

Abstract
Metabolic perturbations that decrease or limit blood glucose—such as fasting or adhering to a ketogenic diet—reduce epileptic seizures significantly. To date, the critical links between altered metabolism and decreased neuronal activity remain unknown. More generally, metabolic changes accompany numerous CNS disorders, and the purines ATP and its core molecule adenosine are poised to translate cell energy into altered neuronal activity. Here we show that nonpathological changes in metabolism induce a purinergic autoregulation of hippocampal CA3 pyramidal neuron excitability. During conditions of sufficient intracellular ATP, reducing extracellular glucose induces pannexin-1 hemichannel-mediated ATP release directly from CA3 neurons. This extracellular ATP is dephosphorylated to adenosine, activates neuronal adenosine A1 receptors, and, unexpectedly, hyperpolarizes neuronal membrane potential via ATP-sensitive K+ channels. Together, these data delineate an autocrine regulation of neuronal excitability via ATP and adenosine in a seizure-prone subregion of the hippocampus and offer new mechanistic insight into the relationship between decreased glucose and increased seizure threshold. By establishing neuronal ATP release via pannexin hemichannels, and hippocampal adenosine A1 receptors coupled to ATP-sensitive K+ channels, we reveal detailed information regarding the relationship between metabolism and neuronal activity and new strategies for adenosine-based therapies in the CNS.
Introduction
ATP and adenosine play dual roles in the nervous system. These purines participate in both cell energy and extracellular signaling and thus link metabolism and neuronal activity (Kato et al., 2004). Furthermore, ATP and adenosine are key signals between neurons and glia (Fellin et al., 2006), interact directly with other neurotransmitter systems (Ferré et al., 2007; Sichardt and Nieber, 2007), and are influenced dynamically by changes in ongoing physiology (Dunwiddie and Masino, 2001; Dulla et al., 2005, 2009). A more comprehensive understanding of the physiological or cellular mechanisms regulating ATP and adenosine holds promise for a variety of acute and chronic neurological disorders, particularly adenosine-augmenting therapies for brain injury, epilepsy, and neurodegenerative diseases (Boison, 2006; Li et al., 2008). To date, however, adenosine receptor-based strategies to stop seizures and enhance neuronal survival have been hampered by unacceptable side effects (Dunwiddie, 1999; Elzein and Zablocki, 2008).
In hippocampus and cerebral cortex, tonic extracellular adenosine inhibits neuronal activity presynaptically and postsynaptically via the adenosine A1 receptor (A1R) subtype (Dunwiddie and Masino, 2001). Recent evidence revealed gap junction hemichannels located on astrocytes (Fellin et al., 2006) and vesicular ATP release from astrocytes as sources of extracellular ATP (Pascual et al., 2005), confirmed the extracellular dephosphorylation of astrocyte-derived ATP to adenosine (Pascual et al., 2005), and demonstrated the significant influence of astrocyte-derived adenosine on brain activity via A1Rs (Fellin et al., 2006). Notably, hemichannels can reduce ischemic damage (Schock et al., 2008) and precipitate preconditioning (Lin et al., 2008), conditions in which A1Rs can play a critical inhibitory and neuroprotective role (Heurteaux et al., 1995; Johansson et al., 2001). Together, these findings underscore the signaling partnerships between neurons and glia, ATP and adenosine, and metabolism and neuronal activity, and highlight the therapeutic importance of understanding of the dynamics of ongoing metabolism, neuronal excitability, and the regulation of extracellular ATP and adenosine.
Whereas altered metabolism is a hallmark of many neurological conditions (Sas et al., 2007; Rezin et al., 2009), a cause-and-effect relationship is often unknown. One clear exception is observed during adherence to a high-fat low-carbohydrate (ketogenic) diet, a metabolic therapy recognized for decades to offer significant anticonvulsant benefits, particularly in pediatric and medically refractory epilepsy (Wilder, 1921; Neal et al., 2008). Ketogenic metabolism is characterized by moderate hypoglycemia, mitochondrial biogenesis and increased cell energy molecules (DeVivo et al., 1978; Nakazawa et al., 1983; Bough et al., 2006), yet the critical neuronal mechanism(s) underlying the success of dietary therapy remains enigmatic. To characterize and quantify the relationship between cell energy and neuronal excitability, we manipulated intracellular and extracellular energy and mimicked key aspects of ketogenic metabolism during whole-cell patch-clamp recordings in acute hippocampal slices. Here we report that clinically relevant conditions of reduced extracellular glucose with sufficient or increased intracellular ATP induce a novel autocrine regulation of CA3 pyramidal neuron excitability. We show that this effect is consequent to direct ATP release via pannexin-1 hemichannels, extracellular adenosine acting at A1Rs, and activation of ATP-sensitive K+ (KATP) channels.
Materials and Methods
Slice preparation.
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Trinity College animal use committee. Sprague Dawley rats or C57BL/6 mice [wild type or lacking A1Rs (Johansson et al., 2001)] aged 4–8 weeks of either sex were anesthetized with isoflurane and decapitated (age within this range and sex had no significant effect on the amplitude of reduced extracellular glucose ([glucose]e)-induced outward current) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Standard slice preparation and recording conditions were used, similar to our previous publications (Masino et al., 2002; Kawamura et al., 2004). Briefly, three to five coronal hippocampal slices of 300 μm thickness were made in ice-cold artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 3 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 11 glucose, 26 NaHCO3 (osmolarity 320 mOsm, pH 7.4, when saturated with 95% O2 plus 5% CO2) with a vibrating slice cutter (Series 1000, Vibratome). The slices were incubated in aCSF saturated with 95% O2 plus 5% CO2 for 30–40 min at 37°C, then kept at room temperature until the recording. The slice was fixed in a recording chamber (∼0.4 ml volume, RC-26, Warner Instruments) under nylon mesh attached to a U-shaped platinum frame and submerged in and continuously perfused with aCSF at a flow rate of 2 ml min−1 at 32−34°C.
Extracellular glucose was reduced but never removed completely (reduced from standard aCSF with 11 mm to either 3 or 7 mm). Standard [glucose]e in aCSF is maintained at a higher concentration than in vivo (in which it is closer to 3 mm; Shram et al., 1997); using extracellular recordings we confirmed that switching to 3 mm [glucose]e in slices is not pathological (no more than a 12.25% decrease in amplitude measured at 25 min; n = 4, data not shown).
Reduced [glucose]e was achieved by replacing glucose with sucrose. Only one change in [glucose]e or one manipulation of any kind (i.e., pharmacological characterization, ATP manipulation) was tested in each slice. CA3 pyramidal cells or astrocytes were visually identified with an infrared-differential interference contrast (IR-DIC) videomicroscope (Zeiss Axioskop2 FS plus, Carl Zeiss; CoolSnap HQ camera, PhotoMetrics).
