdotal evaluation with domain experts. Our results indicate that applying these concepts in the context of systems for visualizing protein-protein interaction networks may accelerate the discovery of new connections among quantitative proteomic data, interacting proteins, and canonical signaling pathways. While a controlled study may still be needed to verify and quantify the benefits of individual aspects of our methods, we believe an anecdotal evaluation with domain experts is a preferable approach in an iterative design setting, with no predefined requirements since it can provide fast, easy access to usability information on high-level analysis tasks.
Evaluation was performed on the analysis of phosphoproteomic experiments with the help of four proteomic researchers interested in research of the T-cell and Mast cell. Our collaborators artificially stimulate cells and measure the amount of phosphorylation that occurs on proteins as a result. Phosphorylation is an important cellular process by which a phosphate is added to a protein or other molecule. A protein can be phosphorylated in multiple places, called phosphorylation sites.
In a single experiment setting, phosphorylation measurements over multiple time-points can provide causality hints. More importantly, however, researchers can run separate experiments before and after inhibiting an investigated protein. By comparing changes in measured phosphorylation values they can hypothesize about the role of the investigated protein in the cellular pathway.
Finding 1: Visually combining experimental data and known protein interactions enhances analysis
We augment previous results from [2] and [18] with similar findings in our own specific analysis setting. We show that coupling new experimental data with protein interaction data extracted from public databases within a unified visual analysis can shorten the analysis process of a new experimental dataset from weeks to days. In addition to the straightforward time gain, shorter time intervals between individual data observations lets researchers integrate them more efficiently into a cohesive hypothesis.
By using the prototype, our collaborators quickly discovered a meaningful biological fact that eluded them in previous analyses of a T-cell related phosphoproteomic dataset. Our user started by browsing through the list of experimentally measured proteins, displayed as seen in Figure 1 on the right hand side of our prototype. She then decided to take a closer look into the protein Slp76, because of the variation reflected by its heatmap. Double-clicking on the list item opened a detailed exploration view, as shown in Figure 1. The visualization revealed that this protein was known to interact with the protein VAV. Metadata available within the software then revealed that the particular measurement could indeed be related to that specific interaction. In addition, a novel phosphorylation site was detected on SHP1. An interaction with SLP76 and meta-data about this interaction were easily accessible in the software and led to the hypothesis that SHP1 negatively regulates SLP76.
These insights may have been eventually produced using our collaborators' previous strategy of manually querying each experimentally measured protein and gathering information about them. The integration of experimental data and the protein interaction network reduced the time needed to make this discovery.
Finding 2: Canonical pathway-driven layout is intuitive for proteomic researchers
Structuring dynamically extracted protein interactions around a familiar canonical pathway (see Figure 1) provides an intuitive visualization that helps proteomic researchers orient themselves and learn the interaction network quickly. A proteomic experiment revealing hundreds to thousands of protein modification sites overwhelms users with the many unfamiliar proteins. Becoming familiar with the proteins in such an experiment is greatly facilitated by placing those proteins within signaling pathway-structured protein interaction networks.
This pathway-structured method was motivated by negative feedback on an initial prototype that used a standard force-directed network layout. This feedback suggested that generic network-drawing algorithms fail to place proteins in positions that are meaningful either from a biological or a pathway-conventions standpoint (receptors can end up near the nucleus). Moreover, proteomic researchers were overwhelmed by the unstructured node-link diagrams such methods produce and tried to map the new visualization to the signaling pathway they were using before. This was also found by [2] and [10] to be an important issue in systems that employ traditional graph drawing algorithms to display protein interaction networks. Our work differs from theirs by introducing a novel visualization paradigm to address this problem.
In a broader visualization context, integrating dynamic connectivity information into static diagrams is a potentially useful concept because it facilitates the integration of new information into existing thinking schemas. We demonstrate its perceived usefulness in a proteomic context. More targeted research is needed to establish whether the perceived benefits translate to actual task improvement, and to identify other areas of application.
