

Abstract
Approximately 5 – 15% of all CRCs have an activating BRAF somatic mutation, which may be associated with a distinct risk profile compared to tumors without BRAF mutations. Here, we measured the prevalence and epidemiologic correlates of the BRAF V600E somatic mutation in cases collected as a part of a population-based case-control study of colorectal cancer in northern Israel. The prevalence of BRAF V600E was 5.0% in this population, and the mutation was more likely to be found in tumors from cases who were of Ashkenazi Jewish descent (OR = 1.87, 95% C.I., 1.01 – 3.47), female (OR = 1.97, p = 1.17 – 3.31) and older (73.8 years vs. 70.3 years, p < 0.001). These results were similar when restricting to only tumors with microsatellite instability. Whether or not smoking was associated with a BRAF somatic mutation depended on gender. While men were less likely to have a tumor with a BRAF somatic mutation, men who smoked were much more likely to have a tumor with a somatic BRAF mutation (ORinteraction = 4.95, 95% C.I., 1.18 – 20.83) than women who never smoked. We note the strong heterogeneity in the reported prevalence of the BRAF V600E mutation in studies of different ethnicities, with a lower prevalence in Israel than other Western populations, but a higher prevalence among Jewish than non-Jewish Israeli cases. Epidemiologic studies of colorectal cancer should incorporate somatic characteristics to fully appreciate risk factors for this disease.
Keywords: Colorectal cancer, somatic mutations, BRAF, epidemiology
Introduction
The heterogeneity of colorectal carcinogenesis gives rise to distinct tumor phenotypes with corresponding differences in disease prognosis and potentially even treatment (1-6). Examination of these phenotypes in population-based studies has led to an appreciation of distinct epidemiologic risk factors corresponding to the tumor phenotypes. For example, the concordant expansion or contraction of nucleotide repeats in microsatellite markers is indicative of a microsatellite instable (MSI-high) tumor with defective mismatch repair. The MSI-high phenotype, corresponding to 10 – 20% of all colorectal cancers (CRC), is associated with epidemiologic risk factors not appreciated in studies that do not consider MSI status of the tumor. These risk factors include female gender, estrogen withdrawal, smoking and NSAID use (6-14).
Approximately 5 – 15% of all CRCs have an activating BRAF somatic mutation, the vast majority of which are a substitution of glutamic acid for valine at codon 600, or V600E. These BRAF–mutated CRCs are comprised predominantly of sporadic rather than familial tumors that often arise from a methylator pathway (15-18). BRAF mutations are clinically useful in distinguishing tumors with MSI-high resulting from Lynch syndrome from sporadic MSI-high tumors (19-21). Studies of BRAF mutations in CRC find that BRAF positive tumors may be associated with age, family history of CRC, female gender and ethnicity (17, 22). Multiple studies have noted that CRC patients with BRAF mutations have a shorter survival than CRC patients with BRAF-wild type tumors (23-25), further indicating the importance of this subgroup of CRC.
To try to better understand the epidemiologic risk factors for clinically relevant subtypes of colorectal cancer, we genotyped the BRAF V600E somatic mutation in tumors collected as a part of the Molecular Epidemiology of Colorectal Cancer (MECC) study, a population-based study in northern Israel. We found that there is a distinct risk profile of gender, smoking and ethnicity beyond that which is found when only considering microsatellite instability status of the tumor.
Materials and Methods
Study population
The Molecular Epidemiology of Colorectal Cancer (MECC) study is a population-based, matched case-control study that includes 2126 incident CRC cases and corresponding matched controls. The MECC study participants have previously been described (26). Eligible cases include any person newly diagnosed with colorectal cancer between March 31, 1998 and April 1, 2004 in Northern Israel. Eligible cases were invited to participate and interviewed. Potential controls were matched for exact year of birth, sex, and primary residence. Individuals previously diagnosed with cancer of the colorectum were not eligible to participate. The study was approved by the Institutional Review Boards at the University of Michigan and Carmel Medical Center in Haifa. Written, informed consent was required for eligibility.
DNA Extraction from Tumor Slides
DNA was extracted from tumor slides as previously described (26). Briefly, tumor DNA was microdissected from unstained, recut slides of paraffin-embedded tumors. Areas for microdissection were circled by one pathologist (JKG) and the hematoxylin and eosin stained slide was used as a template. Following dissection from slides, xylene was added to remove paraffin and the DNA was precipitated with ethanol. Following centrifugation, the supernatant was discarded and the pellet was lyophilized. The pellet was resuspended in 100 μl Proteinase K buffer (50mM tris and 200 ng/μl Proteinase K). The samples were incubated overnight at 37°C and then denatured at 95°C.
