

Abstract
To examine the role of T cell receptor (TCR) in γδ T cells in adaptive immunity, a macaque model was used to follow Vγ2Vδ2+ T cell responses to mycobacterial infections. These phosphoantigen-specific γδ T cells displayed major expansion during Mycobacterium bovis Bacille Calmette-Guérin (BCG) infection and a clear memory-type response after BCG reinfection. Primary and recall expansions of Vγ2Vδ2+ T cells were also seen during Mycobacterium tuberculosis infection of naïve and BCG-vaccinated macaques, respectively. This capacity to rapidly expand coincided with a clearance of BCG bacteremia and immunity to fatal tuberculosis in BCG-vaccinated macaques. Thus, Vγ2Vδ2+ T cells may contribute to adaptive immunity to mycobacterial infections.
The majority of circulating γδ T cells in humans express a TCR heterodimer comprised of Vγ2 and Vδ2 segments. Recent in vitro studies have demonstrated that human Vγ2Vδ2+ T cells recognize small organic phosphate antigens (1–6) from microbes and other nonpeptide molecules such as alkylamines and aminobisphosphonates (7, 8). The recognition of these nonpeptide antigens by Vγ2Vδ2+ T cells does not require antigen processing or presentation by major histocompatibility complex (MHC) or CD1 molecules (9). Mycobacterium tuberculosis–induced tuberculosis remains a leading cause of morbidity and mortality among infectious diseases. Immune correlates of protection against tuberculosis remain poorly characterized in humans. Detailed analyses of Vγ2Vδ2+ T cell responses and their potential role in microbial infections have not been well documented in humans [see review, (10)], despite compelling evidence that murine γδ T cells provide protection against a range of infections (11–16). The capability of Vγ2Vδ2+ T cells to mount a memory response after microbial reinfection or reactivation has not been demonstrated. Relevant studies in mice cannot be performed because murine γδ T cells do not express the homolog of the Vγ2Vδ2+ TCR, and there is no functional equivalent for these cells, so far, identified in mice. We reasoned that nonhuman primates could provide a good model in which to determine the nature of the immune responses of Vγ2Vδ2+ T cells in a mycobacterial infection.
To examine a primary γδ T cell response during mycobacterial infection, macaques were inoculated with BCG (17) and assessed for changes in their γδ T cell repertoire (18). Striking expansions of circulating Vγ2Vδ2+ T cells were detected in the blood of these monkeys after BCG inoculation, whereas there was no apparent increase in other circulating Vγ+ or Vδ+ T cells (Fig. 1A). The percent of peripheral blood CD3+ T cells that were Vγ2Vδ2+ and the absolute number of circulating Vγ2Vδ2+ T cells increased by up to 25-fold and as much as a 200-fold, respectively (Fig. 1A). The expansion of Vγ2Vδ2+ T cells was most evident 3 to 5 weeks after BCG inoculation (Fig. 1A). The expansion of circulating Vγ2Vδ2+ T cells in the infected macaques indicated the development of primary response of these cells during a mycobacterial infection.
Fig. 1.

BCG infection induced a major expansion of Vγ2Vδ2+ T cells in the macaques. (A) Changes in relative and absolute numbers of CD3+ peripheral blood lymphocytes (PBL) that are Vγ+, Vδ+, or Vγ2Vδ2+ T cells after BCG inoculation. (B) A major expansion of Vγ2Vδ2+ T cells in pulmonary alveoli and intestinal mucosae but not in lymph nodes of the macaques. Mm, rhesus monkeys (Macaca mulatta). The monkeys, 3 to 15 years of age, were inoculated intravenously with 106 CFU of BCG.
