

Abstract
The effect of targeted inactivation of the gene encoding N-deacetylase/N-sulfotransferase-1 (Ndst1), a key enzyme involved in the biosynthesis of heparan sulfate (HS) chains, on the inflammatory response associated with allergic inflammation in a murine model of OVA-induced acute airway inflammation was investigated. OVA-exposed Ndst1f/fTekCre+ (mutant) mice deficient in endothelial and leukocyte Ndst1 demonstrated significantly decreased allergen-induced airway hyperresponsiveness and inflammation characterized by a significant reduction in airway recruitment of inflammatory cells (eosinophils, macrophages, neutrophils, and lymphocytes), diminished IL-5, IL-2, TGF-β1, and eotaxin levels, as well as decreased expression of TGF-β1 and the angiogenic protein FIZZ1 (found in inflammatory zone 1) in lung tissue compared with OVA-exposed Ndst1f/fTekCre− wild-type littermates. Furthermore, murine eosinophils demonstrated significantly decreased rolling on lung endothelial cells (ECs) from mutant mice compared with wild-type ECs under conditions of flow in vitro. Treatment of wild-type ECs, but not eosinophils, with anti-HS Abs significantly inhibited eosinophil rolling, mimicking that observed with Ndst1-deficient ECs. In vivo, trafficking of circulating leukocytes in lung microvessels of allergen-challenged Ndst1-deficient mice was significantly lower than that observed in corresponding WT littermates. Endothelial-expressed HS plays an important role in allergic airway inflammation through the regulation of recruitment of inflammatory cells to the airways by mediating interaction of leukocytes with the vascular endothelium. Furthermore, HS may also participate by sequestering and modulating the activity of allergic asthma-relevant mediators such as IL-5, IL-2, and TGF-β1.
Heparan sulfate (HS),3 a type of glycosaminoglycan (GAG), is widely distributed and is usually found covalently bound to core proteins of HS proteoglycans (HSPGs). HSPGs constitute a major component of the extracellular matrix (ECM) and are present on the plasma membrane of all animal cells studied so far (1). By virtue of their ability to bind to growth factors and their receptor tyrosine kinases, chemokines, interleukins, enzymes, enzyme inhibitors, lipases and apolipoproteins, ECM and plasma proteins (1), HSPGs play a critical role in various physiological aspects, including normal development, cell signaling and morphogenesis, cellular cross-talk, organization of basement membrane barriers, nutritional metabolism, injury and wound repair (1), and microbial adhesion to host cells (2). The extent of sulfation and the distribution of the sulfate substitutions on the HS backbone appear to play a vital role in regulating normal physiology. For example, a systemic deletion of N-deacetylase/N-sulfortransferase-1 (Ndst1), a key enzyme responsible for the addition of sulfate to the HS chains (3), has been shown to be lethal for mice due to forebrain defects (4). Other defects include skeletal malformation (5), anophthalmia (6), lung hypoplasia, and respiratory distress (7, 8). In fact, the importance of HS and its proper N-sulfation for normal lung function is further evident from the finding that Ndst1 is essential for the maturation of type II pneumocytes, and its inactivation leads to neonatal respiratory distress syndrome (8) Furthermore, recent studies have demonstrated that Ndst1-dependent HS is essential for proper functioning of bone morphogenetic protein by regulating binding and internalization of the protein during lung development (9). Utilizing mice deficient in endothelial and leukocyte Ndst1 produced by targeted inactivation of the gene coding for Ndst1, Wang et al. demonstrated that endothelial HS plays an important role during inflammation, promoting neutrophil recruitment by acting as a ligand for L-selectin during neutrophil rolling on vascular endothelial cells (EC) and mediating chemokine transcytosis by binding and presenting chemokines at the lumenal surface of the endothelium (10). In addition to inflammation, Fuster et al. demonstrated a pathological role for HS in tumor-induced angiogenesis, but not physiologic angiogenesis, during wound repair (11). Pathological angiogenesis was found to be inhibited in these mice due to significantly decreased binding of the mutant EC to fibroblast growth factor-2 and vascular endothelial growth factor-164, as well as due to altered sprouting in response to stimulation with these factors (11). In contrast, altering HS in leukocytes had no effect on inflammation and did not appear to play a crucial role in lymphocyte development (10, 12).
While the studies described above demonstrate an important role for HS in development, tumor angiogenesis, and neutrophil-mediated inflammation, the importance of these sulfated sugars in the pathogenesis of allergic airway inflammation, including asthma, has not been determined. Studies have shown that proteoglycan metabolism is altered in patients with asthma, as fibroblasts from afflicted individuals produce significantly higher levels of hyaluronan, perlecan, versican, decorin, and biglycan. Furthermore, proteoglycan levels correlated with severity of airway hyperresponsiveness (AHR) in these patients, suggesting that increased proteoglycan deposition in the bronchial mucosa may lead to AHR (13). More recent studies have demonstrated that enzymes such as eosinophil peroxidase and myeloperoxidase released extracellularly by activated leukocytes bind to and react with proteoglycans, including HSPGs, resulting in the localized release of hypobromous acid, which in turn causes degradation of the ECM GAGs that may contribute to tissue damage associated with airway inflammatory diseases such as asthma (14). In the present study, we provide the first documentation that Ndst1 and endothelial-expressed HS play a role in the recruitment and trafficking of eosinophils in a murine model of allergic airway inflammation using the previously described endothelial- and leukocyte-selective Ndst1-deficient mice (Ndst1f/fTekCre+) (10, 11).
Materials and Methods
Mice
Ndst1f/fTekCre+ mice that were deficient in endothelial and leukocyte Ndst1 were generated as described in previous studies (10, 11). Briefly, C57BL/6 mice with a conditional mutation in exon 2 of the Ndst1 gene (Ndst1f/f) were bred with transgenic mice expressing Cre recombinase under the control of the Tek promoter-enhancer (TekCre). Male Ndst1f/fTekCre+ mice were bred with female Ndst1f/f mice, yielding mutant Ndst1f/fTekCre+ offspring and wild-type (WT) littermate controls (Ndst1f/fTekCre−). Offspring were genotyped using genomic DNA isolated from tail clippings as described (10, 11). All studies involving mice were performed following standards and procedures approved by the local Institutional Animal Care and Use Committee.
Model of allergic airway inflammation
Ndst1f/fTekCre+ mice and their WT littermates were immunized with 20 μg of OVA (grade V; Sigma-Aldrich) in 2 mg of aluminum hydroxide gel (alum) i.p. as described previously (15). Control mice received alum alone. Fourteen days later, OVA-sensitized mice were exposed to aerosolized OVA (20 mg/ml) in saline administered with the help of a nebulizer for 30 min each day for 5 days, while the control mice received nonpyrogenic saline (Baxter International) at corresponding time points.
Measurement of airway responsiveness
Three hours after the last aerosol challenge, airway responsiveness was measured in the allergen-challenged Ndst1f/fTekCre+ and WT mice as well as corresponding controls using a whole body plethysmograph (Buxco) as described (16). Mice were first exposed to saline followed by increasing concentrations of methacholine (MCh, 3–25 mg/ml in saline; Sigma-Aldrich) using an Aerosonic ultrasonic nebulizer (DeVilbiss Healthcare). Each exposure was for 3 min, and the enhanced pause (Penh) in breathing after each nebulization was monitored and recorded every 10 s over a 5-min period and averaged for each MCh concentration. Results were expressed as a percentage of the baseline Penh values following exposure to saline.
Analysis of bronchoalveolar lavage fluid (BALF)
After measurement of airway responsiveness, mice were sacrificed and BALF was collected. Cells in the BALF were recovered by centrifugation at 400 × g and total as well as differential cell counts were determined from cytocentrifuged slides stained with Leukostat or Wright-Giemsa (Thermo Fisher Scientific) based on morphological and histological criteria. Cell viability was assessed by trypan blue exclusion. The BALF supernatant was centrifuged at 1000 × g to remove cellular debris and stored at −70°C until further evaluation.