Evaluation
Overall, this layout method was preferred by our collaborators over two network visualizations they tried before: [18] and our earlier interaction network prototype. At the time of their use, both systems used traditional graph drawing algorithms and were criticized for their lack of structure.
Conclusions drawn from specific user comments were: the familiar pathway model that seeds the exploration is visually appealing and reduces the initial ball-of-strings shock associated with most network visualizations; it helps users orient by providing a familiar context; it gives protein placements more meaning and ensures that well known proteins are placed in familiar locations.
A problem identified in early testing was that growing the pathway from the user-specified proteins alone omitted many experimentally observed proteins from the network due to the lack of connections between these proteins and the known pathway within the protein interaction database. This problem was addressed by growing the network not only from the pathway proteins but also from the proteins indicated by the experiment, thus ensuring their inclusion within the network. However, some experimental proteins will still not be connected. These proteins are placed in the lower right corner of the representation, essentially forming an island of proteins revealed in the experiment but not known to be connected to the user-provided signaling pathway skeleton (Figure 4).
This approach has its benefits, as one test case revealed. After loading a large phosphoproteomic dataset onto the well established insulin pathway, a user immediately observed that many of the experimental phospho-proteins were connected to the signaling pathway, while the island of unconnected proteins was fairly small. This increased the users confidence in both the experimental results and the visualization.
Finding 3: Global and local exploration modes (multilayer, multiscale views)
We found that researchers prefer to explore an interaction network by using a local view of each protein, looking only at its direct interactors at a time (Figure 5, left). This initial hypothesis guided our design choices and was validated during our usage evaluations.
During the testing stages much time was spent in local view instead of global view. This finding suggests that protein network analysis benefits from views that isolate one protein and its interaction from the rest of the network. Current interaction network visualization frameworks lack Focus+Context capabilities, and little research exists to address this issue.
Evaluation
Our evaluation revealed that the exploration plane was indeed the most popular mode of protein-network exploration. A second demonstration and usage session with a separate proteomic group led to the same conclusion. The global view was used to apply filters, browse through the data and jump-start exploration. It also created an important first impression of the visualization as a whole and kept the users engaged. For reasoning about connectivity however, researchers rarely looked directly at interactions in the global view, even though zooming and panning were available. The navigation plane was used instead.
Our observations of proteomic workflows during development and evaluations suggest that current proteomic analysis happens mostly at interaction level. This explains why our Focus+Context method was preferred over traditional global exploration: a single protein and its interactors can be viewed without clutter from any other network elements; all interactors are visible at once without panning; the space can be distorted to make room for additional glyphs and information associated with the proteins; and both views – global and local – are visible at the same time, with an emphasis on the local view.
Given the synergy between local and global viewing, with a stronger emphasis on local exploration for accurate, analysis tasks, we believe our method to be adequate. The local view is in the focus, while the entire global view is maintained in the background as a mental anchor. The user can switch between views immediately using an intuitive operation that requires minimal mental transformations.
Probably the main contribution of this result is that techniques for the exploration of networks concurrently at varying degrees of detail are suited for proteomic analysis tasks and should be included in specialized systems. While we also present a novel technique that we believe works well in this domain, other Overview+Detail paradigms, such as the ones described in our related work section, may also produce good results. We discuss this in more depth in section 5.3.
We note that we used unfiltered interactions directly from proteomic databases. This resulted in dense networks. Curating interactions that are placed in the pathway could allow all information to be visible at the same time as seen in some networks presented in [2]. In this case zooming and panning may be sufficient for interacting with the network.
Finding 4: Comparative displays of multiple experiments help identify important pathway players
Our test cases showed that the ability to load and compare multiple experimental results, for example from cells containing deleted or mutated proteins, helped researchers link cell behavior to experimental results. Also, researchers found it useful to have the experimental data permanently visible to drive the exploration.
Evaluation
Our first prototype did not present the user with an explicit list of experimental proteins. Instead they were marked on the network. Our users argued that they prefer to be able to go through their experimentally derived proteins systematically, preferably in a list. We then added an experimental proteins list in one of our submenus. Further testing showed that our users refered to that list throughout their analysis. We thus concluded that having it permanently displayed and linked to all the views would speed up their analysis process.
