

Abstract
Transgenic mice that overexpress PKCα in the epidermis (K5-PKCα mice) exhibit acute CXCR2-mediated intraepidermal neutrophilic inflammation and a strong epidermal hyperplasia in response to application of 12-O-tetradecanoyl-phorbol-13-acetate (TPA). We now show that hyperplasia is independent of infiltrating neutrophils. Further, when K5-PKCα mice were initiated with 7, 12-dimethylbenz[a]anthracene (DMBA) and promoted with a low dose of TPA, 58% of K5-PKCα mice developed skin papillomas that progressed to carcinoma while wildtype mice did not develop tumors. We confirmed that CXCR2 is expressed by keratinocytes and demonstrated that transformation by oncogenic ras (a hallmark of DMBA initiation) or TPA exposure induced all CXCR2 ligands. Ras induction of CXCR2 ligands was mediated by autocrine activation of EGFR and NF-κB and potentiated by PKCα. Oncogenic ras also induced CXCR2 ligands in keratinocytes genetically ablated for CXCR2. However, ras transformed CXCR2 null keratinocytes formed only small skin tumors in orthotopic skin grafts to CXCR2 intact hosts while transformed wildtype keratinocytes produced large tumors. In vitro, CXCR2 was essential for CXCR2 ligand-stimulated migration of ras transformed keratinocytes and for ligand activation of the ERK and Akt pathways. Both migration and activation of ERK and Akt were restored by CXCR2 reconstitution of CXCR2 null keratinocytes. Thus, activation of CXCR2 on ras transformed keratinocytes has both pro-migratory and pro-tumorigenic functions. The upregulation of CXCR2 ligands following initiation by oncogenic ras and promotion with TPA in the mouse skin model provides a mechanism to stimulate migration by both autocrine and paracrine pathways and contribute to tumor development.
Keywords: PKC, inflammation, skin, cancer, CXCR2, chemokines
INTRODUCTION
The essential contribution of inflammation to tumor development and progression has gained more attention based on clinical and experimental evidence. Accumulating data have ascribed both positive and negative influences of inflammatory pathways on tumor development and progression and have assigned these diverse influences to particular subsets of inflammatory cells, cancer stages and target tissues (1). Furthermore, controversy exists regarding the relative importance of interactions of tumor-stromal inflammatory cells versus the role of soluble mediators released during those interactions (2;3). Many of these insights have been derived from studies of squamous cell tumor induction by DMBA and TPA application on mouse skin. For example, mice deficient for TNFα or its receptors are resistant to chemically induced skin carcinogenesis (4;5), a result attributed to decreased inflammation in the dermis, notably neutrophil recruitment. Further, skin resident αβT cells and γδT cells have opposite effects on the sensitivity to DMBA/TPA chemical skin carcinogenesis (6). In the K14-HPV16 mouse skin cancer model, B lymphocytes recruit mast cells where they contribute to the angiogenic response of the stroma (7). The facilitating role of neutrophils and macrophages on tumor angiogenesis has also been reported in a matrix inserted surface transplantation model using oncogene transformed HaCat cells (8).
The recruitment of leukocytes is critically regulated by small cytokine-like soluble proteins called chemokines under both homeostatic and inflammatory conditions. Mouse neutrophils express a receptor homologous to human IL-8 (CXCL8) receptor or CXCR2. Mouse CXCR2 binds several CXCL-8 like CXC chemokines most notably KC (CXCL1) and macrophage inflammatory protein 2 (MIP-2/CXCL2/3) and GCP-2 (CXCL6). KC and MIP-2 are related to the three human GRO chemokines (CXCL1/3). We and others have previously demonstrated that transgenic mice that overexpress the conventional PKCα in basal keratinocytes exhibit an acute intraepidermal inflammatory response when mice were topically exposed to the PKC activator 12-O-tetradecanoyl-phorbol-13-acetate (TPA) (9;10). Driving the inducible cutaneous inflammation is a PKCα dependent upregulation of CXCR2 ligands attracting neutrophils into the epidermis, forming intraepidermal neutrophilic microabscesses, sloughing of the epidermis and a regenerative hyperplastic response emanating from the intact hair follicle epithelium (9;11).