Vγ2Vδ2+ T cells compose the majority of the human γδ T cell population in the circulation. We next examined whether macaque Vγ2Vδ2+ T cells in tissue compartments had expanded during a BCG infection. An increase in pulmonary Vγ2Vδ2+ T cells was evident after intravenous BCG inoculation (Fig. 1B). Expansion of the Vγ2Vδ2 T cell subpopulation was also apparent within intestinal mucosae (Fig. 1B). Despite this expansion in blood and tissue sites, only a subtle expansion of Vγ2Vδ2+ T cells was observed in organized lymph nodes of the same monkeys. These findings suggest that the Vγ2Vδ2+ T cells in mucosal but not peripheral lymph node tissues could expand in response to a mycobacterial infection. The expansion of Vγ2Vδ2+ T cells in these mucosal areas may result from their local proliferation in response to the BCG infection. In addition, Vγ2Vδ2+ T cells may also accumulate in these tissue sites through the recruitment from the circulating pool of γδ T cells.
The hallmarks of memory T cell responses include their antigen-specific persistence after the initial infection and the rapid and prolonged recall responses upon reinfection. To determine whether Vγ2Vδ2+ T cell responses possessed the capacity for immunological memory, macaques that had previously been infected with BCG were inoculated again with BCG and were assessed for memory Vγ2Vδ2+ T cell responses. As early as 4 to 6 days after the second BCG inoculation, a marked expansion of Vγ2Vδ2+ T cells was apparent in the blood of the monkeys (Fig. 2). The Vγ2Vδ2+ T cell expansions during the BCG reinfection were 2 to 9 times as large as those seen during primary infection (Fig. 2). The expansion of these cells persisted for as long as 7 months after the second BCG inoculation (Fig. 2). Analyses of TCR sequences revealed that some of the Vγ2Vδ2+ T cell clones that expanded during primary BCG infection were also identified in the Vγ2Vδ2+ T cell subpopulation that expanded after reinfection [see supplemental material (19)]. These molecular analyses provide evidence that Vγ2Vδ2+ T cells that underwent polyclonal expansion during a primary BCG infection can mount a memory or recall response after a secondary BCG infection.
Fig. 2.

BCG reinfection induced a recall expansion of circulating Vγ2Vδ2+ T cells. The left panel shows a rapid increase in γδ+ T cells in CD3+ PBL after a second BCG inoculation, whereas the right panel shows that the responses of circulating Vγ2Vδ2+ T cells after BCG reinfection were greater in magnitude and longer in duration than the response of these cells after the first BCG infection. BCG reinfection of the monkeys that recovered from the primary active infection was done by intravenous inoculation of them with 108 CFU of BCG 3 to 5 months after the first BCG inoculation.
Given that Vγ2Vδ2+ T cells contribute to adaptive immune responses, it seems likely that the clonally expanded cells that emerge after BCG infection were involved in the control of active BCG replication in the macaques. To address this possibility, we monitored active BCG infection by quantitating BCG colony counts in the blood (20) and determined the kinetics of Vγ2Vδ2+ T cell expansions during the course of the BCG infection. The expansion of Vγ2Vδ2+ T cells was associated with a clearance of detectable BCG bacteremia (Fig. 3). Our recent studies suggest that the decline of BCG colony-forming units (CFU) in the blood is dependent on the immune competence, because the enhanced T cell suppression during simian immunodeficiency virus–BCG (SIV-BCG) coinfection results in a prolonged BCG bacteremia in the blood (20, 21). Thus, the kinetics of BCG CFU counts and Vγ2Vδ2+ T cells in BCG-infected macaques suggest that Vγ2Vδ2+ T cells contribute to the immune containment of avirulent mycobacterial infections in macaques, and these findings are consistent with the antimicrobial activities of γδ T cells observed in mice (11–16).
Fig. 3.