Measurement of BALF cytokines and chemokines
The relative production of Th1 (IL-2 and IFN-γ) and Th2 (IL-4, IL-5, and IL-13) type cytokines as well as that of TGF-β1 in the BALF was measured by ELISA using commercially available reagents/kits. Ab pairs and recombinant cytokines for generation of standard curves for IL-2, IL-4, IL-5, and IFN-γ were obtained from BD Pharmingen, while reagents for IL-13 were obtained from R&D Systems. BALF TGF-β1 and eotaxin levels were measured using kits (R&D Systems). All ELISAs were performed according to manufacturers’ recommendations. OD of the color developed in each case was measured with the help of a microplate reader (Molecular Devices; or FLUOstar OPTIMA from BMG Labtech), and the concentration of each cytokine/chemokine in the BALF was determined against a standard curve generated.
Lung histology
Following BALF collection, a section of the lungs of control and OVA-exposed Ndst1f/fTekCre+ and WT mice were collected, fixed in 4% paraformaldehyde (Sigma-Aldrich), and paraffin-embedded. Tissue sections (5 μm thick) were deparaffinized and hydrated through a sequential alcohol series before staining. To examine the lungs for infiltrated cells, tissue sections were stained with Harris modified hematoxylin and Shandon instant eosin (Thermo Fisher Scientific). For all immunohistological analyses, tissue sections were subjected to Ag retrieval followed by quenching of endogenous peroxidase activity before staining with specific Abs. Sections were briefly counterstained (<30 s) with hematoxylin. Appropriate Vectastain ABC kits using biotinylated secondary Abs (Vector Laboratories) and the peroxidase AEC (3-amino-9-ethylcarbazole) substrate kit (Vector Laboratories) were used for detection. Stained slides were analyzed using either a Leica DME light microscope or a Nikon Microphot EPI-FL microscope (× 10 or × 40 magnification), and images were captured with an Olympus DP71 camera. Lung tissue eosinophils were evaluated by staining for eosinophil-specific major basic protein (MBP) with rat mAbs against murine MBP (2 μg/ml; provided by Dr. J. Lee, Mayo Clinic Arizona, Scottsdale, AZ). The MBP-positive cells in five randomly chosen nonoverlapping microscopic fields were counted and results were expressed as the average number of cells per field. Lung tissue macrophages were evaluated using rat anti-mouse F4/80 (10 μg/ml; AbD Serotec). All airways and all F4/80-positive cells around the airways were counted on each slide and results were expressed as the number of F4/80-positive cells per airway, which was determined by dividing the total number of F4/80-positive cells by the total number of airways. TGF-β1 expression was detected with polyclonal Abs against TGF-β1 (4 μg/ml; Santa Cruz Biotechnology). Found in inflammatory zone 1 (FIZZ1) protein expression was evaluated using goat polyclonal anti-mouse FIZZ1 Ab (resistin-like α [E19], 2 μg/ml; Santa Cruz Biotechnology). Total FIZZ1 protein expression was quantitated from a minimum of five captured images per slide per mouse using the ImageJ image analysis system (downloaded from www.nist.gov/lispix/imlab/prelim/dnld.html) as described previously (17) and was expressed as average FIZZ1-positive area. Type II alveolar epithelial cells were identified based on morphology, and the numbers of FIZZ1-positive type II alveolar epithelial cells were counted in five randomly chosen microscopic fields with no visible airways or blood vessels and expressed as the average number of cells per field.
Measurement of TGF-β1 in lung tissue by Western blotting
Lung tissue collected from control and OVA-exposed Ndst1f/fTekCre+ and WT mice was lysed in RIPA buffer (Santa Cruz Biotechnology) and estimated for total protein (BCA protein assay kit; Pierce). Lysates (60 μg protein/lane/sample) were electrophoresed on 10–20% Tricine gels (Invitrogen), transferred to polyvinylidene difluoride membranes (Millipore), blocked (5% nonfat milk in PBS with 0.1% Tween 20), and exposed to rabbit Abs against TGF-β1 (Cell Signaling Technology) followed by HRP-conjugated anti-rabbit IgG (Cell Signaling Technology). Bound Abs were detected using Immobilon Western chemiluminescent HRP substrate (Millipore). HRP-conjugated anti-mouse β-actin (Santa Cruz Biotechnology) was used to monitor levels of β-actin expression as an internal control. Bands were visualized on x-ray films, which were scanned and subsequently analyzed using ImageJ to quantitate the density (pixels) of the bands.
Isolation of murine eosinophils
Eosinophils were isolated from peripheral blood of IL-5 transgenic mice by discontinuous density gradient centrifugation followed by negative selection. Briefly, peripheral blood obtained by cardiac puncture was first incubated with hetastarch (6% in normal saline) for 50 min to sediment erythrocytes. The upper plasma layer was collected and centrifuged on a discontinuous Percoll gradient consisting of 57% (v/v), 61% (v/v), and 68.5% (v/v) Percoll (Amersham Biosciences) in 150 mM NaCl at 2000 rpm for 20 min at room temperature. The top layer containing lymphocytes and a few eosinophils was carefully removed and the middle layer containing mostly eosinophils with a small percentage of contaminating lymphocytes was collected in a separate tube. Cells were washed once with 1% BSA in PBS, resuspended in the same buffer, and incubated for 30 min at room temperature with anti-CD90.2 and anti-CD45R (eBioscience) that bind to T and B lymphocytes, respectively. Cells were washed as described earlier and incubated with goat anti rat IgG-coated microbeads (Miltenyi Biotec) for 15 min at room temperature, using 20 μl of beads for every 107 cells. Cells were washed and passed through MACS separation columns (Miltenyi Biotec) connected to an OctoMACS separation Unit (Miltenyi Biotec). The flow through containing unbound eosinophils was collected and assessed for viability and purity, which was typically >95%.
In vitro laminar flow assay
The interaction of murine eosinophils with murine lung EC (MLEC) from Ndst1f/fTekCre+ mice and WT littermates was assessed in an in vitro parallel plate laminar flow chamber as described in our previous studies (10, 18). Confluent cultures of MLEC gelatin-coated glass coverslips (11) were treated with murine TNF-α (50 ng/ml; BD Pharmingen) for 6 h at 37°C before dynamic flow studies. MLECs were then treated with media alone, a phage display anti-HS Ab AO4B08, that interacts with the N-, 2-O-, and 6-O-sulfated saccharide motif, as well as an internal 2-O-sulfate group (19) or a non-GAG binding control Ab MPB49 (both Abs provided by Dr. T. van Kuppevelt, Radboud University Nijmegen Medical Center, The Netherlands) at a 1/100 dilution for 30 min at room temperature before placement in the flow chamber. MLEC-coated coverslips were exposed to flow conditions (wall shear stress ~1.0 dyn/cm2) by perfusing warm media (RPMI 1640) through a constant infusion syringe pump (Harvard Apparatus). Murine eosinophils (2 × 105 cells) treated with media alone, AO4B08, or MPB49 for 30 min at room temperature were perfused into the flow chamber for a period of 5 min. The interaction of the injected cells with the MLEC-coated coverslips was observed using a Leitz Wetzlar inverted microscope as previously described (18, 20). The images were recorded for subsequent offline analysis to manually determine the number of interacting cells. Cells demonstrating multiple discrete interruptions in flow and moving more slowly were considered as rolling cells.
Leukocyte trafficking in lung vessels
Lung sections from OVA-exposed Ndst1f/fTekCre+ and WT mice were transplanted into skin-fold chambers in nude mice and allowed to undergo revascularization for 10–14 days as described previously (21, 22). The transplanted lung sections were periodically observed with the help of a Leica DM LB intravital microscope (IVM; Leica Microsystems) equipped with a Prosilica ethernet camera (model no. GC138OH) until they were fully revascularized. Nude mice bearing revascularized lung allografts from control and OVA-challenged Ndst1f/fTekCre+ and WT mice were administered i.v. with acridine orange (0.5 mg/mouse; Sigma-Aldrich) to fluorescently label circulating leukocytes in vivo (23). The interaction of the labeled leukocytes with the vascular endothelium of the revascularized lung microvessels was made visible by stroboscopic epi-illumination using a xenon lamp, and all images recorded using StreamPix digital video recording software (NorPix). This was followed by off-line analysis of recorded digital images. Leukocytes visibly interacting with the lung microvascular endothelium (postcapillary venules) and passing at a slower rate than the main bloodstream were considered as rolling cells and were quantitated by manually counting the number of rolling cells passing through a reference point in a vessel segment (200 μm) for a certain duration of time and expressed as the number of rolling cells per minute. Rolling in four to eight lung microvessels was analyzed per mouse on an average.