Microsatellite Instability Analyses
Microsatellite instability (MSI) analyses were performed as previously described (27). Normal and tumor DNA were extracted from microdissected DNA and analyzed for the consensus panel of seven markers (28). Briefly, forward primers for Bat 25, Bat 26, BAT40, TGFβII, D2S125 and D5S346 and reverse primers for D17S250 were labeled with γ32P-ATP, and included in a 20 μl PCR reaction that included 1 μl of microdissected DNA. PCR products were run on 6% polyacrylamide gels for approximately 3 hours at 65 watts, and exposed to film at -80° C for twelve to twenty hours. Films were double scored and entered as stable, instable, or loss of heterozygosity (LOH); markers with LOH were not counted in MSI calculations. The threshold for MSI-high for less than 7 markers was ≥ 30%. Where data for all seven markers were available, tumors were designated as MSI-high if there was instability at three or more markers, MSI-low if there was instability at one or two markers, and microsatellite stable (MSS) if stable at all markers. Where data were available for less than all seven markers, tumors had to have data for at least three markers to be scored, one of which had to be a mononucleotide marker (BAT25, BAT26, BAT40).
Identification of BRAF Mutations
Mutations in BRAF codon 600 were identified by direct sequencing of exon 15 of BRAF following PCR amplification of DNA extracted from paraffin embedded samples. PCR reactions included 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 200 μM dNTPs, 100 ng both forward and reverse primer, 1.5 U AmpliTaq Gold (Applied Biosystems), and 2μl microdissected tumor DNA in a total volume of 50 μl. Samples were denatured for 5 minutes at 95°C and were passed through 40 cycles of amplification, which consisted of 60 seconds of denaturation at 95°C, 1 minute of primer annealing at 56°C, and 1 minute of elongation at 72°C. The DNA sequences of the primers are forward: 5′TCATAATGCTTGCTCTGATAGGA and reverse: 5′GGCCAAAAATTTAATCAGTGGA (21). A primer sequence with a smaller product (Forward: 5′ TTCTTCATGAAGACCTCAC and Reverse: 5′ CCATCCACAAAATGGATCC) was used on a small subset of samples that failed to amplify with the original primer set. All sequencing for BRAF was performed on an ABI 3700 sequencer in the University of Michigan Sequencing Core Facility. Mutations were detected by using Mutation Surveyor™ software (Softgenetics, Inc., State College, PA). All chromatograms were also manually reviewed to confirm the presence or absence of mutations.
Statistical Analyses
We used a case-only study design to evaluate the association between somatic BRAF mutations and classical risk factors for colorectal cancer. Chi-square analyses, Fisher's exact test and unconditional logistic regression were used for analyzing factors associated with the presence or absence of BRAF V600E somatic mutations. Factors evaluated included Ashkenazi versus non-Ashkenazi ethnicity, family history of colorectal cancer in a first-degree relative, weekly use of nonsteroidal anti-inflammatory drugs/aspirin for at least 1 year, use of hormone replacement therapy, smoking status and pack-years of smoking. Stepwise forward regression was used for multivariate model building for prediction of the presence of a BRAF V600E mutation using a threshold p-value of 0.10. Polychotomous logistic regression was used to model the risk of colorectal cancer cases with and without a BRAF mutation compared to controls using the proc catmod command. All analyses used SAS version 9.1 (SAS Institute, Cary, NC).
Results
Paraffin-embedded tumors were available for 1737 MECC cases, MSI analyses were available for 1592 MECC cases and both BRAF and MSI analyses were available for 1297 MECC tumors. Cases with and without BRAF status available did not differ on any baseline characteristics (age, gender, ethnicity) or tumor characteristics (stage, grade, location). Of these tumors, 65 (5.0%) carried the BRAF V600E mutation and 147 (11.3%) were MSI-high (Table 1). As expected, tumors with the BRAF V600E mutation were 18.1 times more likely to be MSI-high than those tumors with wild type BRAF (95% C.I., 10.5 – 31.2). Cases with BRAF mutated tumors were more likely to be female, of Ashkenazi Jewish ethnicity and older than subjects with BRAF wild type tumors (Table 1). There was no difference in smoking history or duration, use of NSAIDS/aspirin or hormone replacement therapy (HRT) or family history of CRC between subjects with and without BRAF mutated tumors (Table 1). These characteristics were not simply due to the strong association between BRAF mutation and the MSI-high phenotype. When restricting the sample to only MSI-high CRCs, age, ethnicity and gender continue to be associated with presence of a BRAF V600E somatic mutation with a similar or stronger magnitude of association (Table 2).