In our final evaluation, the typical analysis workflow consisted of systematically going through the experimental protein list, selecting ones with interesting patterns as suggested by their heatmaps, and opening them in local exploration.
The following test scenario showed the usefulness of this approach: in an experiment, a known T-cell signaling protein ZAP70 was removed from the cell and quantitative phosphoproteomic perturbations were recorded before and after the removal.
Our user started his analysis by examining the heat-maps indicating the fold changes between the two experiments. The heatmap profile signaled an interesting change on the Lck protein, an upstream component of the pathway: the phosphoryaltion of Lck was greatly delayed when the downstream protein ZAP70 was removed. By bringing up Lck in the exploration plane, a direct interaction was discovered that connected Lck to Zap70 and explained the change.
5 Discussion
5.1 General Considerations
Maintaining tight collaboration between researchers from computer science and proteomics let us better understand the requirements and specifications of proteomic visualizations. Our canonical pathway-driven network layout and experimental data-guided network exploration are tangible results of our collaboration.
Good proteomic visualizations should support and automate part of proteomic researchers data analysis workflows. But identifying these workflows is nontrivial and often varies among individual labs and researchers. The novelty of experimental data and constantly evolving proteomic methodologies make it hard for the researchers themselves to describe their workflows clearly. However, the process of workflow discovery, while laborious for both proteomic and computer science researchers, is beneficial for both parties since it identifies where computers can help most.
5.2 Layout
One drawback of the canonical pathway-guided layout is the overhead associated with specifying the canonical signaling pathway within the software. The most laborious step is not so much inputting the structure but searching for correct protein identifiers in the interaction database; this can be time-consuming due to naming ambiguities, multiple matches, missing proteins, and inconsistencies across protein databases. Initial testing revealed that identifying correctly canonical signaling pathway proteins within the protein database is aided by additional cues and metadata such as number of interactions or interacting partners.
The average time required by users to input the pathway skeleton and attach database identifiers was around 20 minutes for medium-sized pathways like those shown in the figures here. This overhead is acceptable if one considers that researchers commonly spend months or years studying a few pathways. Moreover, a canonical signaling pathway skeleton, once constructed, can be used to build multiple networks for different proteomic experiments and parameters.
Proteins imported from databases are by default displayed smaller then pathway proteins and are sometimes not legible without zooming. This mode of display was motivated by our desire to keep the pathway structure in the foreground and by the need to save canvas space and minimize overlaps in dense protein networks. However, the default size settings are adjustable and users can customize them for individual classes of proteins.
Another issue related to protein glyphs is that proteomic researchers often place several icons corresponding to the same protein in various places on the canvas, usually depending on the specific function and context. Many-to-one correspondences between graphical icons and data entities are uncommon in network representations. Our software allows this type of representation by automatically adding numbered suffixes to identical proteins to differentiate them. However, extensive use of this feature tends to clutter the representation with redundant information since interactions are replicated for each copy of the same protein.
Augmenting a pathway image with dynamic data does not always work. While our software is designed to accept any type of image, low image quality or high complexity can make our system unable to extract the pathway structure. Our feature detection algorithm is flexible and can automatically adjust its parameters based on user feedback. However, the image-processing techniques were not the focus of this research and are not state of the art.
5.3 Focus and Context Exploration
The local exploration plane received positive feedback and was used extensively in our test cases. Its simplicity is both an advantage and a limitation. Users can easily understand what the display is showing and how to crawl around the network, while the visualization avoids clutter and in most cases does not require zooming or panning. Showing a single network level, however, can make it difficult to determine the optimal direction for future exploration. Unfortunately, real-life uncurated protein networks have high graph degrees that limit the number of levels we can show without clutter. Possible solutions to this problem are: hyperbolic views, automatically adjusting the number of levels that can be displayed without clutter, or attaching glyphs to nodes that provide cues about interesting exploration directions.