Statistical analysis
The results are expressed as the mean s ± SE. Statistical significance was determined using Student’s t test. A p value of <0.05 was considered as significant.
Results
Ndst1f/fTekCre+ mice exhibit attenuated cellular infiltration of the airways in response to allergen
Ndst1f/fTekCre+ mutant mice and their WT Ndst1f/fTekCre− littermates were allergen-sensitized and -challenged with OVA, and cellular infiltration in the BALF and lung tissue was evaluated. OVA-challenged Ndst1f/fTekCre+ mice exhibited a significant reduction ( p = 0.011) in recruitment of inflammatory cells to the lungs compared with allergen-challenged WT mice as demonstrated by the total number of cells recovered from the BALF (Fig. 1A) and cellular infiltration of lung tissue as visualized by H&E staining (Fig. 1B). Analysis of differential cell counts in the BALF demonstrated not only a marked reduction in eosinophil recruitment, but also a significant reduction in monocyte/macrophage, neutrophil, and lymphocyte infiltration (Fig. 2A). Evaluation of the percentages of the different cells types in the BALF of allergen-challenged Ndst1f/fTekCre+ mice demonstrated that infiltration of eosinophils, but not monocyte/macrophages, neutrophils, or lymphocytes, was preferentially inhibited in these mice compared with allergen-challenged WT mice (44.83 ± 13.33 vs 74.29 ± 3.21% (eosinophils), 6.86 ± 2.49 vs 29.50 ± 12.83% (macrophages), 10.29 ± 2.38 vs 13.00 ± 2.92% (neutrophils), and 8.00 ± 1.46 vs 12.64 ± 7.13% (lymphocytes), respectively). Quantitation of tissue eosinophils by immunohistology using mAbs specific for murine MBP demonstrated a significant reduction in the number of eosinophils (p < 0.05) in the alveolar and peribronchial regions in Ndst1f/fTekCre+ mice (Fig. 2, B and C). Furthermore, a significant reduction was observed in the number of F4/80-stained macrophages (p < 0.05) in the peribronchial region of allergen-challenged Ndst1f/fTekCre+ mice as opposed to allergen-challenged WT mice (Fig. 2, D and E), while there was no difference in the number of macrophages in the alveolar region between the two groups (data not shown).
FIGURE 1.
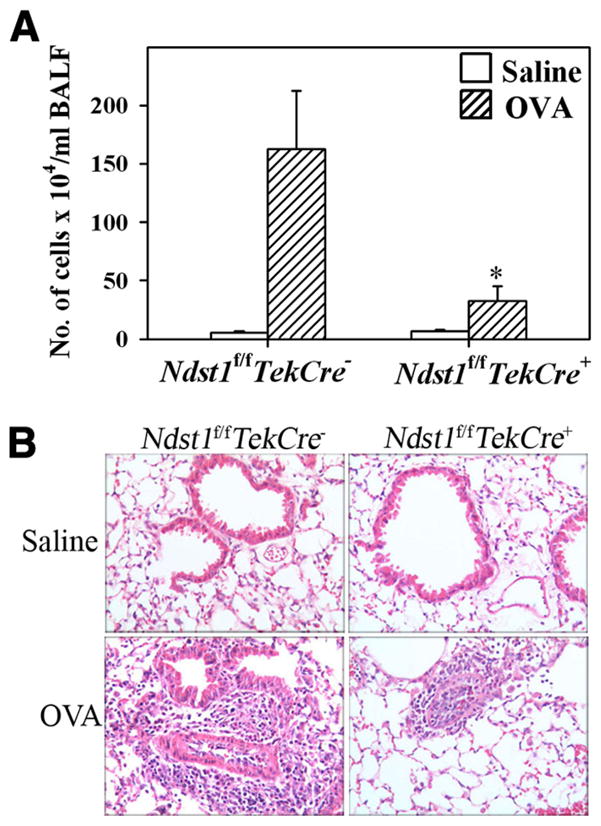
Cellular infiltration of the airways in response to allergen challenge is inhibited in Ndst1f/fTekCre+ mice. Ndst1f/fTekCre+ mice and WT littermates were sensitized and challenged with OVA while control mice received only saline. BALF and lung tissue collected 3 h after the last allergen challenge were processed for evaluation of cellular infiltration. A, Cells recovered from the BALF were counted with a hemocytometer and expressed as the number of cells × 104/ml of BALF (n = 8–11 mice for control groups and 14–16 mice for OVA-challenged groups). *, p = 0.011. B, Cellular infiltration of lung tissue was evaluated by H&E staining of paraformaldehyde-fixed lung tissue sections. Representative images from each group at a magnification of × 20 are shown.
FIGURE 2.

Allergen-challenged Ndst1f/fTekCre+ mice exhibit decreased airway recruitment of eosinophils and macrophages. A, BALF collected from control (saline) and OVA-sensitized and challenged Ndst1f/fTekCre+ mice and WT littermates was evaluated for differential cell counts by microscopic evaluation of cytocentrifuged slides (n = 5–11 mice for control groups and 11–16 mice for OVA-challenged groups). B, Lung tissue eosinophils in these mice were evaluated by immunohistochemical staining for eosinophil-specific MBP using rat mAbs specific for murine MBP. Representative images from each group at a magnification of × 20 are shown. C, MBP-positive cells in five randomly selected nonoverlapping microscopic fields were counted (× 40 magnification) and results were expressed as the average number of cells per field (n = 5 mice/group). D, Lung tissue macrophages in control and OVA-exposed Ndst1f/fTekCre+ mice and WT littermates were evaluated by immunohistology with rat anti-mouse F4/80. Representative images from each group at a magnification of × 20 are shown. E, Airways in a microscopic field were identified and the number of F4/80-positive cells around the airways was counted (× 40 magnification). Results were expressed as the number of F4/80-positive cells per airway, which was determined by dividing the total number of F4/80-positive cells by the total number of airways (n = 4 mice/group). *, p < 0.05.
Allergen-induced AHR is reduced in Ndst1f/fTekCre+ mice
A characteristic feature of asthma in humans is the development of AHR, which has also been established in various murine models of allergic airway inflammation (24). In the present study, airway responsiveness to increasing concentrations of MCh was measured in allergen-challenged and control Ndst1f/fTekCre+ and WT mice by whole body plethysmography (Fig. 3). As expected, at 25 mg/ml MCh, allergen-challenged WT mice exhibited a significant increased in AHR compared with WT saline-exposed mice (p < 0.01). However, AHR in response to increasing concentrations of MCh was only marginally higher in the OVA-challenged Ndst1f/fTekCre+ mice compared with corresponding saline-exposed mice, but it was significantly reduced compared with allergen-challenged WT mice (p = 0.028).
FIGURE 3.
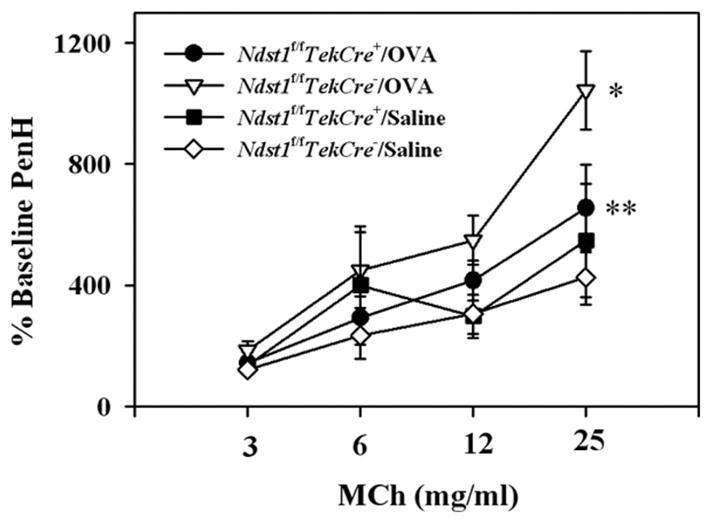
AHR is diminished in allergen-challenged Ndst1f/fTekCre+ mice. Airway responsiveness in control and allergen-challenged Ndst1f/fTekCre+ mice and WT littermates was measured 3 h after the last challenge by whole body plethysmography. Mice were first exposed to saline followed by increasing concentrations of nebulized MCh. The enhanced pause (Penh) in breathing after each nebulization was monitored, and results for each MCh concentration were expressed as a percentage of the baseline Penh values following exposure to saline (n = 5 mice for the control groups and 10–12 mice for the OVA-challenged groups). *, p < 0.01; **, p = 0.028.