Table 1.
Characteristics of samples with MSI status and BRAF genotyping
| BRAF Wild Type N = 1232 | BRAF V600E N = 65 | OR (95% C.I.) | |
|---|---|---|---|
| Age (years) | 70.3 (sd =11.5) | 73.8 (sd = 11.1) | p=0.02 |
| Range | 21 - 99 | 45 - 93 | |
| Pack-years | 33.7 (sd = 28.4) | 36.4 (sd = 25.7) | p=0.68 |
| Never smoked | 693 (94.8) | 38 (5.2) | 0.85 (0.50 – 1.44) |
| Ever smoked | 514 (95.5) | 24 (4.5) | 1.00 |
| Ashkenazi Jewish | 840 (94.2) | 52 (5.8) | 1.87 (1.01 – 3.47) |
| Non-Ashkenazi Jewish | 392 (96.8) | 13 (3.2) | 1.00 |
| Female | 593 (93.4) | 42 (6.6) | 1.97 (1.17 – 3.31) |
| Male | 639 (96.5) | 23 (3.5) | 1.00 |
| HRT Use | 22 (93.1) | 2 (6.9) | 0.94 (0.21 – 4.11) |
| No HRT Use | 417 (92.7) | 33 (7.3) | 1.00 |
| Aspirin/NSAID Use | 326 (95.0) | 17 (5.0) | 1.00 (0.56 – 1.77) |
| No Aspirin/NSAID Use | 863 (95.0) | 45 (5.0) | 1.00 |
| 1° Fam Hx of CRC | 123 (94.6) | 7 (5.4) | 1.08 (0.48 – 2.42) |
| No Fam Hx of CRC | 1103 (95.0) | 58 (5.0) | 1.00 |
| MSI-high | 106 (72.1) | 41 (27.9) | 18.1 (10.5 – 31.2) |
| MSS/MSI-Low | 1126 (97.9) | 24 (2.1) | 1.00 |
Table 2.
Characteristics of MSI-high tumors, by BRAF mutation status
| BRAF WT N = 106 | BRAF V600E N = 41 | OR (95% C.I.) | |
|---|---|---|---|
| Age (years) | 68.0 (sd = 12.1) | 77.7 (sd =9.2) | p <0.0001 |
| Female | 53 (65.4) | 28 (34.6) | 2.15 (1.01 – 4.61) |
| Male | 53 (80.3) | 13 (19.7) | 1.00 |
| Ashkenazi Jewish | 70 (66.7) | 35 (33.3) | 3.00 (1.15 – 7.79) |
| Non-Ashkenazi Jewish | 36 (85.7) | 6 (14.2) | 1.00 |
| Never smoked | 52 (69.3) | 23 (30.7) | 0.71 (0.33 – 1.50) |
| Ever smoked | 51 (76.1) | 16 (23.9) | 1.00 |
| Pack-years | 38.8 (sd=26.3) | 36.1 (sd=26.1) | p = 0.75 |
| HRT Use | 2 (66.6) | 1 (33.3) | 0.74 (0.06 – 8.63) |
| No HRT Use | 34 (59.7) | 23 (40.4) | 1.00 |
| Aspirin/NSAID Use | 25 (69.4) | 11(30.6) | 1.24 (0.54 – 2.85) |
| No Aspirin/NSAID Use | 79 (73.8) | 28 (26.2) | 1.00 |
| 1° Fam Hx of CRC | 9 (69.2) | 4 (30.8) | 1.15 (0.33 – 3.97) |
| No Fam Hx of CRC | 96 (72.2) | 37 (27.8) | 1.00 |
| MSI-high | 106 (72.1) | 41 (27.9) | - |
Despite the differences in epidemiologic characteristics, pathologically the BRAF V600E tumors have very similar characteristics to MSI-high tumors overall (Figure 1). The majority of BRAF V600E tumors were right-sided, with only four tumors located in the rectum (one MSI-high, three MSS). A high proportion of BRAF V600E tumors, regardless of instability status, showed a mucinous histology (52.2% MSS/BRAF V600E, 73.2% MSI-high/BRAF V600E, vs 44.6% MSI-high/BRAF WT, 19.4% MSS/BRAF WT, p < 0.001 for both comparisons within MSI status). However, the number of tumor infiltrating lymphocytes per high-powered field (TIL/hpf) were similar within MSI grouping regardless of BRAF mutation status, shown to be a powerful pathologic predictor of MSI-high in this study population (29).