The decision to place the exploration plane on top of the global view rather than using a separate window was primarily motivated by the desire to save screen real estate. This choice has the disadvantage of occlusion, but we believe this is outweighed by the ability to use the entire display area for exploration while preserving a view of the global layout in the background. This situation arises frequently in protein interaction networks since many proteins are highly connected and need large display areas. The area assigned to the exploration plane is computed dynamically depending on the number of proteins to be displayed, thus minimizing occlusion as much as possible. The transparency of the exploration plane is also adjustable. We also note that the current proliferation in screen real estate, even in common analysis settings, opens the way to placing the two views next to each other. This approach would remove the occlusion problem but the need for frequent changes in focus across views and to spatially relate elements across the two views might lead to an additional cognitive cost.
Our usage observations confirmed our design choice: the global view was used mainly as a visual reference, especially for large networks, and as support for posing visual queries using selectors and filters. These tasks are not significantly hindered by occlusion. Proteins and interactions were rarely looked at closely in the global view, a task that occlusion would affect more.
It is also possible that using some filtering criteria on protein interactions will lead to sparser, more relevant networks like those featured in [2]. These could then be fully legible and explorable at a global view, potentially minimizing the need for a separate exploration view. However, our domain experts have not identified in the biological databases they currently use any such criteria that can be automatically applied.
The placement of interacting proteins within the upper expanded view plane is designed to mimic the placement within the global lower plane while preserving aesthetic criteria such as node overlap. In addition to highlighting in the global view the interacting proteins that are being explored, this allows the user to better relate the exploration views to the global view.
The view during exploration can be either tilted, as in Figure 1, or parallel to the view plane, as in Figure 7. An in-depth analysis of the benefits of each type of projection was out of the scope of this work. We can point out however that several of our users expressed a strong preference for the tilted view. We attribute this preference to the superior visual appeal for a 3D representation rather than a perceptual benefit. Negative comments about distortions caused by the perspective projection seem to support this hypothesis.
Fig. 7.
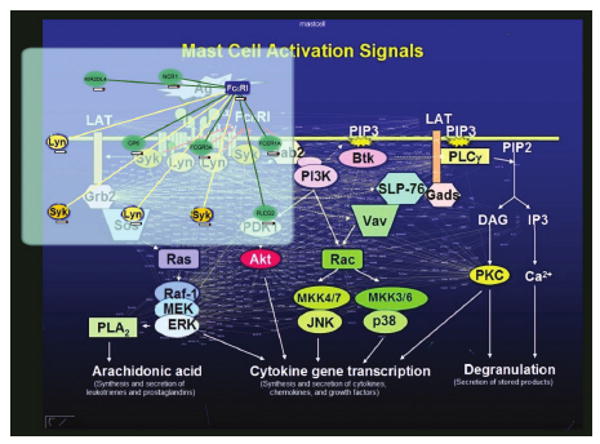
Mast cell image augmented with HPRD proteins and interactions. Focus+Context achieved by transparent plane gliding over the globa view. Exploration plane not tilted.
Finally, as stated in section 4, other techniques for exploring networks at varying degrees of detail might also provide good results. While we have not gathered concrete evidence as to what types of Overview+Detail techniques are preferable, based on results presented in this paper we believe they should exhibit at least two properties. First, the detail view should remove network elements that are not of immediate interest. This is motivated by the observation that proteomics researchers often analyze single proteins and their interactors. Second, the global view should be undistorted and fully visible at all times. Our Focus+Context method evolved from an initial prototype where the user could toggle between the local and global views, but only see one at a time. At that development stage our users expressed the desire to keep the pathway view visible while using the exploration view. This is motivated by Finding 2 whereby the pathway under investigation acts as a mental anchor for the researcher.
6 Conclusions
We have presented several novel visualization methods and paradigms for the analysis and quantitative comparison of multiple proteomic data sets in the context of published protein-protein interaction networks and known signaling pathways. We evaluated the effectiveness of the methods in terms of data insights, hypothesis generation, and improvements in analysis time. We believe that applying the principles presented here to proteomic interaction visualizations will increase adoption rates among proteomic researchers, sharpen system and network learning curves, and accelerate protein network knowledge extraction from massive quantitative proteomic datasets.