Allergen-challenged Ndst1f/fTekCre+ mice exhibit decreased eotaxin levels in BALF
Our studies demonstrate that there is decreased recruitment of eosinophils to the airways (BALF as wells as lung tissue) in allergen-exposed Ndst1f/fTekCre+ mice (Fig. 2, A–C). Since eotaxin is a major chemoattractant for eosinophils, the levels of this chemokine in the BALF of allergen-challenged Ndst1f/fTekCre+ and WT littermates was measured by ELISA (Fig. 4). Eotaxin levels in the BALF of allergen-challenged Ndst1f/fTekCre+ mice were lower than those observed in allergen-challenged WT littermates.
FIGURE 4.
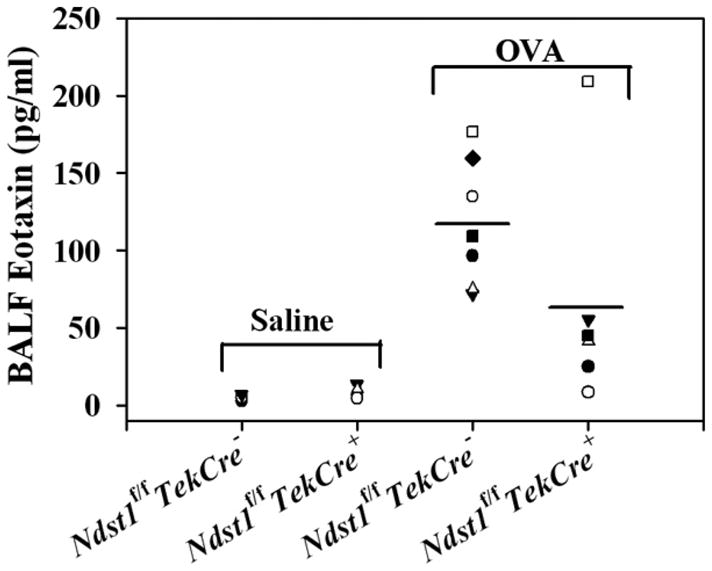
Allergen-challenged Ndst1f/fTekCre+ mice exhibit decreased eotaxin levels. BALF collected from control and allergen-challenged Ndst1f/fTekCre+ mice and WT littermates was used to evaluate eotaxin levels by ELISA. Concentration of eotaxin in the BALF was determined against a standard curve generated (range, 7.8–500 pg/ml) and expressed as pg/ml of BALF (n = 4–5 mice for the control groups and 6–7 mice for the OVA-challenged groups).
IL-5 levels are decreased in the BALF of allergen-challenged Ndst1f/fTekCre+ mice
Allergic airway inflammation is predominantly a Th2-driven phenomenon (25). We evaluated Th1 (IL-2 and IFN-γ) and Th2 (IL-4, IL-5 and IL-13) cytokine levels in the BALF of allergen-challenged Ndst1-deficient mice by ELISA to determine whether the decreased cellular recruitment was associated with a decrease in Th2 cytokines as well. WT mice exposed to OVA had elevated IL-5 levels compared with mice exposed to saline. While IL-5 levels in the BALF of OVA-exposed Ndst1f/fTekCre+ mice were higher than in corresponding control mice, they were significantly lower (p < 0.01) than in OVA-challenged WT mice (Fig. 5). In contrast to IL-5, there was no difference in IL-4 and IL-13 levels between OVA-challenged Ndst1f/fTekCre+ mice and WT littermates. Among the Th1 cytokines tested, there was no difference in IFN-γ levels between OVA-challenged Ndst1f/fTekCre+ and WT mice, whereas a significant reduction in IL-2 levels was observed in OVA-challenged Ndst1f/fTekCre+ mice compared with OVA-challenged WT mice (p < 0.05). Quantitative PCR analysis using mRNA isolated from lung lysates of representative OVA-challenged Ndst1f/fTekCre+ and WT mice and their respective controls demonstrated similar trends for IL-4, IL-5, IL-2, and IFN-γ (data not shown).
FIGURE 5.
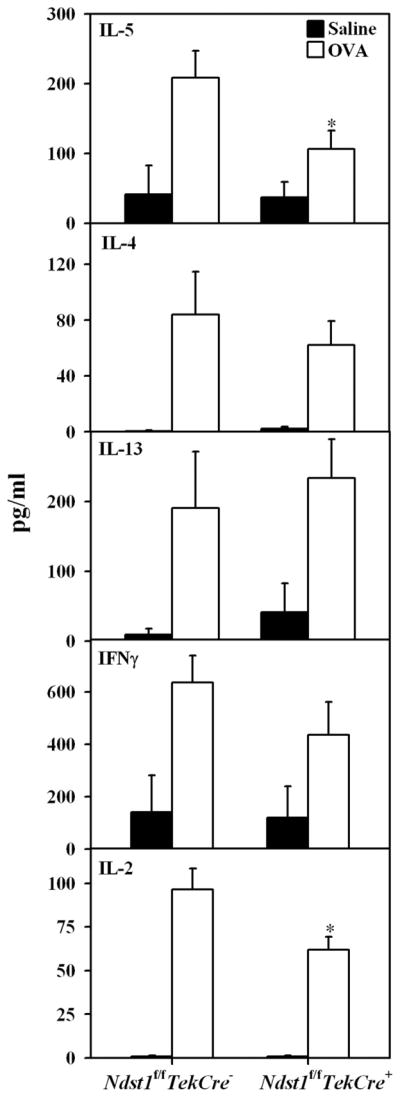
IL-5 and IL-2 levels are decreased in the BALF of allergen-challenged Ndst1f/fTekCre+ mice. IL-5, IL-13, IL-4, IFN-γ, and IL-2 levels in BALF from control and allergen-challenged Ndst1f/fTekCre+ mice and WT littermates were evaluated by ELISA using Ab pairs and recombinant cytokines for generation of standard curves. Cytokine levels were expressed as pg/ml of BALF (n = 4 mice for the control groups and 6–11 mice for the OVA-challenged groups). *, p < 0.01 for IL-5 and p < 0.05 for IL-2.
TGF-β1 levels are decreased in allergen-challenged Ndst1f/fTekCre+ mice
TGF-β1 is an important mediator of allergic airway inflammation. While strong expression is observed in the bronchial epithelium during asthma, inflammatory cells such as eosinophils, macrophages, and fibroblasts are a significant source of TGF-β1 (26). HSPGs are known to bind to TGF-β1 (27). Consequently, the presence of this growth factor in the BALF as well as its level of expression in lung tissue of allergen-exposed Ndst1f/fTekCre+ mice and WT littermates was determined. TGF-β1 levels in the BALF (measured by ELISA) of allergen-exposed Ndst1f/fTekCre+ mice was significantly lower (p < 0.05) compared with allergen-challenged WT littermates (Fig. 6A). Additionally, TGF-β1 expression in the lung tissue determined by Western blotting and immunohistology was lower in these mice demonstrating decreased TGF-β1 expression not only in the bronchial epithelium but also by peribronchial inflammatory cells compared with their WT counterparts (Fig. 6, B and C).
FIGURE 6.