Figure 1.
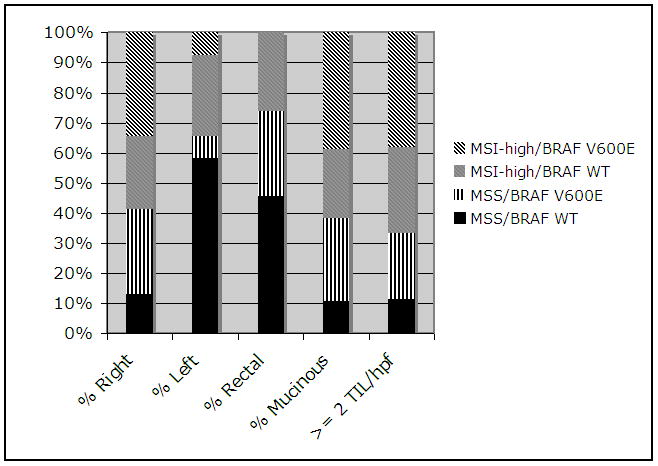
Relationship between clinicopathologic characteristics of MECC tumors, MSI and BRAF somatic mutation
It is necessary to be attentive to the role of gender in understanding the relationship between smoking history and the somatic profile of the tumor. Table 3 shows the relationship between smoking and BRAF V600E somatic mutation stratified by MSI status of the tumor and gender. Women who smoke are less likely to have a BRAF V600E somatic mutation, despite the fact they are twice as likely overall to have a BRAF somatic mutation. Men are more likely to have a somatic BRAF mutation if they smoke, and this effect is stronger in MSI-H tumors. A case-only analysis shows that female CRC cases are approximately twice as likely to have a BRAF mutation than male cases. A multivariate logistic model evaluating only cases shows that the interaction between smoking and gender is highly significant (Table 4) after adjusting for age, MSI status of the tumor, ethnicity, gender and smoking.
Table 3.
Gender, smoking and BRAF mutation status
| Male: MSS | Female: MSS | Male: MSI-HIGH | Female: MSI-HIGH | |||||
|---|---|---|---|---|---|---|---|---|
| BRAF WT | BRAF V600E | BRAF WT | BRAF V600E | BRAF WT | BRAF V600E | BRAF WT | BRAF V600E | |
| Never smoked | 208 | 3 | 433 | 12 | 20 | 2 | 32 | 21 |
| Ever smoked | 369 | 7 | 94 | 1 | 31 | 11 | 20 | 5 |
| OR (95% C.I.) | 1.32 (0.34 – 5.14) | 0.38 (0.05 – 2.99) | 3.55 (0.71 – 17.7) | 0.38 (0.12 – 1.17) | ||||
Table 4.
Case-only multivariate model: risk of BRAF V600E CRC
| OR (95% C.I.) | |
|---|---|
| MSI-HIGH | 19.24 (10.88 – 34.02) |
| Age (per year) | 1.02 (1.00 – 1.05) |
| Ethnicity (Ashkeanzi Jewish ethnicity vs. non-Ashkenazi Jewish ethnicity) | 1.65 (0.81 – 3.34) |
| Gender (Male) | 0.30 (0.11 – 0.82) |
| Smoking (ever vs. never) | 0.44 (0.17 – 1.17) |
| Smoking*gender | 4.95 (1.18 – 20.83) |
Recognizing that the overall case-control study is a design matched on gender and that the risk of cancer by gender cannot be estimated directly, it is interesting to note that gender is associated with BRAF V600E CRC compared to controls (Table 5). Next, we used polychotomous logistic regression to estimate the risk of BRAF-related CRC compared to controls (Table 6). As a note, pack-years was modeled as zero (a non-smoker), less than 27 pack-years (the median) and greater than or equal to 27 pack-years of smoking. Female gender was associated with BRAF-positive cancer (OR = 3.66, 95% C.I. 2.35 – 5.69), but not BRAF-negative cancer (OR = 0.97, 95% C.I. = 0.88 – 1.07). Ashkenazi Jewish ethnicity and history of CRC in a first degree relative were associated with an increase in risk of CRC regardless of BRAF status. Pack-years of smoking was associated with risk of BRAF-positive cancer, and an interaction term modeling pack-years and gender was highly significant. As indicated by the case-only analyses, CRC female cases were much less likely to have a history of smoking compared to controls, and this effect was stronger in BRAF positive cases.