Whole-cell transmembrane current recordings.
Patch electrodes (3–5 MΩ) were fabricated from borosilicate glass capillaries of 1.5 mm outer diameter with a programmable puller (P-97, Sutter Instruments) and filled with an intracellular solution containing the following (in mm): 117 K-gluconate, 3.0 KCl, 1.0 EGTA, 3.0 MgCl2, 10.0 HEPES (free acid), 1.0 CaCl2, 2.0 Mg2+-ATP, pH 7.3, with KOH. When changing [ATP], Mg2+-ATP was replaced by KCl and MgCl2. [ATP] in the pipette was typically 2.0 mm but was also held at 0.5, 1.0, and 5.0 mm as described in specific experiments below. Only one [ATP] was tested per neuron. The 2.0 mm intracellular adenosine concentration solution was made by adding 2.0 mm adenosine to the 0.5 mm ATP intracellular solution. A cesium-based intracellular solution containing the following (in mm): 117 Cs-methanesulfonate, 3.0 CsCl, 1.0 EGTA, 3.0 MgCl2, 10.0 HEPES, 1.0 CaCl2, 2.0 Mg2+-ATP was used to obtain the I–V curve shown below (see Figure 7). To visually identify the astrocytes (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), Lucifer yellow was included in the cesium-based intracellular solution (0.5 mg/ml) and visualized by excitation with a 495 nm xenon lamp with a fluorescent microscope (Zeiss Axioskop2 FS plus and LAMBDA DG-4, Sutter Instruments).
Figure 7.
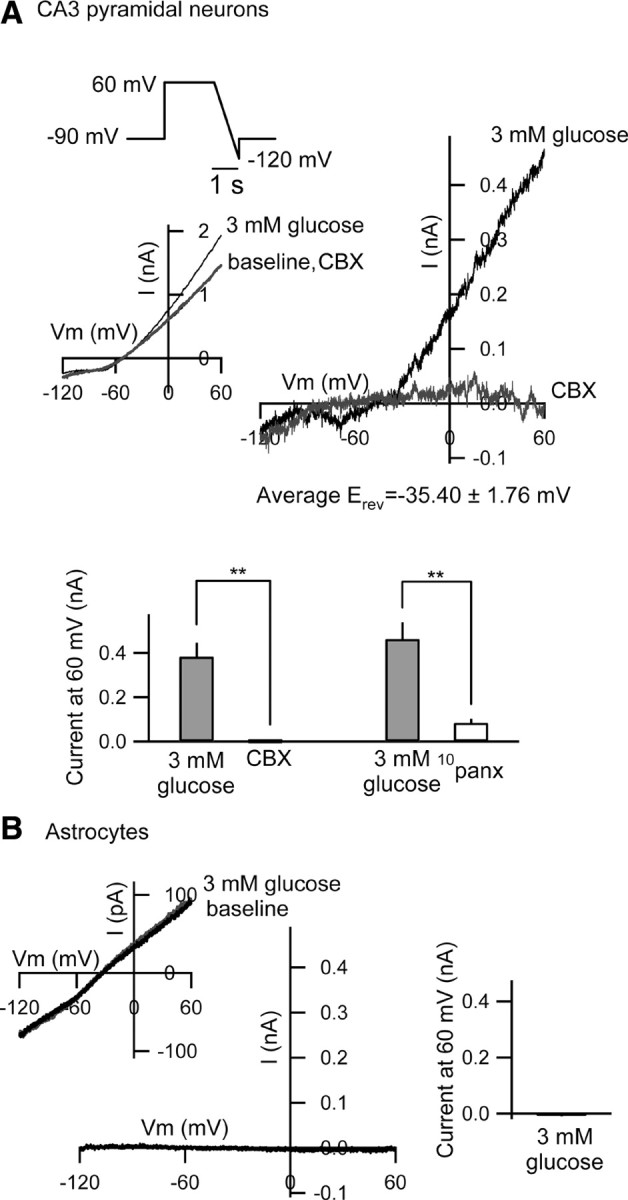
I–V relationship of the reduced [glucose]e-induced current. A, The I–V curve generated with reduced [glucose]e with cesium-based, 2 mm [ATP]i solution in the presence of tolbutamide (500 μm) in rat CA3 pyramidal neurons. The ramp voltage command (+60 to −120 mV, 180 mV/s) was applied before, during reduced [glucose]e and after antagonist application. The E rev was −35.40 ± 1.76 mV (n = 8), and this I–V curve was suppressed significantly by CBX (100 μm, n = 4) or 10panx (100 μm, n = 4). Ramp responses in baseline and after CBX are largely overlapping; **p < 0.01. B, In rat astrocytes recorded in the stratum radiatum, reduced [glucose]e causes no current with cesium-based, 2 mm [ATP]i solution in the presence of tolbutamide (500 μm). The ramp voltage command was as in A. Ramp responses in baseline and after 3 mm glucose are largely overlapping. Right graph shows the summary of the current at 60 mV (n = 4). Cell identification of the astrocytes is shown in supplemental Figure 4, available at www.jneurosci.org as supplemental material.
After obtaining a cell-attached configuration (1–10 GΩ seal resistance), the whole-cell recording mode was established with a brief negative current and pressure pulses. Recording started at least 15 min after the rupture of the patch membrane to allow stabilization of the intracellular milieu. Neurons showing unstable or large (more than ∼50 pA) holding currents were rejected, and holding current was required to be stable for a minimum of 5 min to proceed. Series resistance (<40 MΩ) and membrane capacitance were compensated and checked regularly. Unless otherwise noted, during recording the membrane potential was held at −70 mV corrected for liquid junction potential. To measure input resistance, negative currents of 100 pA were applied for 1 s every 30 s (Fig. 1). For measuring the I–V curve of the outward currents, the ramp voltage commands (−50 mV to −150 mV at 100 mV/s; duration time, 1 s) were applied before and during reduced [glucose]e application (see Fig. 4 A). Below (see Fig. 7), ramp voltage commands (+60 to −120 mV, 180 mV/s; duration time 1 s) were applied every 1 min due to the very slight inward current. The transmembrane current and potential were recorded with a MultiClamp 700A amplifier (Molecular Devices) with the high-cutoff filter at 2 kHz. The membrane current was digitized (16-channel A/D board, National Instruments) and analyzed on-line using Igor Pro 5 with Igor NIDAQ Tools MX (WaveMetrics).
Figure 1.