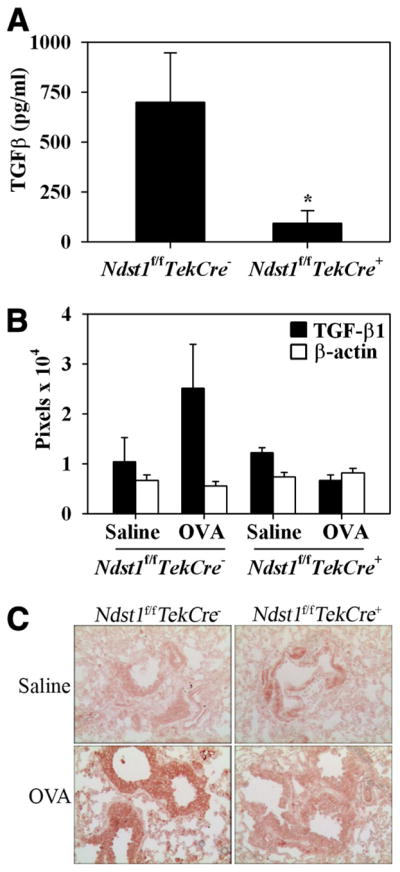
Allergen-challenged Ndst1f/fTekCre+ mice exhibit decreased TGF-β1 levels in BALF and lung tissue. A, TGF-β1 levels were measured in BALF from control and allergen-challenged Ndst1f/fTekCre+ mice and WT littermates by ELISA. TGF-β1 levels in OVA-challenged Ndst1f/fTekCre+ mice and WT littermates after subtracting TGF-β1 levels in corresponding control mice are shown as pg/ml of BALF (n = 7 each for control and OVA-challenged WT littermates and 8 each for control and OVA-challenged Ndst1f/fTekCre+ mice). *, p < 0.05. B, TGF-β1 levels in total lung tissue lysates were determined by Western blotting using rabbit Abs against TGF-β1. Blots were probed with HRP-conjugated anti-mouse β-actin to monitor levels of β-actin expression as an internal control. Bands were visualized on x-ray films and scanned and analyzed using ImageJ to quantitate the density (pixels) of the bands (n = 4 mice/group). C, Distribution of TGF-β1 in lung tissue from control and allergen-challenged Ndst1f/fTekCre+ mice and WT littermates was determined by immunohistochemistry with polyclonal Abs against TGF-β1. Representative images from each group at a magnification of × 20 are shown. *, p < 0.05.
Ndst1f/fTekCre+ mice express diminished levels of FIZZ1 in response to allergen challenge
FIZZ1 is a secreted protein that is associated with allergic pulmonary inflammation and early stages of airway remodeling (28, 29). FIZZ1 protein expression in lung tissue of OVA-challenged Ndst1f/fTekCre+ mice and WT littermates as well as in unchallenged control mice was evaluated by immunohistochemistry. Control mice from both groups expressed very low levels of FIZZ1 protein (Fig. 7A, upper panels). Allergen-challenged WT mice demonstrated intense FIZZ1 expression in the bronchial epithelium as well as in alveolar type II epithelial cells (Fig. 7A, lower left panel) in contrast to allergen-exposed Ndst1f/fTekCre+ mice (Fig. 7A, lower right panel). FIZZ1 protein expression in the bronchial epithelium and type II epithelial cells of allergen-exposed Ndst1f/fTekCre+ mice was significantly diminished (p < 0.05) not only in terms of the intensity of FIZZ1 expression (Fig. 7B, upper panel), but also in terms of the relative number of type II epithelial cells expressing FIZZ1 (Fig. 7B, lower panel).
FIGURE 7.

Expression of FIZZ1 is inhibited in allergen-challenged Ndst1f/fTekCre+ mice. A, FIZZ1 expression in lung tissue of OVA-challenged Ndst1f/fTekCre+ mice and WT littermates as well as in unchallenged control mice was evaluated by immunohistochemistry using goat polyclonal Abs against murine FIZZ1. Representative images from each group at a magnification of ×20 are shown. B, Total FIZZ1 expression was quantitated from captured images (five fields per mouse) at ×10 magnification using ImageJ image analysis system and expressed as average FIZZ1-positive area per field. FIZZ1-positive type II alveolar epithelial cells in five randomly selected microscopic fields with no visible airways or blood vessels were counted (×40 magnification) and expressed as the average number of cells per field. A negligible number of FIZZ1-positive type II alveolar epithelial cells were present in saline-exposed control mice (n = 5 each for control and OVA-challenged WT littermates and 4 each for control and OVA-challenged Ndst1f/fTekCre+ mice). *, p < 0.05.
Endothelial HS supports murine eosinophil rolling in vitro and in vivo
Allergic airway inflammation is characteristically associated with increased recruitment of inflammatory leukocytes, especially eosinophils, to the airways, a process that is orchestrated in part by increased cellular trafficking mediated by cell adhesion molecules on both leukocytes and ECs. To evaluate the role of HS in eosinophil recruitment, the first and rate-limiting step of the adhesion cascade, that is, the ability of murine eosinophils to roll on MLEC from Ndst1f/fTekCre+ and WT littermates, was determined using an in vitro parallel plate laminar flow chamber assay (Fig. 8A). First, peripheral blood murine eosinophils exhibited significantly reduced (p < 0.001) rolling on MLECs from Ndst1f/fTekCre+ mice (Fig. 8A, right panel) compared with WT MLECs (Fig. 8A, left panel). Second, pretreatment of eosinophils with function-blocking anti-HS Ab AO4B08 did not have any effect on eosinophil rolling on Ndst1f/fTekCre+ or WT MLECs (Fig. 8A), whereas pretreatment of MLECs with this Ab significantly inhibited eosinophil rolling on WT MLECs (Fig. 8A, left panel; p < 0.001), but it had no further effect on the already reduced rolling observed on MLECs from Ndst1f/fTekCre+ mice (Fig. 8A, right panel). Treatment of eosinophils or MLECs with a non-GAG-binding control Ab MPB49 did not have any effect on eosinophil rolling on Ndst1f/fTekCre+ or WT MLECs. These data suggest that HS on MLECs, but not eosinophils, is required for eosinophil rolling under conditions of flow.
FIGURE 8.

Rolling of murine eosinophils on MLECs and circulating leukocytes on vascular endothelium of Ndst1f/fTekCre+ mice. A, Single-cell suspensions of murine eosinophils (n = 2 mice in duplicate) were infused into a flow chamber holding coverslips coated with monolayers of TNF-α-stimulated lung EC from Ndst1f/fTekCre+ mice or WT littermates (n = 2 mice/group) that were treated with media alone, anti-HS Ab AO4B08, or control Ab MPB49 at a wall shear stress of ~1.0 dyn/cm2 for 5 min and the interactions of the injected cells with the coated coverslips were recorded. In some cases, the eosinophils were treated with AO4B08 or MPB49 before infusion into the flow chamber. Interaction of the injected cells with the MLEC-coated coverslips was recorded for subsequent offline analysis. Slow-flowing cells demonstrating multiple discrete interruptions in flow were considered as rolling cells. Results were expressed as the number of rolling cells per minute. B, The interaction of acridine orange-labeled circulating leukocytes with the endothelium of revascularized microvessels of lung allografts from allergen-challenged Ndst1f/fTekCre+ mice and WT littermates mice was evaluated by IVM. Leukocytes visibly interacting with the lung microvascular endothelium and passing at a slower rate than the main blood stream were considered as rolling cells and were quantitated by manually counting the number of rolling cells passing through a reference point in a 200-μm vessel segment and expressed as the number of rolling cells per minute (n = 4–8 lung microvessels/mouse, 2 mice/group). *, p < 0.001.
Since allergen-challenged Ndst1f/fTekCre+ mice demonstrated decreased cellular infiltration of the airways, we next examined leukocyte trafficking in lung microvessels of allergen-challenged Ndst1f/fTekCre+ and WT littermates by IVM. Acridine orange-labeled circulating leukocytes demonstrated significantly reduced (p < 0.001) leukocyte-endothelial interactions (rolling) in microvessels of revascularized lung allografts from allergen-challenged Ndst1f/fTekCre+ mice compared with lung microvessels of allergen-challenged WT littermates (Fig. 8B).