Table 5.
Gender and BRAF in case-control comparisons and case-only analyses
| Female | Male | Odds Ratio | |
|---|---|---|---|
| BRAF Positive Cases | 42 (64.6) | 23 (35.4) | |
| Controls | 998 (49.4) | 1021 (50.6) | OR= 1.87, 95% Cl = (1.12-3.13) |
| BRAF Positive Cases | 42 (64.6) | 23 (35.4) | |
| BRAF Negative Cases | 591 (48.2) | 635 (51.8) | OR= 1.96, 95% Cl = (1.17-3.30) |
Table 6.
Case-control multivariable model: risk of BRAF V600E CRC
| BRAF-negative CRC vs. controls | BRAF-positive CRC vs. controls | |
|---|---|---|
| Gender: Female vs. male | 0.97 (0.88 – 1.07) | 3.66 (2.35 – 5.69) |
| Age | 0.99 (0.99 – 1.00) | 1.02 (1.00 – 1.03) |
| Ashkenazi Jewish ethnicity | 1.30 (1.20 – 1.41) | 2.22 (1.59 – 3.09) |
| Family hx CRC | 1.35 (1.19 – 1.53) | 1.55 (1.03 – 2.34) |
| Pack-years | 0.99 (0.92 – 1.05) | 1.78 (1.35 – 2.35) |
| Pack-years*gender | 0.82 (0.73 – 0.93) | 0.36 (0.23 – 0.57) |
Discussion
Defining the somatic fingerprint of a tumor may be a powerful way to precisely define subgroups of cancer that correspond to distinct risk factors. Here we evaluated risk factors associated with one mutation, BRAF V600E, in incident cases of colorectal cancer from northern Israel. This has provided insight into how to interpret epidemiologic studies of CRC, particularly with regard to heterogeneity in reported risk factors for CRC between populations.
Notably, the frequency of the BRAF V600E mutation varies widely between population groups, both within this study (5.8% in the Ashkenazim vs 3.2% in the non-Ashkenazim) and reported by other groups (Samowitz et al., 9.5%; English et al., 16.3%, here, 5.0% overall). Heterogeneity of rates between studies may reflect an underlying genetic predisposition to BRAF mutated tumors that differ by ethnic group, the sensitivity of the assay, specific environmental influences present at different levels in different populations, a combination of both, or chance. Sanger sequencing may have lower analytic sensitivity for somatic mutations than Pyrosequencing (30)), although another study of BRAF mutations reported a higher frequency mutations while using Sanger sequencing (12% in CRC cases, (25)). There is no reason to think that the detection method would be associated with epidemiologic risk factors within our study. It is especially intriguing to consider the hypothesis that specific somatic alterations are associated with distinct environmental exposures. There are several examples of this in cancers, most notably in the distinct KRAS mutational profile in never smokers with lung adenocarcinoma (31). This is further underscored by the distinct histology of BRAF-mutated tumors, with a high proportion of the tumors showing a mucinous histology and overwhelmingly right-sided regardless of MSI status, possibly indicating a specific etiologic pathway. Combining comprehensive epidemiologic data with high-throughput sequencing of somatic tissue could elucidate previously unappreciated environmental and genetic risk factors for cancers.
There is a two-fold difference in the odds of a BRAF CRC between those of Ashkenazi Jewish ethnicity and those who are not Ashkenazi Jewish. Incidence rates of CRC vary widely in Israel, with the highest rates found in the Ashkenazim and the lowest in the Arab population; Israeli Jews are at intermediate risk (32). English et al. describe a higher incidence of BRAF mutated tumors in the Anglo-Saxon population compared to the southern European population of Australia, with similar incidence rates of CRC without BRAF. Given that the risk profile likely differs by somatic profile of the tumor, it is essential to consider the somatic profile to appreciate differences in CRC rates between different populations.