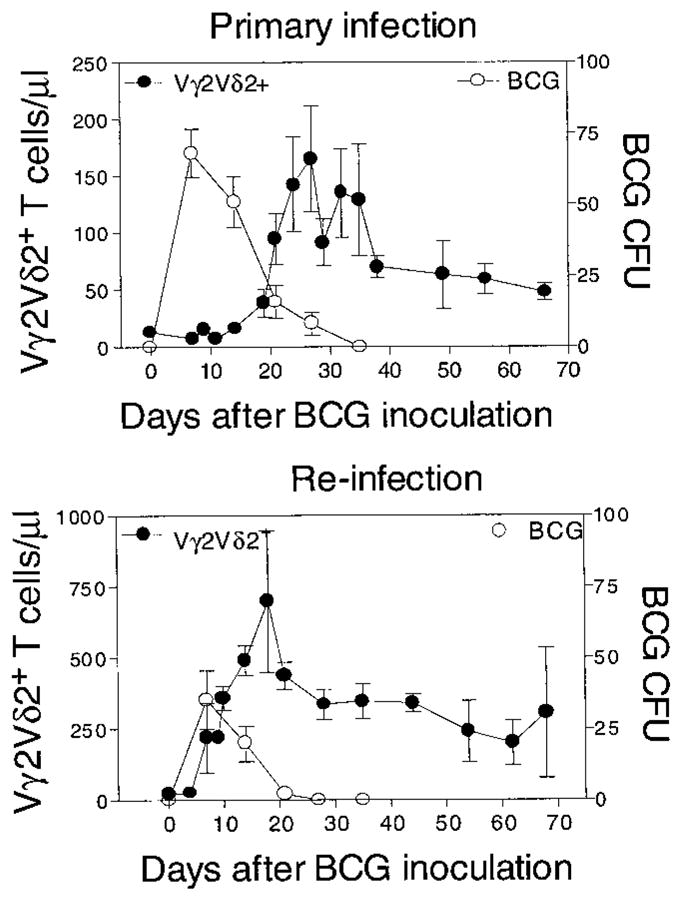
Vγ2Vδ2+ T cell expansion was associated temporally with the decreasing trend of active BCG infection. Shown is the kinetics of peripheral blood BCG CFUs (ml) and Vγ2Vδ2+ T cell expansions after the first (top) and second (bottom) BCG infection. Data shown are the means of values with the error bars of SEM from four infected animals.
Lastly, we sought to determine whether the adaptive immune responses of Vγ2Vδ2+ T cells seen in the BCG infection could also be identified in M. tuberculosis infection (19). After a M. tuberculosis aerosol challenge (exposure to aerosolized M. tuberculosis), the naïve macaques exhibited a primary expansion of Vγ2Vδ2+ T cells in the bronchoalveolar lavage (BAL) fluid (Fig. 4A). A rapid recall expansion of Vγ2Vδ2+ T cells was also apparent in BAL fluid after a M. tuberculosis aerosol challenge of the BCG-vaccinated macaques (Fig. 4A). Similar to what was observed after the pulmonary BCG exposure (unpublished observations), M. tuberculosis aerosol challenge did not induce an apparent expansion of Vγ2Vδ2+ T cells in the blood of naïve and BCG-vaccinated macaques in the acute infection. The rapid recall expansion of alveolar Vγ2Vδ2+ T cells in the BCG-vaccinated, M. tuberculosis-infected macaques coincided with the development of protective immunity to the acutely fatal form of tuberculosis (Fig. 4B). The BCG-vaccinated macaques remained clinically healthy during a 2.5-month period of follow up after M. tuberculosis challenge. In contrast, the unvaccinated macaques developed lethargy, anorexia, and wasting; they subsequently died and they showed evidence of miliary tuberculosis 4 to 6 weeks after M. tuberculosis aerosol challenge. We realize that the BCG-vaccinated, M. tuberculosis-infected monkeys may progress to a subclinical form of tuberculosis, because they did exhibit detectable M. tuberculosis and its mRNA in BAL cells (Fig. 4B). Nevertheless, this potential outcome would not negate our observation that a rapid recall response of Vγ2Vδ2+ T cells coincided with immune protection against the acutely fatal tuberculosis in monkeys.
Fig. 4.