Discussion
In the present study, we evaluated the role of HS in a murine model of OVA-induced allergic airway inflammation using genetically modified mice deficient in endothelial and leukocyte Ndst1. In WT mice, exposure to aerosolized OVA resulted in the development of a classical asthmatic response characterized by eosinophil infiltration into the airways, AHR, elevated levels of the Th2 cytokine IL-5, TGF-β1, eotaxin, as well as FIZZ1 in the airways. In contrast, several features of the allergic disease were significantly attenuated in allergen-exposed Ndst1-deficient mice with an overall suppression of the asthma phenotype. Taken together, the findings indicate a direct role for HS in the pathogenesis of allergic airway disease, including asthma, which has not been previously studied genetically.
The reduced airway recruitment of eosinophils in Ndst1-deficient mice was associated with lower eotaxin levels in the BALF (Fig. 4). Previous studies have demonstrated that the recruitment of neutrophils during inflammation is in part dependent on the binding of neutrophil-active chemokines (MIP-2 and CXCL1) to endothelial-expressed HS in mice (10). However, recent studies have demonstrated that eotaxin binds selectively to heparin but not to HS (30), suggesting that the reduced eotaxin levels observed in allergen-challenged Ndst1-deficient mice may be due to reduced infiltration of the airways by inflammatory cells that release eotaxin such as Th2 cells, alveolar macrophages, and/or eosinophils (Th2 cytokine-mediated) themselves that might be more directly regulated by HS. Allergen-challenged Ndst1-deficient mice also demonstrated significantly lower levels of IL-5 in the BALF compared with WT mice, while there was no difference between the two groups in IL-4 or IL-13 levels (Fig. 5). Apart from being involved in eosinophil maturation, sustenance, and activation, IL-5 has been shown to be required for the development of airway eosinophilia as well as AHR (31, 32). Interestingly, studies have shown that IL-5 can bind to heparin and HS, and they suggest that IL-5-HS binding modulates IL-5 activity through sequestration of this cytokine (33). Furthermore, HS preparations with highly N-sulfated domains were especially efficient at inhibiting IL-5-heparin binding in these studies, clearly underscoring the importance of N-sulfation of HS chains in their interaction with IL-5. In Ndst1-deficient mice, where N-sulfation of endothelial and leukocyte HS is altered, IL-5-HS interactions may be impaired, resulting in decreased eosinophil maturation and infiltration into the airways.
Analysis of the Th1 cytokines IFN-γ and IL-2 demonstrated that IL-2 levels were significantly lower in allergen-challenged Ndst1-deficient mice compared with allergen-challenged WT mice, while IFN-γ levels remained comparable (Fig. 5). In an immunoregulatory role, IL-2 is required for the expansion of CD4+CD25+ regulatory T cells, which in turn suppress T cell activation (34) and play a role in asthma by controlling Th2-driven responses (35). While regulatory T cell levels in the airways of allergen-challenged Ndst1-deficient mice were not evaluated, the suppression of Th2 responses observed in the present study may be due to the overall reduction in recruitment of inflammatory cells, including IL-2-producing cells, accounting for the decreased IL-2 levels in the airways. However, IL-2 is thought to have opposing functions during an inflammatory response, as it is also a potent inducer of T cell proliferation, differentiation, and development of memory cells (36). IL-2-HS interactions can be important in this context. Previous studies have shown that IL-2 binds to heparin-like GAGs, and this interaction is thought to be a mechanism for retaining IL-2 close to its sites of secretion, giving rise to localized concentration gradients in the tissues (37). Additionally, more recent studies using function-blocking anti-HS Abs (same as the one used in the present study (AOB08)) have established that IL-2 binds to HS in the rat spleen (38). The decreased binding of IL-2 to HS due to altered N-sulfation of leukocyte- and endothelial-expressed HSPGs in Ndst1-deficient mice may inhibit T cell proliferation, differentiation, and such, contributing to reduced Th2 responses observed in these mutant mice.
Another cytokine that is widely implicated in the development of allergic airway inflammation and asthma in mice, as well in humans, is TGF-β1 (25, 39). In the present study, TGF-β levels in the BALF and lung tissue of allergen-challenged Ndst1-deficient mice were significantly lower than in WT littermates. In addition to mediating airway remodeling in response to chronic repetitive allergen exposure (40, 41), studies have demonstrated strong staining for TGF-β1 in the bronchial epithelium and peribronchial inflammatory cells associated with early changes of remodeling, even in response to short periods of exposure to OVA in acute models similar to the one used in the present study (42, 43).TGF-β1 is secreted by a wide variety of cell types, including eosinophils and macrophages (44). Reduced infiltration of the airways by the latter cell types is likely to contribute to the decreased levels of TGF-β1 observed in the BALF of allergen-challenged Ndst1 mice. Furthermore, TGF-β1 is known to bind to HS (27) and HSPGs have also been shown to participate in TGF-β1 availability at sites of inflammation by mediating binding between fibronectin and latent TGF-β-binding proteins, which play a major role in the storage of latent TGF-β1 in the ECM (45). Decreased TGF-β1 in peribronchial inflammatory cells as well as in total lung lysates of allergen-challenged Ndst1-deficient mice may be due to reduced binding of TGF-β1 to HS in these mice.
FIZZ1 is expressed in bronchial epithelial and alveolar type II cells during hypoxia (46) and allergic inflammation (28, 29). It has recently been shown to induce proliferation of ECs (47) and stimulate fibroblast differentiation associated with expression of α-SMA and collagen type I, characteristic of early stages of airway remodeling seen in asthma (29, 48). Furthermore, in a murine model of allergic airway inflammation, FIZZ1 expression was found to significantly correlate with the percentage of vascularity and vascular endothelial growth factor expression, further demonstrating a role for this molecule in angiogenesis associated with airway remodeling (47). In the present study, FIZZ1 expression in the bronchial epithelium and by type II alveolar epithelial cells was significantly inhibited in allergen-challenged Ndst1-deficient mice as opposed to corresponding WT mice. FIZZ1 does not appear to bind to heparin or HSPG in direct binding assays (data not shown). Thus, the reduction in FIZZ1 expression observed in the lungs of allergen-challenged mutant mice is likely due to the overall decrease in levels of one or more inflammatory mediators that could regulate FIZZ1 expression rather than due to reduced FIZZ1-HS binding. While Th2 cytokines such as IL-4 and IL-13 are known to regulate FIZZ1 gene expression (49), it is unlikely that these cytokines regulate FIZZ1 expression in allergen-challenged mice, as the levels of both IL-4 and IL-13 in Ndst1-deficient mice are similar to those observed in allergen-challenged WT littermates. Additional studies are required to further understand the role of HS in regulating FIZZ1 expression in allergen-challenged mice.
Overall, the reduction in recruitment of inflammatory cells to the airways in Ndst1-deficient mice appears to significantly suppress the pathological indications associated with allergic airway inflammation. Airway recruitment of inflammatory leukocytes during allergic inflammation involves chemokine-induced trafficking mediated by cell surface selectins, integrins, and their ligands, which facilitate leukocyte rolling followed by firm adhesion on vascular endothelium (50, 51). While endothelial- and leukocyte-deficient Ndst1 mice exhibit impaired neutrophil infiltration in inflammation models partially due to altered L-selectin-dependent trafficking (reduced rolling velocity) on Ndst1-deficient ECs (10), the critical requirement of HS in the regulation of eosinophil trafficking is not known. In the present study, purified murine eosinophils exhibited significantly reduced rolling on MLECs from Ndst1-deficient mice compared with WT MLECs under conditions of physiologic shear stress in vitro. The importance of HS in mediating eosinophil rolling was further substantiated by treatment of WT MLECs with function-blocking anti-HS Abs that significantly inhibited eosinophil rolling on these cells, mimicking that observed on untreated Ndst1-deficient MLECs. Importantly, treatment of murine eosinophils with anti-HS Abs failed to inhibit eosinophil rolling on Ndst1-deficient or WT MLECs, suggesting that the surface expression of HS on vascular endothelium, rather than on eosinophils, regulates eosinophil rolling. IVM studies demonstrating significantly reduced leukocyte rolling in lung microvessels of allergen-challenged Ndst1-deficient mice compared with lung microvessels of corresponding WT littermates further confirmed that endothelial-expressed HS promotes leukocyte trafficking in the lung microcirculation. These data clearly demonstrate that endothelial HS is required for rolling of leukocytes, specifically eosinophils, under conditions of flow, a process that is also known to be dependent on the engagement of L-selectin (52, 53). Indeed, HS has been shown to function as an endothelial ligand for monocyte and neutrophil L-selectin (10, 54, 55). It is therefore likely that the inhibition of leukocyte recruitment, including eosinophils, in Ndst1-deficient mice is due to decreased leukocyte-endothelial interactions as a result of altered HS expression on vascular endothelium.