The picture becomes complicated when evaluating MSI-high/BRAF mutated tumors with respect to smoking. Women are twice as likely to have a tumor with a BRAF mutation, but this is not strongly associated with smoking. Upon closer inspection, men who smoke have a significantly higher risk of colorectal cancer with a BRAF mutation (OR = 4.95, 95% C.I. = 1.18 – 20.83, p-value interaction = 0.03, after adjusting for age, ethnicity, MSI status of tumor). When evaluating risk factors in a multinomial model that includes controls, gender is strongly associated with risk of BRAF –mutated CRC, but much less likely to have a history of smoking regardless of mutational status. This suggests that tumors with BRAF somatic mutations arise from a different pathway in women. Slattery et al. (12) hypothesize that estrogen hormone replacement therapy may reduce the risk of MSI-high colorectal cancer in women by reducing the likelihood of estrogen receptor methylation, as loss of ER expression, possibly due to gene silencing by methylation, was shown to be ubiquitous in colon cells that give rise to cancer (33). The low prevalence of HRT use in the MECC study does not allow an adequate comparison in this population. The exact pathways of how smoking and estrogen may affect colorectal carcinogenesis are unknown. Large studies such as the MECC study are needed to continue to address these questions in a meaningful way.
There are several limitations to this study. First, despite the large sample size, BRAF mutations are rare and the number in this population is relatively small (5.0%). This limits the study to detect only large effects. We do find significant associations with many epidemiologic risk factors, including a significant interaction between male gender and smoking and BRAF tumor status. The smoking rate in Israeli women is low and thus the study is less generalizable to other female populations where smoking is more prevalent. However, our findings in this case-only study are consistent with case-control studies showing that smoking in females is not strongly associated with risk of colon cancer.
Here, we show that the prevalence of BRAF V600E mutations, while relatively rare (∼ 5% of all CRCs in the Israeli population), is associated with distinct risk factors in the Israeli population. In contrast to other populations, we note striking differences in the prevalence of this somatic mutation. Epidemiologic studies should consider somatic alterations to best understand the risk factors for this common complex disease.
Acknowledgments
This work was funded by RO1 CA81488. LSR was funded under a NRSA Kirschstein Fellowship CA110622.
References
- 1.Barratt PL, Seymour MT, Stenning SP, et al. DNA markers predicting benefit from adjuvant fluorouracil in patients with colon cancer: a molecular study. Lancet. 2002;360:1381–1391. doi: 10.1016/s0140-6736(02)11402-4. [DOI] [PubMed] [Google Scholar]
- 2.Farrington SM, McKinley AJ, Carothers AD, et al. Evidence for an age-related influence of microsatellite instability on colorectal cancer survival. Int J Cancer. 2002;98:844–850. doi: 10.1002/ijc.10264. [DOI] [PubMed] [Google Scholar]
- 3.Hemminki A, Mecklin JP, Jarvinen H, Aaltonen LA, Joensuu H. Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology. 2000;119:921–928. doi: 10.1053/gast.2000.18161. [DOI] [PubMed] [Google Scholar]
- 4.Jernvall P, Makinen MJ, Karttunen TJ, Makela J, Vihko P. Microsatellite instability: impact on cancer progression in proximal and distal colorectal cancers [see comments] Eur J Cancer. 1999;35:197–201. doi: 10.1016/s0959-8049(98)00306-2. [DOI] [PubMed] [Google Scholar]
- 5.Lee S, Cho NY, Choi M, Yoo EJ, Kim JH, Kang GH. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol Int. 2008;58:104–13. doi: 10.1111/j.1440-1827.2007.02197.x. [DOI] [PubMed] [Google Scholar]
- 6.Samowitz WS, Curtin K, Ma KN, et al. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol Biomarkers Prev. 2001;10:917–23. [PubMed] [Google Scholar]
- 7.Newcomb PA, Storer BE. Postmenopausal hormone use and risk of large-bowel cancer. J Natl Cancer Inst. 1995;87:1067–71. doi: 10.1093/jnci/87.14.1067. [DOI] [PubMed] [Google Scholar]
- 8.Newcomb PA, Taylor JO, Trentham-Dietz A. Interactions of familial and hormonal risk factors for large bowel cancer in women. Int J Epidemiol. 1999;28:603–608. doi: 10.1093/ije/28.4.603. [DOI] [PubMed] [Google Scholar]