This model system of inducible cutaneous inflammation offered an opportunity to address several questions related to tumor development. The relatively pure neutrophilic infiltration provided an opportunity to address the role of that cell type in the regenerative response. Epidermal regeneration from hair follicle cells tested the source of initiated cells. The dependence of the model on CXCR2 mediated responses provided a setting to question the function of this receptor and its ligands in tumor formation. The results indicate that CXCR2 and its ligands contribute to neoplastic development unexpectedly through an autocrine response loop that may prove a novel target for therapy.
MATERIALS AND METHODS
Mice and treatments
Mouse studies were performed under a protocol approved by the National Cancer Institute and National Institutes of Health (NIH) Animal Care and Use Committee. The construction and characterization of K5-PKCα mice on an FVB/N background were previously described (9). K5-PKCα mice were crossed to homozygous mice deficient in CXCR2. F1 mice K5-PKCα/CXCR2+/− were backcrossed to CXCR2+/− to generate K5-PKCα mice deficient for CXCR2 (11). 12-O-tetradecanoylphorbol-13-acetate (TPA, Calbiochem) was dissolved in acetone, and one microgram was applied in 200 μl to the shaved dorsal skin of experimental animals.
Purified rat anti–mouse Ly-6G and Ly-6C (Gr-1, IgG2b, κ isotype) and purified rat IgG2b, κ isotype-matched control antibody were purchased from BD Biosciences. Mice were injected intra-peritoneally with 100μg of the anti-Gr1 antibody or isotype control 16 hours prior to TPA application. Depletion of neutrophils reported earlier (12) was confirmed by differential white blood counts 12h after TPA (Supplemental figure 1A), myeloperoxydase immunostaining of skin sections collected at 12 and 24h after TPA (Supplemental figure 1B) and H&E staining of paraffin embedded skin sections (data not shown). Approximately 1h prior to sacrifice, animals were injected with 250–300μl of a 3 mg/ml solution of BrdU in sterile PBS. Samples were fixed in formalin or IHC zinc fixative (BD Biosciences) and embedded in paraffin for H&E analysis and immunohistochemistry. For BrdU staining sections were treated as recommended by manufacturer (Roche) using anti-bromodeoxyuridine-POD antibody (clone BMG 6H8). Apoptosis and CD31 staining were performed by the Pathology/Histotechnology Laboratory (NCI core facility) using respectively ApopTag kit (Millipore), anti-PECAM-1/CD31 antibody (Santa Cruz, sc-1506) and anti–myeloperoxidase (DAKO) on zinc fixed sections. The microvascular density was determined by counting the number of CD31 stained cells in at least five fields (x400 magnification, Nikon Eclipse E400) per tumor originating from CXCR2 WT or CXCR2 KO v-rasHa-transduced keratinocytes. The antibodies against the mouse filaggrin and loricrin have been described previously (13).
Tumor induction experiments
The backs of 7-week-old mice were shaved 2 days before initiation with 7,12-dimethylbenz[a]anthracene (DMBA). Initiation was accomplished by a single topical application of 100 μg DMBA in 0.2 ml acetone. Promoter treatments with TPA (1 μg (1.6 nmoles) in 0.2 ml acetone, twice weekly) were begun 1 week after initiation and continued for 20 weeks. Skin tumors induced by the initiation-promotion protocol were removed and both frozen and formalin fixed at times from 20–52 weeks after initiation. Tumor type (squamous papilloma or squamous cell carcinoma) was verified by examination of H&E-stained sections of tumors
Nude mouse grafting
On day 3 in culture, CXCR2+/+ and CXCR2−/− primary keratinocytes were infected with the v-rasHa retrovirus, and trypsinized and used for grafting on day 8 as described previously (14). Two million keratinocytes were mixed with 5 million SENCAR mouse dermal fibroblasts (cultured for 1 week) and grafted onto the back of nude mice on a prepared skin graft site (14). Tumor dimensions were measured weekly using calipers, and approximate tumor volumes were determined by multiplying tumor height × length × width.
Cell culture
Primary mouse keratinocytes and hair follicle buds were isolated from newborn transgenic and wildtype littermate epidermis as described (14). EGFR deficient keratinocytes were isolated from pups obtained from EGFR heterozygous breeding pairs (15).