Collectively, our data demonstrate that endothelial HS plays a crucial role in allergic airway inflammation. In response to allergen challenge, mice deficient in leukocyte and endothelial Ndst1 exhibited diminished AHR associated with decreased IL-5, IL-2, TGF-β, eotaxin, and FIZZ1 levels in the lungs attributable to significantly reduced infiltration of the airways by inflammatory cells. Furthermore, endothelial-expressed HS plays an important role in promoting leukocyte rolling in allergen-challenged lung microvessels, a prerequisite for their subsequent migration across the vascular endothelium to sites of inflammation. Furthermore, since HS binds to IL-5, IL-2, and TGF-β1, the glycan may participate by sequestering and modulating the activity of these cytokines during allergic airway inflammation including asthma.
Acknowledgments
The authors thank Cari M. Calhoun and Yana G. Greenberg for their technical assistance.
Footnotes
This work was supported by National Institutes of Health grants U19-AI70535, HL0793041, AI35796 (to P.S.), and HL57345 (to J.D.E.), and a Career Development Award from the Department of Veterans Affairs (to M.M.F.).
Abbreviations used in this paper: HS, heparan sulfate; GAG, glycosaminoglycan; HSPG, heparan sulfate proteoglycan; ECM, extracellular matrix; Ndst1, N-deacetylase/N-sulfotransferase-1; EC, endothelial cells; AHR, airway hyperresponsiveness; WT, wild type; MCh, methacholine; BALF, bronchoalveolar lavage fluid; MBP, major basic protein; FIZZ1, found in inflammatory zone 1; MLEC, murine lung endothelial cells; IVM, intravital microscopy.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 2.Rostand KS, Esko JD. Microbial adherence to and invasion through proteoglycans. Infect Immun. 1997;65:1–8. doi: 10.1128/iai.65.1.1-8.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kjellén L. Glucosaminyl N-deacetylase/N-sulphotransferases in heparan sulphate biosynthesis and biology. Biochem Soc Trans. 2003;31:340–342. doi: 10.1042/bst0310340. [DOI] [PubMed] [Google Scholar]
- 4.Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132:3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pallerla SR, Pan Y, Zhang X, Esko JD, Grobe K. Heparan sulfate Ndst1 gene function variably regulates multiple signaling pathways during mouse development. Dev Dyn. 2007;236:556–563. doi: 10.1002/dvdy.21038. [DOI] [PubMed] [Google Scholar]
- 6.Pan Y, Woodbury A, Esko JD, Grobe K, Zhang X. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- 7.Ringvall M, Ledin J, Holmborn K, van Kuppevelt T, Ellin F, Eriksson I, Olofsson AM, Kjellen L, Forsberg E. Defective heparan sulfate biosynthesis and neonatal lethality in mice lacking N-deacetylase/N-sulfotransferase-1. J Biol Chem. 2000;275:25926–25930. doi: 10.1074/jbc.C000359200. [DOI] [PubMed] [Google Scholar]
- 8.Fan G, Xiao L, Cheng L, Wang X, Sun B, Hu G. Targeted disruption of NDST-1 gene leads to pulmonary hypoplasia and neonatal respiratory distress in mice. FEBS Lett. 2000;467:7–11. doi: 10.1016/s0014-5793(00)01111-x. [DOI] [PubMed] [Google Scholar]
- 9.Hu Z, Wang C, Xiao Y, Sheng N, Chen Y, Xu Y, Zhang L, Mo W, Jing N, Hu G. NDST1-dependent heparan sulfate regulates BMP signaling and internalization in lung development. J Cell Sci. 2009;122:1145–1154. doi: 10.1242/jcs.034736. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Fuster MM, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 11.Fuster MM, Wang L, Castagnola J, Sikora L, Reddi K, Lee PHA, Radek KA, Schuksz M, Bishop JR, Gallo RL, et al. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 2007;177:539–549. doi: 10.1083/jcb.200610086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garner OB, Yamaguchi Y, Esko JD, Videm V. Small changes in lymphocyte development and activation in mice through tissue-specific alteration of heparan sulphate. Immunology. 2008;125:420–429. doi: 10.1111/j.1365-2567.2008.02856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westergren-Thorsson G, Chakir J, Lafrenière-Allard MJ, Boulet LP, Tremblay GM. Correlation between airway responsiveness and proteoglycan production by bronchial fibroblasts from normal and asthmatic subjects. Int J Biochem Cell Biol. 2002;34:1256–1267. doi: 10.1016/s1357-2725(02)00058-4. [DOI] [PubMed] [Google Scholar]
- 14.Rees MD, McNiven TN, Davies MJ. Degradation of extracellular matrix and its components by hypobromous acid. Biochem J. 2007;401:587–596. doi: 10.1042/BJ20061236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuberi RI, Hsu DK, Kalayci O, Chen HY, Sheldon HK, Yu L, Apgar JR, Kawakami T, Lilly CM, Liu FT. Critical role for galectin-3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am J Pathol. 2004;165:2045–2053. doi: 10.1016/S0002-9440(10)63255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho JY, Miller M, Baek KJ, Han JW, Nayar J, Lee SY, McElwain K, McElwain S, Friedman S, Broide DH. Inhibition of airway remodeling in IL-5-deficient mice. J Clin Invest. 2004;113:551–560. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 18.Rao SP, Wang Z, Zuberi RI, Sikora L, Bahaie NS, Zuraw BL, Liu FT, Sriramarao P. Galectin-3 functions as an adhesion molecule to support eosinophil rolling and adhesion under conditions of flow. J Immunol. 2007;179:7800–7807. doi: 10.4049/jimmunol.179.11.7800. [DOI] [PubMed] [Google Scholar]
- 19.Kurup S, Wijnhoven TJM, Jenniskens GJ, Kimata K, Habuchi H, Li J-p, Lindahl U, van Kuppevelt TH, Spillmann D. Characterization of anti-heparan sulfate phage display antibodies AO4B08 and HS4E4. J Biol Chem. 2007;282:21032–21042. doi: 10.1074/jbc.M702073200. [DOI] [PubMed] [Google Scholar]
- 20.Sriramarao P, DiScipio RG, Cobb RR, Cybulsky M, Stachnick G, Castenada D, Elices M, Broide DH. VCAM-1 is more effective than MAdCAM-1 in supporting eosinophil rolling under conditions of flow. Blood. 2000;95:592–601. [PubMed] [Google Scholar]
- 21.Sikora L, Johansson ACM, Rao SP, Hughes GK, Broide DH, Sriramarao P. A murine model to study leukocyte rolling and intravascular trafficking in lung microvessels. Am J Pathol. 2003;162:2019–2028. doi: 10.1016/S0002-9440(10)64334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao SP, Sikora L, Hosseinkhani MR, Pinkerton KE, Sriramarao P. Exposure to environmental tobacco smoke induces angiogenesis and leukocyte trafficking in lung microvessels. Exp Lung Res. 2009;35:119–135. doi: 10.1080/01902140802449729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sikora L, Rao SP, Sriramarao P. Selectin-dependent rolling and adhesion of leukocytes in nicotine-exposed microvessels of lung allografts. Am J Physiol. 2003;285:L654–L663. doi: 10.1152/ajplung.00448.2002. [DOI] [PubMed] [Google Scholar]
- 24.Taube C, Dakhama A, Gelfand EW. Insights into the pathogenesis of asthma utilizing murine models. Int Arch Allergy Immunol. 2004;135:173–186. doi: 10.1159/000080899. [DOI] [PubMed] [Google Scholar]