Reduced [glucose]e causes hyperpolarization in rat CA3 pyramidal neurons. A, The membrane potential trace (top) and the time course of the changes in the input resistance (bottom) in the CA3 pyramidal neuron recorded with standard (2 mm) ATP in the intracellular solution. Negative currents of 100 pA were applied every 30 s to measure input resistance. The [glucose]e was switched from 11 to 3 mm at the horizontal bar (reduced glucose). B, C, Summaries of the effect of low-glucose extracellular solution on the membrane potentials (B) and the input resistances (C). *p < 0.05; **p < 0.01; n = 5.
Figure 4.

Reduced [glucose]e opens K+ channels in rat CA3 pyramidal neurons. A, I–V relationship of the outward current generated with reduced [glucose]e. The ramp voltage command (−50 mV to −150 mV at 100 mV/s) was applied before (1) and during (2) reduced [glucose]e application. The reversal potential (E rev) of the outward current was −92.75 ± 1.67 mV (n = 4). The I–V relationship showed inward rectification and the average E rev was similar to the equilibrium potential for K+: −92.89 mV. B, BaCl2 (1 mm) reversed (left trace and right panel; n = 5) or prevented (pretreatment, right panel; n = 4) completely the outward current caused with reduced glucose.
Drugs and their application.
All chemicals and drugs were purchased from Sigma. Pannexin-1 mimetic blocking peptide 10panx (WRQAAFVDSY, with C-terminal amidation) was synthesized by Biomatik. As described in our previous publications (Masino and Dunwiddie, 1999; Dulla et al., 2005, 2009), all drugs were dissolved in aCSF at 100 times the desired final concentration and applied via syringe pump (Model A-99, Razel Scientific Instruments) to reach the final bath concentration. In all figures, the point indicated as the onset of drug application is the calculated time when the solution first begins to mix into the volume of the slice chamber. For pretreatments, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), carbenoxolone (CBX), octanol, BaCl2, or tolbutamide was perfused at least for 10 min before testing the effects of reduced [glucose]e. After application of any antagonist or blocker, no additional neurons were recorded from that slice. Because there was never more than one manipulation per slice, no more than one neuron from any slice was included in the final dataset.
Data and statistical analysis.
The recorded membrane potential or current was analyzed off-line with Igor Pro 5. The application of reduced [glucose]e and drugs is indicated at the time point these solutions began to enter the slice chamber. The amplitude of the outward current was measured as the difference between baseline before changing [glucose]e and the peak current (in reduced [glucose]e) and as the difference between the baseline current and the current remaining after reversal by an antagonist. When no significant change in current was detected after the pretreatment with antagonists, the amplitude of the outward current was measured at 12 min after reduced [glucose]e. This time point was standardized and determined by the average time to peak current with 3 mm [glucose]e applied during 2 mm [ATP]i (739.39 ± 69.68 s; n = 9). The mean spontaneous postsynaptic current (PSC) frequency was quantified during a 1 min baseline period, during reduced [glucose]e(either at the peak of the outward current or 12 min after reduced [glucose]e in pretreatment experiments when no change in current was observed), and after blocker application to compare spontaneous PSC frequencies. The PSCs were counted with using an Igor Pro procedure. All detected events were monitored and checked visually to ensure that all detected events had typical PSC waveforms. All data are expressed as mean ± SE (SEM). Amplitudes of reduced [glucose]e-induced outward currents were compared with one-way ANOVA; spontaneous PSC frequencies were compared with paired t test. p < 0.05 was considered significant.
Results
Reduced glucose and A1R-mediated inhibition
During whole-cell recording of visualized CA3 pyramidal neurons in rat hippocampal slices we manipulated intracellular ATP ([ATP]i) and/or [glucose]e. CA3 is a region with recurrent excitatory collaterals, used often for in vitro models of epilepsy and central to some types of seizure disorders. While most in vitro studies of hypoglycemia use a nominally glucose-free solution—often combined with hypoxia or anoxia to mimic conditions during an ischemic stroke—we used a nonpathological and physiologically relevant condition of reduced [glucose]e.
Upon reducing [glucose]e to 3 mm during current-clamp recordings we observed a significant and reversible hyperpolarization and concomitant decrease in input resistance (Fig. 1). We tested the influence of intracellular ATP on this low-glucose-induced hyperpolarization by loading whole-cell patch pipettes with a range of [ATP]i (0.5–5.0 mm; standard solution contains 2.0 mm). During voltage-clamp recordings (holding potential −70 mV), we found that [ATP]i bidirectionally determines the effects of reduced [glucose]e (Fig. 2 A,B). [ATP]i concentrations of ≥1.0 mm all resulted in a significant outward current, while when [ATP]i was reduced to 0.5 mm, reduced [glucose]e caused a large but transient inward current (Fig. 2 A,B). A linear relationship held for [ATP]i between 0.5 and 2.0 mm but not for 5.0 mm [ATP]i; at this concentration the amplitude of the outward current was reduced (Fig. 2 A,B). During normal [ATP]i (2.0 mm) the effects of reducing [glucose]e are dose-dependent, with an inverse relationship between [glucose]e and the amplitude of the outward current (Fig. 2 C). In addition, spontaneous PSC frequency was inhibited in a dose-dependent manner by both [ATP]i (Fig. 2 D; supplemental Fig. 2A, available at www.jneurosci.org as supplemental material) and [glucose]e (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Unlike the amplitude of the outward current, the linear relationship between [ATP]i and the inhibition of PSC frequency persisted with 5.0 mm [ATP]i, suggesting insensitivity to [ATP]i and thus mediation by different mechanisms. EPSCs but not IPSCs are sensitive to adenosine in CA3 pyramidal neurons (Thompson et al., 1992; Kawamura et al., 2004). Thus, presynaptic inhibition (measured as decreased PSC frequency) and postsynaptic hyperpolarization (measured as increased outward current amplitude) are both altered significantly and consistently by small changes in [ATP]i and [glucose]e.
Figure 2.