Acknowledgments
This work has been supported in part by NIH grant R24AI072073.
Biographies
Radu Jianu

Radu Jianu received the Dipl. Ing. degree in Computer Science in 2005 from Polytechnic University in Timisoara, Romania. In 2007 he received the MS degree at the Computer Science Department at Brown University where he currently pursues a PhD degree in Computer Science. His research interests are mainly in graphics and visualization with specific applications in genomic and proteomic biology.
Kebing Yu

Kebing Yu received the Bachelor of Science degree in Chemistry from Peking University in Beijing, China in 2004. Then he joined the Ph.D program in Chemistry at Brown University. His research focuses on bioinformatic analysis on large-scale proteomic data. He also has interests in developing LC/MS based analytical methods to study cell signaling pathways.
Lulu Cao

Lulu Cao received her B.S. degree in Chemistry from University of Science and Technology of China in 2004. She is currently pursuing her PHD degree in Chemistry Program in Brown University. Her research mainly concentrates on the utilization of tandem mass spectrometer to analyze the posttranslational modifications of complex signaling proteins and further elucidate the signaling pathways of biological cellular processes.
Vinh Nguyen

Vinh Nguyen received a BS degree in Biochemistry, Cell Biology in 2004 from the University of California, San Diego. In 2005, he received a MS degree in Biology from the University of California, San Diego. He is currently pursuing a PhD degree in Molecular, Cellular Biology at Brown University where his research focuses on using a phosphoproteomic approach to study the T cell activated signaling pathway.
Arthur R. Salomon

Arthur R. Salomon received his Ph.D degree in Chemistry from Stanford University in the lab of Prof. Chaitan Khosla studying the mechanism of action of the highly selective anti-cancer agent apoptolidin. Dr. Salomon did Post-Doc work at the Genomics Institute of the Novartis Research Foundation in the lab of Prof. Peter Schultz where he developed novel phosphoproteomic technologies. Dr. Salomon is now assistant professor in the department of moelcular biology, cell biology and biochemistry where he applies phosphoproteomic technologies by mass spectrometry to the problem of cellular signaling pathway elucidation.
David H. Laidlaw

David H. Laidlaw received the PhD degree in computer science from the California Institute of Technology, where he also did post-doctoral work in the Division of Biology. He is a professor in the Computer Science Department at Brown University. His research centers on applications of visualization, modeling, computer graphics, and computer science to other scientific disciplines. He is a senior member of the IEEE and the IEEE Computer Society.
Contributor Information
Radu Jianu, Email: jr@cs.brown.edu, Department of Computer Science, Brown University, Providence, RI.
Kebing Yu, Department of Biology, Brown University, Providence, RI.
Lulu Cao, Department of Biology, Brown University, Providence, RI.
Vinh Nguyen, Department of Biology, Brown University, Providence, RI.
Arthur R. Salomon, Department of Biology, Brown University, Providence, RI
David H. Laidlaw, Email: dhl@cs.brown.edu, Department of Computer Science, Brown University, Providence, RI.