Rapid recall expansion of Vγ2Vδ2+ T cells after M. tuberculosis aerosol challenge was associated with immunity to acutely fatal tuberculosis in BCG-vaccinated monkeys. (A) M. tuberculosis aerosol challenge induced primary and recall expansions of alveolar Vγ2Vδ2+ T cells in four naïve (left) and four BCG-vaccinated (right) macaques, respectively. Both percentage and absolute numbers of alveolar Vγ2Vδ2+ T cells are shown after the M. tuberculosis aerosol challenge. (B) A reduced number of viable BCG (CFU/ml) and Ag85 mRNA in BAL cells of BCG-vaccinated monkeys. The naïve macaques died from acutely fatal tuberculosis on days 25, 28, 38, and 41 after the M. tuberculosis aerosol challenge. Necropsy showed miliary tuberculosis in the lung, liver, spleen, and kidney of the dead macaques. The BCG-vaccinated macaques remained clinically healthy during a 2.5-month follow up after M. tuberculosis aerosol challenge (P < 0.001). The eight monkeys, all of which were 2 years of age, were challenged with 400 to 500 M. tuberculosis organisms by aerosol route (21). The BCG vaccination was done by intravenous inoculation of 106 CFU 6 weeks before the M. tuberculosis aerosol challenge. Quantitation of M. tuberculosis Ag85B mRNA was done with the use of real-time quantitative polymerase chain reaction (PCR) (21).
Our studies provide strong evidence that Vγ2Vδ2+ T cells, like αβ+ T cells, contribute to adaptive immune responses in mycobacterial infections. The adaptive immune responses of Vγ2Vδ2+ T cells are indeed driven by BCG nonpeptide antigens (19). The contribution of these cells to vaccine protection against tuberculosis was demonstrated in the juvenile rhesus model. BCG-mediated protection against the fatal form of tuberculosis has been reported in children, although its protective efficacy for chronic pulmonary tuberculosis in adults and monkeys is controversial (22–29). It seems that the antigen specificity, TCR diversity, and recall features of Vγ2Vδ2+ T cells separate this γδ T cell subset from innate cells including peripheral mononuclear cells (PMN), monocytes, and natural killer (NK) cells as well as those γδ T cells that express invariant γδ TCR (30, 31). The unique ability of Vγ2Vδ2+ T cells to mount rapid and large expansions in mycobacterial infections suggests that vaccine-elicited Vγ2Vδ2+ T cell immunity may be both possible and useful. Thus, Vγ2Vδ2+ T cells may broadly contribute to both innate and acquired immunity against microbial infections.
Acknowledgments
Supported by NIH RO1 grants HL64560 and RR13601 (to Z.W.C.). We thank members at Viral Pathogenesis for technical support and W. R. Jacobs at Albert Einstein College of Medicine for providing M. tuberculosis H37Rv used in the study.