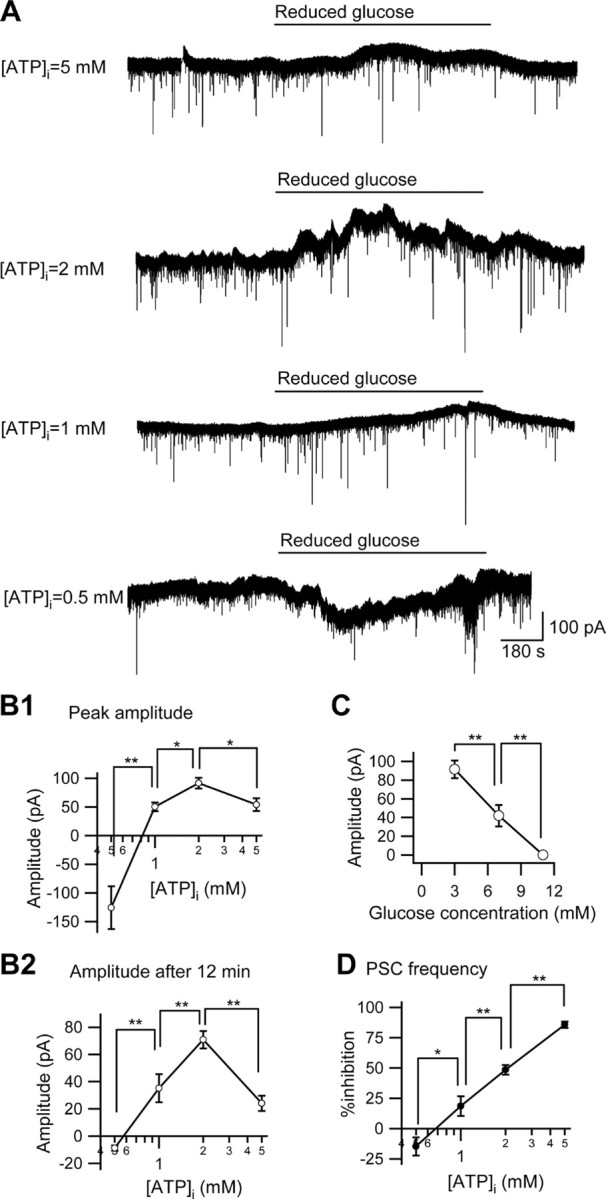
The effect of different concentrations of intracellular ATP or extracellular glucose on the reduced [glucose]e-induced outward current in CA3 pyramidal neurons. A, The membrane current traces of four different intracellular ATP concentrations ([ATP]i = 5, 2, 1, and 0.5 mm) with reducing [glucose]e from 11 to 3 mm (reduced glucose). The holding potential was −70 mV. B1, B2, Summaries of the significant effect of intracellular ATP concentration on the peak current amplitude (B1) and the amplitude 12 min after changing the glucose concentration (B2) in reduced glucose-induced outward currents ([ATP]i = 0.5 mm, n = 4; [ATP]i = 1 mm, n = 5; [ATP]i = 2 mm, n = 9; [ATP]i = 5 mm, n = 4). C, Summary of the significant dose-dependency of the effects of reduced [glucose]e with [ATP]i = 2 mm (3 mm [glucose]e, n = 9; 7 mm [glucose]e, n = 5). D, PSC frequency was decreased significantly by reduced [glucose]e with a dose-dependent relationship to [ATP]i. *p < 0.05; **p < 0.01.
Hypoglycemia is known to increase extracellular adenosine (Fowler, 1993), and presynaptic and postsynaptic A1Rs cause decreased spontaneous excitatory PSC frequency and hyperpolarization, respectively, in CA3 pyramidal neurons (Kawamura et al., 2004). We determined the role of A1Rs in this reduced [glucose]e-induced outward current by applying a selective A1R antagonist, DPCPX (1.0 μm). As expected, DPCPX itself caused a small inward current and increase of PSC frequency, due to blocking tonic activation of presynaptic and postsynaptic A1Rs (Fig. 3 A; supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). DPCPX reversed or prevented completely the outward current observed during reduced [glucose]e (Fig. 3 A), and also blocked the decrease in PSC frequency (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Genetic knock-out confirmed these pharmacological results: reduced [glucose]e induced a significant outward current in CA3 pyramidal neurons recorded from wild-type mouse slices but had no effect on those recorded from A1R knock-out mouse slices (Fig. 3 B). Thus, during conditions of sufficient or high [ATP]i, reduced [glucose]e caused a significant A1R-mediated regulation of neuronal activity. Note that current traces are more stable in the presence of an antagonist or in the A1R knock-out mouse slices, perhaps due to eliminating the dynamics of the autocrine mechanism with a pharmacological or genetic block.
Figure 3.

Reduced [glucose]e-induced outward current is mediated by activation of A1Rs. A, DPCPX (1 μm) reversed significantly (top) or prevented (bottom) completely the reduced [glucose]e-induced outward current in CA3 pyramidal neurons recorded from Sprague Dawley rat hippocampal slices. Average data shown in right panel for reversal (top right; n = 5) and prevention (bottom right; n = 4). B, Reduced [glucose]e caused a significant DPCPX-sensitive outward current in CA3 neurons recorded from wild-type (WT) mouse (top left) but had no effect on CA3 neurons recorded from the A1R receptor knock-out (A1R-KO) mouse (bottom left). Average data shown in right panel for WT mice (n = 6) and A1R-KO mice (n = 8). NS, Not significantly different; **p < 0.01.
The reversal potential and shape of the current–voltage (I–V) curve of the outward current-induced by reduced [glucose]e indicated that inwardly rectifying K+ channels were opened (Fig. 4 A). Initially, to test the coupling of A1Rs to K+ channels in CA3, we applied barium chloride (BaCl2), a nonselective K+ channel antagonist, either before or after the application of reduced [glucose]e while recording with a patch pipette loaded with 2.0 mm [ATP]i. BaCl2 (1.0 mm) itself caused a small inward current, likely due to tonic activation of A1Rs and their coupling to postsynaptic K+ channels (Fig. 4 B). BaCl2 reversed or prevented the effects of reduced [glucose]e (Fig. 4 B), identifying K+ channels as the source of outward current induced by reduced [glucose]e.
A1Rs couple to KATP channels in CA3 pyramidal neurons
Postsynaptic activation of A1Rs is associated traditionally with G-protein-coupled inwardly rectifying K+ (GIRK) channels in CA1 pyramidal neurons, but detailed studies have not been published in CA3 pyramidal neurons, and BaCl2 nonselectively blocks both GIRK and KATP channels (Guatteo et al., 1998). The amplitude of outward current induced with reduced [glucose]e was attenuated in 5.0 mm [ATP]i (Fig. 2 A,B), indicating that high [ATP]i may be inhibiting the effects of reduced [glucose]e. In contrast to the postsynaptic effect in the CA3 neuron, the inhibition of PSC frequency was related linearly to [ATP]i (Fig. 2 D). Brain KATP channels can link to A1Rs (Andoh et al., 2006), and these K+ channels are expressed functionally in CA3 pyramidal neurons (Griesemer et al., 2002).