References
- 1.Adai A, Date S, Wieland S, Marcotte E. LGL: creating a map of protein function with an algorithm for visualizing very large biological networks. Journal of Molecular Biology. 2004;340(1):179–190. doi: 10.1016/j.jmb.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 2.Barsky A, Munzner T, Gardy J, Kincaid R. Cerebral: visualizing multiple experimental conditions on a graph with biological context. IEEE Transactions on Visualization and Computer Graphics. 2008;14(6):1253–1260. doi: 10.1109/TVCG.2008.117. [DOI] [PubMed] [Google Scholar]
- 3.Botstein D, Kohn K. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Molecular Biology of the Cell. 1999;10(8):2703–2734. doi: 10.1091/mbc.10.8.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson R, Harel D. Drawing graphs nicely using simulated annealing. ACM transactions on graphics. 1996;15(4):301–331. [Google Scholar]
- 5.Dwyer T, Koren Y, Marriott K. IPSep-CoLa: An incremental procedure for separation constraint layout of graphs. IEEE Transactions on Visualization and Computer Graphics. 2006;12(5):821–828. doi: 10.1109/TVCG.2006.156. [DOI] [PubMed] [Google Scholar]
- 6.Frick A, Ludwig A, Mehldau H. A fast adaptive layout algorithm for undirected graphs. Proceedings of the DIMACS International Workshop on Graph Drawing; London, UK. 1994. pp. 388–403. [Google Scholar]
- 7.Frishman Y, Tal A. Online dynamic graph drawing. IEEE Transactions on Visualization and Computer Graphics. 2008:727–740. doi: 10.1109/TVCG.2008.11. [DOI] [PubMed] [Google Scholar]
- 8.Fruchterman T, Reingold E. Graph drawing by force-directed placement. Software: Practice and Experience. 1991;21(11):1129–1164. [Google Scholar]
- 9.Hu Z, Mellor J, Wu J, Yamada T, Holloway D, DeLisi C. VisANT: data-integrating visual framework for biological networks and modules. Nucleic acids research. 2005;33(Web Server Issue):W352. doi: 10.1093/nar/gki431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jourdan F, MelanÇon G. Tool for metabolic and regulatory pathways visual analysis. Proceedings of SPIE. 2003;5009:46. [Google Scholar]
- 11.Michal G. On representation of metabolic pathways. BioSystems. 1998;47(1-2):1–7. doi: 10.1016/s0303-2647(98)00019-7. [DOI] [PubMed] [Google Scholar]
- 12.Munzner T. H3: Laying out large directed graphs in 3D hyperbolic space. IEEE Symposium on Information Visualization, 1997 Proceedings; 1997. pp. 2–10. [Google Scholar]
- 13.Munzner T, Guimbretiére F, Robertson G. Constellation: a visualization tool for linguistic queries fromMindNet. 1999 IEEE Symposium on Information Visualization, 1999.(Info Vis' 99) Proceedings; 1999. pp. 132–135. [Google Scholar]
- 14.Peri S, Navarro J, Amanchy R, Kristiansen T, Jonnalagadda C, Surendranath V, Niranjan V, Muthusamy B, Gandhi T, Gronborg M, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome research. 2003;13(10):2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plumlee M, Ware C. Zooming versus multiple window interfaces: Cognitive costs of visual comparisons. ACM Transactions on Computer-Human Interaction (TOCHI) 2006;13(2):179–209. [Google Scholar]
- 16.Robertson G, Mackinlay J, Card S. Cone trees: animated 3D visualizations of hierarchical information. Proceedings of the SIGCHI conference on Human factors in computing systems: Reaching through technology; New York, NY, USA. 1991. pp. 189–194. [Google Scholar]
- 17.Sarkar M, Brown M. Graphical fisheye views of graphs. Communications of ACM. 1994;37(12):73–84. [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, Baliga N, Wang J, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D T, Bier E, Stone M, Pier K, Buxton W. Toolglass and magic lenses: the see-through interface. Proceedings of SIGGRAPH. 93:73–80. [Google Scholar]
- 20.Teoh S, Ma K. RINGS: A technique for visualizing large hierarchies. Lecture Notes in Computer Science. 2002;2528:268–275. [Google Scholar]
- 21.Tunkelang D. A practical approach to drawing undirected graphs. Technical report. 1994 [Google Scholar]
- 22.von Mering C, Jensen L, Snel B, Hooper S, Krupp M, Foglierini M, Jouffre N, Huynen M, Bork P. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic acids research. 2005;33(Database Issue):D433. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xenarios I, Salwinski L, Duan X, Higney P, Kim S, Eisenberg D. DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions. Nucleic acids research. 2002;30(1):303. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, Cesareni G. MINT: a Molecular INTeraction database. FEBS letters. 2002;513(1):135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]