- 9.Newcomb PA, Zheng Y, Chia VM, et al. Estrogen plus progestin use, microsatellite instability, and the risk of colorectal cancer in women. Cancer Res. 2007;67:7534–9. doi: 10.1158/0008-5472.CAN-06-4275. [DOI] [PubMed] [Google Scholar]
- 10.Slattery ML, Benson J, Berry TD, et al. Dietary sugar and colon cancer. Cancer Epidemiology, Biomarkers & Prevention. 1997 Sep;6(9):677–85. 677–685. [PubMed] [Google Scholar]
- 11.Slattery ML, Boucher KM, Caan BJ, Potter JD, Ma KN. Eating patterns and risk of colon cancer. American Journal of Epidemiology. 1998;148:4–16. doi: 10.1093/aje/148.1.4-a. [DOI] [PubMed] [Google Scholar]
- 12.Slattery ML, Potter JD, Curtin K, et al. Estrogens reduce and withdrawal of estrogens increase risk of microsatellite instability-positive colon cancer. Cancer Res. 2001;61:126–130. [PubMed] [Google Scholar]
- 13.Campbell PT, Curtin K, Ulrich C. Mismatch repair polymorphisms and risk of colon cancer, tumor microsatellite instability, and interactions with lifestyle factors. Gut. 2008 doi: 10.1136/gut.2007.144220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chia VM, Newcomb PA, Bigler J, Morimoto LM, Thibodeau SN, Potter JD. Risk of microsatellite-unstable colorectal cancer is associated jointly with smoking and nonsteroidal anti-inflammatory drug use. Cancer Res. 2006;66:6877–83. doi: 10.1158/0008-5472.CAN-06-1535. [DOI] [PubMed] [Google Scholar]
- 15.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci U S A. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 17.Samowitz WS, Albertsen H, Herrick J, et al. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–45. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 18.Samowitz WS, Albertsen H, Sweeney C, et al. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst. 2006;98:1731–8. doi: 10.1093/jnci/djj468. [DOI] [PubMed] [Google Scholar]
- 19.Domingo E, Espin E, Armengol M, et al. Activated BRAF targets proximal colon tumors with mismatch repair deficiency and MLH1 inactivation. Genes Chromosomes Cancer. 2004;39:138–42. doi: 10.1002/gcc.10310. [DOI] [PubMed] [Google Scholar]
- 20.Domingo E, Niessen RC, Oliveira C, et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24:3995–8. doi: 10.1038/sj.onc.1208569. [DOI] [PubMed] [Google Scholar]
- 21.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 22.English DR, Young JP, Simpson JA, et al. Ethnicity and risk for colorectal cancers showing somatic BRAF V600E mutation or CpG island methylator phenotype. Cancer Epidemiol Biomarkers Prev. 2008;17:1774–80. doi: 10.1158/1055-9965.EPI-08-0091. [DOI] [PubMed] [Google Scholar]
- 23.Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–6. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–9. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 25.Zlobec I, Bihl MP, Schwarb H, Terracciano L, Lugli A. Clinico-pathological and protein characterization of BRAF and K-RAS mutated colorectal cancer and implications for prognosis. Int J Cancer. 2009 doi: 10.1002/ijc.25042. [DOI] [PubMed] [Google Scholar]
- 26.Poynter JN, Gruber SB, Higgins PD, et al. Statins and the risk of colorectal cancer. N Engl J Med. 2005;352:2184–92. doi: 10.1056/NEJMoa043792. [DOI] [PubMed] [Google Scholar]
- 27.Greenson JK, Bonner JD, Ben Yzhak O, et al. Phenotype of microsatellite unstable colorectal carcinomas: well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am J Surg Pathol. 2003;27:563–570. doi: 10.1097/00000478-200305000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Research. 1998;58:5248–5257. [Review] [119 refs] [PubMed] [Google Scholar]
- 29.Greenson JK, Huang SC, Herron C, et al. Pathologic predictors of microsatellite instability in colorectal cancer. Am J Surg Pathol. 2009;33:126–33. doi: 10.1097/PAS.0b013e31817ec2b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–21. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riely GJ, Kris MG, Rosenbaum D, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731–4. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fireman Z, Sandler E, Kopelman Y, Segal A, Sternberg A. Ethnic differences in colorectal cancer among Arab and Jewish neighbors in Israel. Am J Gastroenterol. 2001;96:204–207. doi: 10.1111/j.1572-0241.2001.03476.x. [DOI] [PubMed] [Google Scholar]
- 33.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–40. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]