To determine whether KATP channels were involved in the outward current induced with reduced [glucose]e we applied tolbutamide (500 μm), a selective KATP channel blocker. Tolbutamide alone had no effect on the holding potential (Fig. 5 A), suggesting that tonic A1R activation in CA3 pyramidal neurons is not coupled specifically to KATP channels. However, tolbutamide significantly reversed or prevented the outward current caused by reduced [glucose]e (Fig. 5 A). The decrease in PSC frequency with reduced [glucose]e was not inhibited by tolbutamide (Fig. 5 B) or BaCl2 (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), indicating that the decreased PSC frequency is due to a distinct non-K+ channel presynaptic mechanism. Together, the results shown in Figures 3 –5 demonstrate that the reduced [glucose]e-induced outward current is mediated by the opening of postsynaptic KATP channels linked to the activation of A1Rs.
Figure 5.

Postsynaptic KATP channels are opened by reduced [glucose]e. A, Tolbutamide (500 μm) reversed (left trace and right panel; n = 5) or prevented (pretreatment, right panel; n = 4) significantly the outward current caused by reduced glucose. B, Tolbutamide did not prevent (right) or reverse (left traces and middle) the decrease in PSC frequency with reduced glucose. Left traces “1” through “3” are time-extended traces taken at the indicated points in A. NS, Not significantly different; *p < 0.05; **p < 0.01.
Direct ATP release from pannexin-1 hemichannels
Increased extracellular adenosine activating A1Rs could be consequent to the dephosphorylation of extracellular ATP or direct adenosine release via nucleoside transporters. Previous research indicates that the direction of adenosine transport is predominantly inward and serves to remove adenosine from the extracellular space (Dunwiddie and Diao, 1994). In contrast, ATP has been shown to efflux directly from astrocytes, either via hemichannels or vesicular release (Fellin et al., 2006). To determine physiologically whether the increase in A1R activity was caused by direct efflux of adenosine or of ATP, we loaded cells with a combination of 2.0 mm adenosine and 0.5 mm ATP. As we determined previously, in reduced [glucose]e 0.5 mm [ATP]i causes an inward current and 2.0 mm [ATP]i causes an outward current (Fig. 2 A,B). Thus this test combination provided some cell energy (0.5 mm [ATP]i) and an adenosine concentration equivalent to the ATP concentration shown to produce the biggest outward current during reduced [glucose]e (2 mm [ATP]i) (Fig. 2 A,B). Upon intracellular loading with 2.0 mm adenosine and 0.5 mm ATP, reduced [glucose]e caused small transient outward current, significantly lower in amplitude that observed with 2.0 mm [ATP]i (Fig. 6 A). No significant current was observed after 15 min, unlike with 2.0 mm [ATP]i (Fig. 6 A), indicating that the larger and sustained outward current observed during reduced [glucose]e with 2.0 mm [ATP]i was not caused by the direct release of adenosine. A similar pattern was found with PSCs: upon intracellular loading with 2.0 mm adenosine and 0.5 mm ATP, PSC frequency was not different from that found with 0.5 mm [ATP]i alone (0.50 ± 0.11 Hz vs 0.48 ± 0.10 Hz, n = 7, nonsignificant). This small and transient effect of 2.0 mm intracellular adenosine is in contrast to the larger and sustained inhibitory effect of reduced [glucose]e with 2.0 mm [ATP]i (Fig. 2 D), indicating that reduction of PSC frequency was not caused by the direct release of adenosine.
Figure 6.

Autocrine regulation due to ATP release through pannexin-1 hemichannels during reduced [glucose]e. A, With 2 mm intracellular adenosine ([Ado]i) and 0.5 mm [ATP]i, reduced [glucose]e caused a small, transient outward current. The peak amplitude with 2 mm [Ado]i was significantly smaller than that with 2 mm [ATP]i (left), and unlike with 2 mm [ATP]i, no significant current was observed after 15 min (2 mm [ATP]i, n = 9; 2 mm [Ado]i and 0.5 mm [ATP]i, n = 7; right). B, CBX (10 or 100 μm) reversed (top trace and lower left; n = 5 per group) or prevented (lower right; n = 4 per group) significantly the outward current caused with reduced glucose. C, Octanol (1 mm) reversed (top trace and lower left; n = 5) or prevented (lower right; n = 4) significantly the outward current caused with reduced glucose. D, 10panx (100 μm) reversed (top trace and lower left; n = 5) or prevented (lower right; n = 4) significantly the outward current caused with reduced glucose. NS, Not significantly different; **p < 0.01.
To determine pharmacologically whether CA3 pyramidal neurons were releasing ATP directly, we applied both selective and nonselective gap junction hemichannel antagonists. We could both reverse and prevent the outward current caused by reduced [glucose]e significantly with either CBX (10 or 100 μm)(Fig. 6 B) or octanol (1.0 mm)(Fig. 6 C). The decrease in spontaneous PSC frequency was also inhibited significantly by CBX (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material) or octanol (supplemental Fig. 3D, available at www.jneurosci.org as supplemental material), suggesting that presynaptic and postsynaptic A1R activation is subsequent to ATP released by gap junction hemichannels during reduced [glucose]e. Similar results were obtained with a low concentration of CBX (10 μm) known to block pannexin but not connexin hemichannels (Romanov et al., 2007) (Fig. 6 B). To confirm pannexin hemichannel opening in CA3 neurons, we tested a specific inhibitor of pannexin-1 hemichannels (Pelegrin and Surprenant, 2006; Thompson et al., 2008). The peptide 10panx (100 μm) significantly reduced or prevented the outward current (Fig. 6 D) and decrease in spontaneous PSC frequency with reduced [glucose]e (supplemental Fig. 3E, available at www.jneurosci.org as supplemental material). This pharmacology indicates direct ATP release through pannexin-1 hemichannels, during sufficient or high [ATP]i and reduced [glucose]e.
To determine physiologically the identity of the channel opened, we did an I–V analysis of the isolated current recorded during reduced [glucose]e. Opening pannexin hemichannels causes current flow across cell membranes, yet we did not detect any change in membrane current even while preventing the K+ conductance in the presence of DPCPX, BaCl2 or tolbutamide at a holding potential of −70 mV (Figs. 3 A, 4 B, and 5 A, respectively). At a −90 mV holding potential, however, reduced [glucose]e caused a slight inward current in CA3 pyramidal neurons in the presence of tolbutamide (peak inward current 5.49 ± 3.10 pA; p = 0.13). We checked the I–V curve of this slight inward current with cesium-based intracellular solution in the presence of extracellular tolbutamide. Under these conditions we recorded a nonlinear I–V relationship with a reversal potential near −35 mV in CA3 pyramidal neurons (Fig. 7 A) but not in astrocytes in stratum radiatum (Fig. 7 B). This nonlinear I–V relationship and reversal potential are most similar to those recorded from pannexin hemichannels (Romanov et al., 2007). The I–V curve with reduced [glucose]e was suppressed significantly by CBX or by 10panx (Fig. 7 A), indicating that this nonlinear I–V curve stems from pannexin-1 hemichannel opening in CA3 pyramidal neurons.