Retroviral and adenoviral constructs
The v-rasHa replication defective ecotropic retrovirus was prepared using ψ2 producer cells (16). Retrovirus titers were routinely 1 × 107 virus/ml. Keratinocytes were infected with v-rasHa retrovirus (oncogenic ras) on day 3 at a MOI of 1 in medium containing 4 μg/ml Polybrene (Sigma). The mouse CXCR2 cDNA cloned into pEGFP-N1 vector (Clonetch) was a kind gift of Dr. Gao (Laboratory of Molecular Immunology, NIAID, NIH). The Entry clone for CXCR2-eGFP was transferred to the vector pAd/CMV/V5-DEST (Invitrogen) using Gateway LR recombination per the manufacturer’s protocols. Adenoviral clones were verified by restriction analysis, and transfection-ready DNA was made using the Sigma GenElute HP maxiprep kit in the Protein Expression Laboratory (PEL, Core Facility of National Cancer Institute-Frederick). Adenoviruses were amplified in the Viral Technology Laboratory (VTL, Core Facility of National Cancer Institute-Frederick). The IκBαSR adenovirus was introduced into primary keratinocytes using an adenoviral construct driven by a cytomegalovirus (CMV) promoter and empty adenovirus was used as control (A-CMV). Adenoviruses were amplified using QBI 293 cells. Virus was purified over two CsCl gradients and dialyzed against a buffer containing Tris-HCl (pH 7.5), 10% glycerol, and 1 mM MgCl2. Keratinocytes were infected for 30 minutes in serum-free medium with a multiplicity of infection of 5 and 10 viral particles/cell (IκBαsr and CXCR2-EGFP respectively) and 4.0 μg/ml of Polybrene (Sigma) to enhance uptake. Serum containing medium was added to the cells for the next 16 or 48 hours after the infection (CXCR2-EGFP and IκBαsr respectively).
Immunoblotting
Cultured keratinocytes transduced with either control or mCXCR2-GFP adenovirus or an aliquot of cultured cells used for grafting experiments were lysed in MPER lysis buffer (Pierce) supplemented with 200 μM NaVO3, 10 mM NaF and Complete Mini tablets (Roche). Proteins were quantified by the Bradford method (Bio-Rad) and separated by 10% or 18% SDS-PAGE for Akt/ERK or H-ras blotting respectively. Akt and phospho-Akt antibodies (Cell Signaling) were used at 1:1000 overnight, ERK and phospho-ERK antibodies (Cell Signaling) were used at 1:2000 for two hours at room temperature and H-ras antibody (C-20, Santa Cruz Biotechnology) was used at 1:500 overnight at 4 degrees. ECL SuperSignal (Pierce) system was used for detection. The intensities of immunoblots were quantified using ImageJ (NIH), and the relative expression of targeted proteins was normalized.
In Vitro migration assay
On day 3 in culture, CXCR2+/+ and CXCR2−/− primary keratinocytes were infected with the v-rasHa retrovirus then used for migration assay on day 7. Keratinocytes were trypsinized and resuspended in ice-cold 0.2% serum culture medium. 5 × 105 cells were plated in the top chamber of noncoated polyethylene terephthalate (PET) membranes Biocoat control inserts (24-well plate insert with a pore size of 8.0 μm; BD Biosciences). MIP-2 (R&D systems) or TGFα (R&D systems) were added to the bottom well in 0.2% serum culture medium at the concentration indicated. The CXCR2 small-molecule antagonist was synthesized by Dr. Victor E. Marquez and Krishnan Malolanarasimhan (Laboratory of Medicinal Chemistry, NCI) based on the structure previously reported (17). CXCR2 antagonist was added concomitantly to the cells in the top chamber. The cells were incubated for 24 h and those that did not migrate through the pores in the membrane were removed by scraping the membrane with a cotton swab. Cells on the underside of the membrane were stained with Diff-Quick (Dade Behring). Cells migrating across the filters were counted under light microscopy after coding the samples. The results were expressed as chemotaxis index (CI), which represents the fold increase in the number of migrated cells in three 40× fields in response to MIP-2 or TGFα over the spontaneous cell migration in response to control medium.