References and Notes
- 1.Pfeffer K, Schoel B, Gulle H, Kaufmann SH, Wagner H. Eur J Immunol. 1990;20:1175. doi: 10.1002/eji.1830200534. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka Y, et al. Proc Natl Acad Sci USA. 1994;91:8175. doi: 10.1073/pnas.91.17.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burk MR, Mori L, De Libero G. Eur J Immunol. 1995;25:2052. doi: 10.1002/eji.1830250737. [DOI] [PubMed] [Google Scholar]
- 4.Poquet Y, et al. Res Immunol. 1996;147:542. doi: 10.1016/s0923-2494(97)85220-0. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka Y, Morita CT, Nieves E, Brenner MB, Bloom BR. Nature. 1995;375:155. doi: 10.1038/375155a0. [DOI] [PubMed] [Google Scholar]
- 6.Constant P, et al. Science. 1994;264:267. doi: 10.1126/science.8146660. [DOI] [PubMed] [Google Scholar]
- 7.Bukowski JF, Morita CT, Brenner MB. Immunity. 1999;11:57. doi: 10.1016/s1074-7613(00)80081-3. [DOI] [PubMed] [Google Scholar]
- 8.Kunzmann V, Bauer E, Wilhelm M. N Engl J Med. 1999;340:737. doi: 10.1056/NEJM199903043400914. [DOI] [PubMed] [Google Scholar]
- 9.Morita CT, et al. Immunity. 1995;3:495. doi: 10.1016/1074-7613(95)90178-7. [DOI] [PubMed] [Google Scholar]
- 10.Morita CT, Mariuzza RA, Brenner MB. Springer Semin Immunopathol. 2000;22:191. doi: 10.1007/s002810000042. [DOI] [PubMed] [Google Scholar]
- 11.Mombaerts P, Arnoldi J, Russ F, Tonegawa S, Kaufmann SH. Nature. 1993;365:53. doi: 10.1038/365053a0. [DOI] [PubMed] [Google Scholar]
- 12.Ladel CH, et al. Eur J Immunol. 1995;25:838. doi: 10.1002/eji.1830250331. [DOI] [PubMed] [Google Scholar]
- 13.Hiromatsu K, et al. J Exp Med. 1992;175:49. doi: 10.1084/jem.175.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Souza CD, et al. J Immunol. 1997;158:1217. [PubMed] [Google Scholar]
- 15.Sciammas R, Kodukula P, Tang Q, Hendricks RL, Bluestone JA. J Exp Med. 1997;185:1969. doi: 10.1084/jem.185.11.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore TA, Moore BB, Newstead MW, Standiford TJ. J Immunol. 2000;165:2643. doi: 10.4049/jimmunol.165.5.2643. [DOI] [PubMed] [Google Scholar]
- 17.Zhou D, et al. J Immunol. 1999;162:2204. [PubMed] [Google Scholar]
- 18.Chen ZW, et al. J Med Primatol. 2000;29:143. doi: 10.1034/j.1600-0684.2000.290307.x. [DOI] [PubMed] [Google Scholar]
- 19.Supplemental material is available on Science Online at www.sciencemag.org/cgi/content/full/295/5563/2255/DC1
- 20.Shen Y, et al. Infect Immun. 2002;70:869. doi: 10.1128/IAI.70.2.869-877.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Y, et al. J Virol. 2001;75:8690. doi: 10.1128/JVI.75.18.8690-8696.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrigues LC, Diwan VK, Wheeler JG. Int J Epidemiol. 1993;22:1154. doi: 10.1093/ije/22.6.1154. [DOI] [PubMed] [Google Scholar]
- 23.Colditz GA, et al. JAMA. 1994;271:698. [PubMed] [Google Scholar]
- 24.Barclay WR, et al. Am Rev Respir Dis. 1973;107:351. doi: 10.1164/arrd.1973.107.3.351. [DOI] [PubMed] [Google Scholar]
- 25.Colditz GA, et al. Pediatrics. 1995;96:29. [PubMed] [Google Scholar]
- 26.Janicki BW, Good RC, Minden P, Affronti LF, Hymes WF. Am Rev Respir Dis. 1973;107:359. doi: 10.1164/arrd.1973.107.3.359. [DOI] [PubMed] [Google Scholar]
- 27.Ribi E, et al. J Infect Dis. 1971;123:527. doi: 10.1093/infdis/123.5.562. [DOI] [PubMed] [Google Scholar]
- 28.McMurray DN. Clin Infect Dis. 2000;30(suppl 3):S210. doi: 10.1086/313885. [DOI] [PubMed] [Google Scholar]
- 29.Langermans JA, et al. Proc Natl Acad Sci USA. 2001;98:11497. doi: 10.1073/pnas.201404898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukasa A, Born WK, O’Brien RL. J Immunol. 1999;162:4910. [PubMed] [Google Scholar]
- 31.Itohara S, et al. Nature. 1990;343:754. doi: 10.1038/343754a0. [DOI] [PubMed] [Google Scholar]