Together, the data presented herein elucidate a novel and comprehensive autocrine regulation of CA3 pyramidal neuron excitability. Under physiologically relevant conditions of reduced [glucose]e and sufficient [ATP]i, CA3 pyramidal neurons release ATP directly from pannexin-1 hemichannels, extracellular ATP is dephosphorylated to adenosine which acts at A1Rs, and A1Rs couple to KATP channels (Fig. 8).
Figure 8.

Schematic of the purinergic autocrine regulation in CA3 pyramidal neurons. With sufficient [ATP]i (1), reducing [glucose]e (2) induces neuronal ATP release directly via pannexin hemichannels (3); released ATP is dephosphorylated to adenosine (4) to activate adenosine A1 receptors (5) and, ultimately, decrease neuronal excitability by opening KATP channels (6).
Discussion
To date, the relative roles of [ATP]i versus [glucose]e in the dynamic regulation of extracellular adenosine have not been well established, yet are relevant to both normal physiology as well as clinical conditions such as stroke and metabolic maintenance on ketogenic diet therapy. Both of these clinical situations result in reduced [glucose]e, but only one (stroke) appears to be associated with a significant loss of [ATP]i. Here, we demonstrate a novel autocrine regulation of CA3 pyramidal neuron activity upon a metabolic shift characteristic of ketogenic metabolism: reduced [glucose]e coupled with sufficient or high [ATP]i causes a significant hyperpolarization and decreased neuronal excitability—consistent with the anticonvulsant and neuroprotective effects of the ketogenic diet (and fasting (Davis et al., 2008)). Conversely, low [ATP]i and reduced [glucose]e caused pyramidal neuron depolarization—consistent with anoxic depolarization observed in models of ischemia and mild stroke (Yamamoto et al., 1997).
The effects of reduced [glucose]e during sufficient/high [ATP]i are due to increased extracellular adenosine acting at A1Rs. The outward current induced by reduced [glucose]e was prevented or reversed completely by a selective A1R antagonist. None of the electrophysiological consequences of high [ATP]i and reduced [glucose]e were observed in A1R knock-out mouse tissue, validating A1Rs as a critical link between changes in metabolism and altered neuronal excitability. Because the outward current mobilized by reduced [glucose]e was not reproduced by intracellular loading with a concentration of adenosine equal to the most effective concentration of ATP, it was initial evidence that the released molecule is ATP, which would be dephosphorylated rapidly to adenosine extracellularly (Dunwiddie et al., 1997). Involvement of released intracellular ATP was confirmed as these effects were either prevented or reversed completely with both selective and nonselective gap junction hemichannel antagonists, including a pannexin-selective concentration of CBX and the selective peptide blocker 10panx. Furthermore, the I–V curve during blockade of K+ channels showed the I–V curve typical of pannexin hemichannels as reported previously (Romanov et al., 2007).
Together, these data delineate a direct role for pannexin-1 hemichannel-mediated ATP release in autoregulating CA3 neuronal excitability based on metabolic status. Published reports have been mixed on the cellular localization of pannexin-1 hemichannels—either showing an exclusively neuronal localization of pannexin-1 in hippocampus (Litvin et al., 2006) or providing evidence for their functional expression in cultured astrocytes (Iglesias et al., 2009). An autocrine neuronal mechanism, rather than one involving neighboring glia, is supported for several reasons: (1) we manipulated ATP only within the recorded neuron, (2) in that neuron we found a pannexin-dependent current, and (3) we measured no change in current in identified astrocytes in response to the same manipulation (Fig. 7 B). Thus, during reduced [glucose]e with sufficient intracellular ATP and thus adequate cell energy charge, some neuronal ATP is released directly to inhibit neuronal excitability via A1Rs—a neuroprotective pathway to prevent increased excitability, additional ATP demand, and neuronal dysfunction (Abdelmalik et al., 2007).
While it may seem counterintuitive for a cell to release ATP during lowered extracellular glucose, the high [ATP]i-to-[adenosine]i ratio, the rapid dephosphorylation of ATP by ecto-enzymes near A1Rs (Dunwiddie et al., 1997; Cunha et al., 1998), the functional dominance of the inhibitory A1Rs in hippocampus (Johansson et al., 2001), and equilibrative transporters to influx adenosine form a comprehensive set of mechanisms to ensure both the efficiency and efficacy of this autocrine neuronal regulation. Thus, while the released ATP has little effect on overall ATP levels, the resulting increase in the local concentration of extracellular concentration of adenosine hyperpolarizes the neuron directly to reduce further energy demand. The comprehensive autocrine mechanism described here includes ATP released from neuronal pannexin hemichannels, subsequent activation of A1Rs, and ultimately decreased CA3 pyramidal neuron excitability via opening of postsynaptic KATP channels (Fig. 8).
Unexpectedly, we revealed that postsynaptic A1Rs in hippocampal CA3 pyramidal neurons can couple to KATP channels. A1R-mediated hyperpolarization in hippocampus has been shown previously to be coupled to postsynaptic GIRK channels (Trussell and Jackson, 1987), and we verified ongoing GIRK activity in CA3 pyramidal neurons during baseline recording conditions. We confirmed K+ channels as the source of the outward current in the autocrine-mediated decrease in excitability, and determined pharmacologically that the outward current mobilized by reduced [glucose]e is due to the opening of postsynaptic KATP channels, a new finding in hippocampal neurons.
Previously, a different in vitro model of the ketogenic diet (i.e., direct application of ketones to substantia nigra pars reticulata neurons) demonstrated hyperpolarization via KATP channels (Ma et al., 2007), consistent with our present findings. In this study the KATP channels are assumed to be opened by the traditional signal of a decrease in [ATP]i, perhaps in a localized area inside the cell; direct measurements of ATP and cell energy molecules during maintenance on a ketogenic diet reveal a net increase. As an alternate explanation, the KATP channels could also be coupled to A1Rs (Andoh et al., 2006). Here we focus on key aspects of a ketone-based metabolic state—increased ATP and decreased glucose—and record from a brain region and cell type relevant to both experimental seizure models and clinical seizure disorders.
Current trace fluctuations in reduced glucose are minimized in the presence of antagonists or genetic deletion of A1Rs, suggesting a dynamic feedback. These fluctuations may be a hysteresis due to transient microdomains of ATPi and intermittent mobilization of the autocrine response, which would be expected to have ongoing feedback and associated fluctuations; future studies could quantify these effects. Together, the novel autocrine mechanism we delineate here includes ATP released directly from gap junction hemichannels located on CA3 pyramidal neurons, subsequent activation of neuronal A1Rs, and ultimately decreased neuronal excitability via opening of KATP channels (Fig. 8).