Time-lapse-microscopy studies
Keratinocytes were seeded on 12-well plates. On day 3 in culture, CXCR2+/+ and CXCR2−/− primary keratinocytes were infected with the v-rasHa retrovirus then used for imaging on day 7 as described previously (18). Briefly, imaging was performed on an Olympus IX81 research microscope (Olympus America, Melville, NY), equipped with a Proscan motorized stage (Prior Scientific Instruments Ltd., Rockland, MA) and placed in Incubation System, Neue a temperature- and CO2-controlled chamber (LiveCell Biosciences, Camp Hill, PA). Using relief-contrast optics, 10× images were taken of each well every 10 minutes until 100 images were captured. For each variable, triplicate wells were tracked and images managed using Slidebook 4.0 Digital Microscopy Solutions software (Intelligent Imaging Inovations, Denver, CO). Images were transferred to a workstation equipped with Metamorph image analysis software (Molecular Devices Corp., Chicago, IL), where velocities were calculated using the track cell module. Per well, 20 keratinocytes were tracked over the entire time period. From each cell tracked, an average velocity was calculated.
Proliferation (Thymidine incorporation)
Keratinocytes were plated in 24-well tissue culture trays at 5 × 104 cells per well. Cells were treated with varying concentrations of MIP-2 or PBS for 16 h while pulsed for the last 4 h with 1 μCi of [3H]-thymidine (Amersham Pharmacia Biotech, Cleveland, OH). Cells were fixed using methanol and acetic acid (in a 3:1 ratio), solubilized in 5 N NaOH and incorporated counts measured using a scintillation counter.
Transfection and luciferase reporter
The NF-kB-luciferase plasmid DNA was a gift from Dr Zheng-Gang Liu (National Cancer Institute, NIH). Primary keratinocytes were transfected using Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). Three days after ras transduction, transient transfection was carried out for an additional 48h. Cells were then rinsed twice in PBS and harvested in reporter lysis buffer (BD Biosciences Clontech). Luciferase activity was measured by using the luciferase reporter assay kit (BD Biosciences Clontech) according to the manufacturer’s protocol. Results were normalized to the total protein content.
RT-PCR analysis
RNA was isolated from cultured cells with Trizol following manufacturer’s protocol (Invitrogen). cDNA synthesis and real-time PCR analysis was previously described (11). The primer sequences were as follows:
| Gene | Forward Primer | Reverse Primer |
| GAPDH | CATGGCCTTCCGTGTTCCTA | GCGGCACGTCAGATCCA |
| GCP-2/CXCL6 | TGGATCCAGAAGCTCCTGTGA | TGCATTCCGCTTAGCTTTCTTT |
Predesigned Quantitect primers (Qiagen) were used for KC (CXCL1) and MIP-2 (CXCL2/3).
ELISA
KC and MIP-2 levels were quantified from culture supernatants using Quantikine ELISA kit according to the manufacturer’s protocol (R&D Systems).
Statistical analysis
Data were analyzed by prism software and significance values assigned through Student’s t-test or One-Way ANOVA with Tukey post-test. P< 0.05 was considered to be significant.
RESULTS
PKCα overexpression in basal keratinocytes and hair follicles sensitizes mice to skin carcinogenesis
Application of TPA to the skin of K5-PKCα mice causes a neutrophilic infiltrate into the epidermis (9;11). K5-PKCα mice and their wild-type littermates (WT) were compared for susceptibility to 100μg DMBA initiation followed by 1μg (1.6 nmoles) TPA twice weekly for 20 weeks of promotion. By weeks 21, 58% of the K5-PKCα mice developed tumors (with an average tumor multiplicity of 4.6 tumors per tumor-bearing animal) while all WT mice were tumor-free (Table, panel A and Figure 1A). None of the mice in control groups (initiation or promotion only) developed tumors. Sixty eight percent of papillomas converted to carcinomas by week 50 (Table 1, panel A) reflecting the genetic predisposition for malignant skin tumor development on an FVB/N background (19). Previous studies have indicated that mice overexpressing PKCα in the epidermis on a C57/Bl6 background (10) or FVB/N background (20) are not more susceptible to tumor induction than their wild-type counterpart. While those results appear contradictory with the one we are currently reporting, they can be explained by the TPA regimen used for promotion. We have use a much lower dose (1.6 nmoles TPA twice a week) while the study on FVB/N mice used 5 nmoles TPA twice a week (20)and 3 nmoles TPA three times a week for the other study (10), conditions that were favorable for tumor development in wildtype mice. Our results demonstrate that PKCα overexpression in basal keratinocytes and potentially the consequent acute inflammation sensitize mice to skin carcinogenesis using a limiting DMBA initiation-TPA promotion protocol.