- 25.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Ann Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 26.Alcorn JF, Rinaldi LM, Jaffe EF, van Loon M, Bates JHT, Janssen-Heininger YMW, Irvin CG. Transforming growth factor-β1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med. 2007;176:974–982. doi: 10.1164/rccm.200702-334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyon M, Rushton G, Gallagher JT. The interaction of the transforming growth factor-βs with heparin/heparan sulfate is isoform-specific. J Biol Chem. 1997;272:18000–18006. doi: 10.1074/jbc.272.29.18000. [DOI] [PubMed] [Google Scholar]
- 28.Holcomb IN, Kabakoff RC, Chan B, Baker TW, Gurney A, Henzel W, Nelson C, Lowman HB, Wright BD, Skelton NJ, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19:4046–4055. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong L, Wang SJ, Camoretti-Mercado B, Li HJ, Chen M, Bi WX. FIZZ1 plays a crucial role in early stage airway remodeling of OVA-induced asthma. J Asthma. 2008;45:648–653. doi: 10.1080/02770900802126941. [DOI] [PubMed] [Google Scholar]
- 30.Ellyard JI, Simson L, Bezos A, Johnston K, Freeman C, Parish CR. Eotaxin selectively binds heparin: an interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J Biol Chem. 2007;282:15238–15247. doi: 10.1074/jbc.M608046200. [DOI] [PubMed] [Google Scholar]
- 31.Hamelmann E, Wahn U, Gelfand EW. Role of the Th2 cytokines in the development of allergen-induced airway inflammation and hyperresponsiveness. Int Arch Allergy Immunol. 1999;118:90–94. doi: 10.1159/000024037. [DOI] [PubMed] [Google Scholar]
- 32.Hamelmann E, Gelfand EW. IL-5-induced airway eosinophilia: the key to asthma? Immunol Rev. 2001;179:182–191. doi: 10.1034/j.1600-065x.2001.790118.x. [DOI] [PubMed] [Google Scholar]
- 33.Lipscombe RJ, Nakhoul AM, Sanderson CJ, Coombe DR. Interleukin-5 binds to heparin/heparan sulfate: a model for an interaction with extracellular matrix. J Leukocyte Biol. 1998;63:342–350. doi: 10.1002/jlb.63.3.342. [DOI] [PubMed] [Google Scholar]
- 34.Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172:3983–3988. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- 35.Robinson DS, Larché M, Durham SR. Tregs and allergic disease. J Clin Invest. 2004;114:1389–1397. doi: 10.1172/JCI23595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoyer KK, Dooms H, Barron L, Abbas AK. Interleukin-2 in the development and control of inflammatory disease. Immunol Rev. 2008;226:19–28. doi: 10.1111/j.1600-065X.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 37.Najjam S, Gibbs RV, Gordon MY, Rider CC. Characterization of human recombinant interleukin 2 binding to heparin and heparan sulfate using an ELISA approach. Cytokine. 1997;9:1013–1022. doi: 10.1006/cyto.1997.0246. [DOI] [PubMed] [Google Scholar]
- 38.ten Dam GB, Hafmans T, Veerkamp JH, van Kuppevelt TH. Differential expression of heparan sulfate domains in rat spleen. J Histochem Cytochem. 2003;51:727–739. doi: 10.1177/002215540305100604. [DOI] [PubMed] [Google Scholar]
- 39.Doherty T, Broide D. Cytokines and growth factors in airway remodeling in asthma. Curr Opin Immunol. 2007;19:676–680. doi: 10.1016/j.coi.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 40.Boxall C, Holgate ST, Davies DE. The contribution of transforming growth factor-β and epidermal growth factor signalling to airway remodelling in chronic asthma. Eur Respir J. 2006;27:208–229. doi: 10.1183/09031936.06.00130004. [DOI] [PubMed] [Google Scholar]
- 41.Makinde T, Murphy RF, Agrawal DK. The regulatory role of TGF-β in airway remodeling in asthma. Immunol Cell Biol. 2007;85:348–356. doi: 10.1038/sj.icb.7100044. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka H, Masuda T, Tokuoka S, Komai M, Nagao K, Takahashi Y, Nagai H. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm Res. 2001;50:616–624. doi: 10.1007/PL00000243. [DOI] [PubMed] [Google Scholar]
- 43.Locke NR, Royce SG, Wainewright JS, Samuel CS, Tang ML. Comparison of airway remodeling in acute, subacute, and chronic models of allergic airways disease. Am J Respir Cell Mol Biol. 2007;36:625–632. doi: 10.1165/rcmb.2006-0083OC. [DOI] [PubMed] [Google Scholar]
- 44.Bloemen K, Verstraelen S, Van Den Heuvel R, Witters H, Nelissen I, Schoeters G. The allergic cascade: review of the most important molecules in the asthmatic lung. Immunol Lett. 2007;113:6–18. doi: 10.1016/j.imlet.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 45.Chen Q, Sivakumar P, Barley C, Peters DM, Gomes RR, Farach-Carson MC, Dallas SL. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-β (TGF-β) by modulating assembly of latent TGF-β-binding protein-1. J Biol Chem. 2007;282:26418–26430. doi: 10.1074/jbc.M703341200. [DOI] [PubMed] [Google Scholar]
- 46.Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMα, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res. 2003;92:1065–1067. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- 47.Sun Y, Wang J, Li H, Han X. Found in inflammatory zone 1 induces angiogenesis in murine models of asthma. Lung. 2008;186:375–380. doi: 10.1007/s00408-008-9099-1. [DOI] [PubMed] [Google Scholar]
- 48.Chung MJ, Liu T, Ullenbruch M, Phan SH. Antiapoptotic effect of found in inflammatory zone (FIZZ)1 on mouse lung fibroblasts. J Pathol. 2007;212:180–187. doi: 10.1002/path.2161. [DOI] [PubMed] [Google Scholar]
- 49.Stutz AM, Pickart LA, Trifilieff A, Baumruker T, Prieschl-Strassmayr E, Woisetschlager M. The Th2 cell cytokines IL-4 and IL-13 regulate found in inflammatory zone 1/resistin-like molecule α gene expression by a STAT6 and CCAAT/enhancer-binding protein-dependent mechanism. J Immunol. 2003;170:1789–1796. doi: 10.4049/jimmunol.170.4.1789. [DOI] [PubMed] [Google Scholar]
- 50.Broide DH, Sriramarao P. Cellular adhesion in inflammation. In: Adkinson NF Jr, Bochner BS, Busse WW, Holgate ST, Lemanske RF Jr, Simons FER, editors. Middleton’s Allergy: Principles and Practice. 7. Vol. 1. Elsevier; New York: 2008. pp. 149–164. [Google Scholar]
- 51.Rosenberg HF, Phipps S, Foster PS. Eosinophil trafficking in allergy and asthma. J Allergy Clin Immunol. 2007;119:1303–1310. doi: 10.1016/j.jaci.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 52.Sriramarao P, von Andrian UH, Butcher EC, Bourdon MA, Broide DH. L-selectin and very late antigen-4 integrin promote eosinophil rolling at physiological shear rates in vivo. J Immunol. 1994;53:4238–4246. [PubMed] [Google Scholar]
- 53.Maslin CL, Kedzierska K, Webster NL, Muller WA, Crowe SM. Transendothelial migration of monocytes: the underlying molecular mechanisms and consequences of HIV-1 infection. Curr HIV Res. 2005;3:303–317. doi: 10.2174/157016205774370401. [DOI] [PubMed] [Google Scholar]
- 54.Giuffre L, Cordey AS, Monai N, Tardy Y, Schapira M, Spertini O. Monocyte adhesion to activated aortic endothelium: role of L-Selectin and heparan sulfate proteoglycans. J Cell Biol. 1997;136:945–956. doi: 10.1083/jcb.136.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gotte M. Syndecans in inflammation. FASEB J. 2003;17:575–591. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]