As noted, a causal relationship between metabolism and neuronal activity has been well documented in persons with epilepsy (Hallböök et al., 2007; Remahl et al., 2008; Yum et al., 2008; Freeman et al., 2009). Either fasting or adhering to a high-fat, low carbohydrate (ketogenic) diet can offer a highly effective anticonvulsant treatment, particularly in children and even in cases of medically refractory epilepsy (Neal et al., 2008; Zupec-Kania and Spellman, 2008; Kossof and Rho, 2009). Levels of the ketone bodies themselves are not well correlated with the therapeutic effects of the diet (Seymour et al., 1999; Musa-Veloso et al., 2006), but low glucose is thought to be a key factor (Huttenlocher, 1976; Freeman et al., 2009). This study combines two consequences of ketogenic metabolism increasingly considered essential to its therapeutic effects—increased intracellular ATP and low extracellular glucose (Kim and Rho, 2008). We provide cellular evidence, in a relevant brain region, for a powerful link between altered metabolism and an increased inhibitory influence of adenosine, and a new insight into the cooperative role between ATP and adenosine in regulating neuronal activity.
Augmenting activity of A1Rs offers a long-recognized and powerful anticonvulsant and neuroprotective influence and has broad acute and chronic therapeutic potential. The novel autocrine neuronal regulation revealed herein may shed light on cellular mechanisms essential for the paradoxical neuroprotective effects of fasting (Davis et al., 2008) and metabolic (Korde et al., 2005; Pandya et al., 2007) or glycolytic (Garriga-Canut et al., 2006; Stafstrom et al., 2009) inhibition, including improved recovery after spinal cord injury (Plunet et al., 2008), and could provide the critical and coveted neuronal mechanism underlying the anticonvulsant effects of fasting, ketogenic diet therapy, and glycolytic inhibition.
Footnotes
This work was supported by the National Institutes of Health (NS-061290 to S.A.M.), the National Science Foundation (IOS-0843585 to S.A.M.), and Trinity College. We thank Dr. Bertil B. Fredholm for comments on this manuscript.
References
- Abdelmalik PA, Shannon P, Yiu A, Liang P, Adamchik Y, Weisspapir M, Samoilova M, Burnham WM, Carlen PL. Hypoglycemic seizures during transient hypoglycemia exacerbate hippocampal dysfunction. Neurobiol Dis. 2007;26:646–660. doi: 10.1016/j.nbd.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Andoh T, Ishiwa D, Kamiya Y, Echigo N, Goto T, Yamada Y. A1 adenosine receptor-mediated modulation of neuronal ATP-sensitive K channels in rat substantia nigra. Brain Res. 2006;1124:55–61. doi: 10.1016/j.brainres.2006.09.085. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM, Ribeiro JA. Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J Neurosci. 1998;18:1987–1995. doi: 10.1523/JNEUROSCI.18-06-01987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008;86:1812–1822. doi: 10.1002/jnr.21628. [DOI] [PubMed] [Google Scholar]
- DeVivo DC, Leckie MP, Ferrendelli JS, McDougal DB., Jr Chronic ketosis and cerebral metabolism. Ann Neurol. 1978;3:331–337. doi: 10.1002/ana.410030410. [DOI] [PubMed] [Google Scholar]
- Dulla CG, Dobelis P, Pearson T, Frenguelli BG, Staley KJ, Masino SA. Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron. 2005;48:1011–1023. doi: 10.1016/j.neuron.2005.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulla CG, Frenguelli BG, Staley KJ, Masino SA. Intracellular acidification causes adenosine release during states of hyperexcitability in the hippocampus. J Neurophysiol. 2009;102:1984–1993. doi: 10.1152/jn.90695.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV. Adenosine and suppression of seizures. Adv Neurol. 1999;79:1001–1010. [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J Pharmacol Exp Ther. 1994;268:537–545. [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L, Proctor WR. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzein E, Zablocki J. A1 adenosine receptor agonists and their potential therapeutic applications. Expert Opin Investig Drugs. 2008;17:1901–1910. doi: 10.1517/13543780802497284. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Haydon PG. Astrocytes coordinate synaptic networks: balanced excitation and inhibition. Physiology (Bethesda) 2006;21:208–215. doi: 10.1152/physiol.00161.2005. [DOI] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007;30:440–446. doi: 10.1016/j.tins.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Purine release and inhibition of synaptic transmission during hypoxia and hypoglycemia in rat hippocampal slices. Neurosci Lett. 1993;157:83–86. doi: 10.1016/0304-3940(93)90648-5. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Vining EP, Kossoff EH, Pyzik PL, Ye X, Goodman SN. A blinded, crossover study of the efficacy of the ketogenic diet. Epilepsia. 2009;50:322–325. doi: 10.1111/j.1528-1167.2008.01740.x. [DOI] [PubMed] [Google Scholar]
- Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, Roopra A. 2-Deoxy-d-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9:1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- Griesemer D, Zawar C, Neumcke B. Cell-type specific depression of neuronal excitability in rat hippocampus by activation of ATP-sensitive potassium channels. Eur Biophys J. 2002;31:467–477. doi: 10.1007/s00249-002-0241-3. [DOI] [PubMed] [Google Scholar]
- Guatteo E, Federici M, Siniscalchi A, Knöpfel T, Mercuri NB, Bernardi G. Whole-cell patch-clamp recordings of rat midbrain dopaminergic neurons isolate a sulphonylurea- and ATP-sensitive component of potassium currents activated by hypoxia. J Neurophysiol. 1998;79:1239–1245. doi: 10.1152/jn.1998.79.3.1239. [DOI] [PubMed] [Google Scholar]
- Hallböök T, Köhler S, Rosén I, Lundgren J. Effects of ketogenic diet on epileptiform activity in children with therapy resistant epilepsy. Epilepsy Res. 2007;77:134–140. doi: 10.1016/j.eplepsyres.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci U S A. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr Res. 1976;10:536–540. doi: 10.1203/00006450-197605000-00006. [DOI] [PubMed] [Google Scholar]
- Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin1: the molecular substrate of astrocyte “hemichannels”. J Neurosci. 2009;29:7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Giménez-Llort L, Escorihuela RM, Fernández-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hårdemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato F, Kawamura M, Shigetomi E, Tanaka JI, Inoue K. ATP- and adenosine-mediated signaling in the central nervous system: synaptic purinoceptors: the stage for ATP to play its “dual-role”. J Pharmacol Sci. 2004;94:107–111. doi: 10.1254/jphs.94.107. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Gachet C, Inoue K, Kato F. Direct excitation of inhibitory interneurons by extracellular ATP mediated by P2Y1 receptors in the hippocampal slice. J Neurosci. 2004;24:10835–10845. doi: 10.1523/JNEUROSCI.3028-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim do Y, Rho JM. The ketogenic diet and epilepsy. Curr Opin Clin Nutr Metab Care. 2008;11:113–120. doi: 10.1097/MCO.0b013e3282f44c06. [DOI] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem. 2005;94:1676–1684. doi: 10.1111/j.1471-4159.2005.03328.x. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Rho JM. Ketogenic diets: evidence from short- and long-term efficacy. Neurotherapeutics. 2009;6:406–414. doi: 10.1016/j.nurt.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest. 2008;118:571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Lou N, Kang N, Takano T, Hu F, Han X, Xu Q, Lovatt D, Torres A, Willecke K, Yang J, Kang J, Nedergaard M. A central role of connexin 43 in hypoxic preconditioning. J Neurosci. 2008;28:681–695. doi: 10.1523/JNEUROSCI.3827-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvin O, Tiunova A, Connell-Alberts Y, Panchin Y, Baranova A. What is hidden in the pannexin treasure trove: the sneak peak and the guesswork. J Cell Mol Med. 2006;10:613–634. doi: 10.1111/j.1582-4934.2006.tb00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Dunwiddie TV. Temperature-dependent modulation of excitatory transmission in hippocampal slices is mediated by extracellular adenosine. J Neurosci. 1999;19:1932–1939. doi: 10.1523/JNEUROSCI.19-06-01932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Diao L, Illes P, Zahniser NR, Larson GA, Johansson B, Fredholm BB, Dunwiddie TV. Modulation of hippocampal glutamatergic transmission by ATP is dependent on adenosine A1 receptors. J Pharmacol Exp Ther. 2002;303:356–363. doi: 10.1124/jpet.102.036731. [DOI] [PubMed] [Google Scholar]
- Musa-Veloso K, Likhodii SS, Rarama E, Benoit S, Liu YM, Chartrand D, Curtis R, Carmant L, Lortie A, Comeau FJ, Cunnane SC. Breath acetone predicts plasma ketone bodies in children with epilepsy on a ketogenic diet. Nutrition. 2006;22:1–8. doi: 10.1016/j.nut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Nakazawa M, Kodama S, Matsuo T. Effects of ketogenic diet on electroconvulsive threshold and brain contents of adenosine nucleotides. Brain Dev. 1983;5:375–380. doi: 10.1016/s0387-7604(83)80042-4. [DOI] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7:500–506. doi: 10.1016/S1474-4422(08)70092-9. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG. Post-injury administration of mitochondrial uncouplers increases tissue sparing and improves behavioral outcome following traumatic brain injury in rodents. J Neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plunet WT, Streijger F, Lam CK, Lee JH, Liu J, Tetzlaff W. Dietary restriction started after spinal cord injury improves functional recovery. Exp Neurol. 2008;213:28–35. doi: 10.1016/j.expneurol.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Remahl S, Dahlin MG, Amark PE. Influence of the ketogenic diet on 24-hour electroencephalogram in children with epilepsy. Pediatr Neurol. 2008;38:38–43. doi: 10.1016/j.pediatrneurol.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Rezin GT, Amboni G, Zugno AI, Quevedo J, Streck EL. Mitochondrial dysfunction and psychiatric disorders. Neurochem Res. 2009;34:1021–1029. doi: 10.1007/s11064-008-9865-8. [DOI] [PubMed] [Google Scholar]
- Romanov RA, Rogachevskaja OA, Bystrova MF, Jiang P, Margolskee RF, Kolesnikov SS. Afferent neurotransmission mediated by hemichannels in mammalian taste cells. EMBO J. 2007;26:657–667. doi: 10.1038/sj.emboj.7601526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sas K, Robotka H, Toldi J, Vécsei L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci. 2007;257:221–239. doi: 10.1016/j.jns.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Schock SC, Leblanc D, Hakim AM, Thompson CS. ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro . Biochem Biophys Res Comm. 2008;368:138–144. doi: 10.1016/j.bbrc.2008.01.054. [DOI] [PubMed] [Google Scholar]
- Seymour KJ, Bluml S, Sutherling J, Sutherling W, Ross BD. Identification of cerebral acetone by 1H-MRS in patients with epilepsy controlled by ketogenic diet. MAGMA. 1999;8:33–42. doi: 10.1007/BF02590633. [DOI] [PubMed] [Google Scholar]
- Shram NF, Netchiporouk LI, Martelet C, Jaffrezic-Renault N, Cespuglio R. Brain glucose: voltammetric determination in normal and hyperglycaemic rats using a glucose microsensor. Neuroreport. 1997;8:1109–1112. doi: 10.1097/00001756-199703240-00009. [DOI] [PubMed] [Google Scholar]
- Sichardt K, Nieber K. Adenosine A1 receptor: functional receptor-receptor interactions in the brain. Purinergic Signal. 2007;3:285–298. doi: 10.1007/s11302-007-9065-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP. Anticonvulsant and antiepileptic actions of 2-deoxy-d-glucose in epilepsy models. Ann Neurol. 2009;65:435–447. doi: 10.1002/ana.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–1559. doi: 10.1126/science.1165209. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Haas HL, Gähwiler BH. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro . J Physiol. 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Jackson MB. Dependence of an adenosine-activated potassium current on a GTP-binding protein in mammalian central neurons. J Neurosci. 1987;7:3306–3316. doi: 10.1523/JNEUROSCI.07-10-03306.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder RM. The effects of ketonemia on the course of epilepsy. Mayo Clin Proc. 1921;2:307–308. [Google Scholar]
- Yamamoto S, Tanaka E, Shoji Y, Kudo Y, Inokuchi H, Higashi H. Factors that reverse the persistent depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol. 1997;78:903–911. doi: 10.1152/jn.1997.78.2.903. [DOI] [PubMed] [Google Scholar]
- Yum MK, Jung KY, Kang HC, Kim HD, Shon YM, Kang JK, Lee IK, Park KJ, Kwon OY. Effect of a ketogenic diet on EEG: analysis of sample entropy. Seizure. 2008;17:561–566. doi: 10.1016/j.seizure.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Zupec-Kania BA, Spellman E. An overview of the ketogenic diet for pediatric epilepsy. Nutr Clin Pract. 2008;23:589–596. doi: 10.1177/0884533608326138. [DOI] [PubMed] [Google Scholar]