Figure 1.

Two stage skin carcinogenesis in K5-PKCα mice and their wildtype littermates (A) and persistence of epidermal hyperplasia in neutrophil depleted K5-PKCα mice after a single topical treatment with TPA (B–D). A, Mice were initiated by treating with 100μg (390 nmoles) DMBA/0.2ml acetone at zero time and promoted by treating with 1μg (1.6 nmoles)/0.2 ml acetone twice a week from weeks 1–20. Tumors were counted every week. The percent with papilloma is plotted versus time for each group. Tumor multiplicity of 4.6 tumors/tumor bearing mice is not shown. B–D,18 hours prior to TPA application, K5-PKCα mice were injected IP with 100μg anti Ly-6G antibody (C) or isotype control (B) Arrows point to neutrophilic microabscesses present in IgG control group only, right panels represent higher magnification of the epidermis. Skin biopsies were collected three days after TPA treatment and H&E stained. D, Basal BrdU-labeled as well as unlabeled basal nuclei were counted in randomly selected regions. Between 400 and 700 cells were counted for each skin section. Data are reported as mean ± SE. Each group contains five mice and data are representative of two independent experiments.
Table 1.
Tumor formation in two-stage skin carcinogenesis (A) and tumorigenicity of CXCR2 deficient keratinocytes expressing oncogenic rasHa in nude mouse skin grafts (B)
| A | |||||
|---|---|---|---|---|---|
| Initiation | Promotion | Genotype | No. of mice | % with papillomaa | % with carcinomab |
| DMBA | TPA | WT | 31 | 0 | 0 |
| DMBA | TPA | K5-PKCα | 43 | 58 | 68 |
| DMBA | - | WT | 10 | 0 | 0 |
| DMBA | - | K5-PKCα | 11 | 0 | 0 |
| - | TPA | WT | 10 | 0 | 0 |
| - | TPA | K5-PKCα | 11 | 0 | 0 |
% with papilloma calculated at 25 weeks.
% with carcinoma: carcinoma/spindle cell carcinoma pathological evaluation was performed on H&E stains at the sacrifice time or at the termination of the experiment (week 50). 25 mice with tumors at week 25, among them 17 had at least 1 carcinoma by week 50.
PKCα activation causes epidermal hyperplasia independently of acute neutrophil infiltration in the skin
Previous studies have shown that the intraepidermal neutrophilic infiltration after a single TPA treatment of K5-PKCα mice is followed by a prominent regenerative epidermal hyperplasia (9). To test the contribution of neutrophils in the epidermal hyperplasia, K5-PKCα mice were depleted of their circulating neutrophils using Ly-6G antibodies. Ly-6G treatment of K5-PKCα mice prevented the systemic neutrophilia and the subsequent neutrophil infiltration and microabscess formation in the skin after TPA (Supplemental figure 1A and B). After a single TPA application, epidermal hyperplasia and BrdU labeling in neutrophil depleted K5-PKCα mice and K5-PKCα mice receiving isotype control antibody were identical (Figure 1, B, C and D). Collectively these results demonstrate that PKCα activation causes epidermal hyperplasia independently of acute infiltration by neutrophils.
Oncogenic ras upregulates CXCR2 ligands in keratinocytes through activated PKCα and EGFR
The induction of the CXCR2 ligand CXCL8 by H-rasV12 in Hela cells has been reported previously (21). In the DMBA/TPA skin carcinogenesis model virtually all tumors are initiated through mutational activation of H-ras raising the possibility that initiated cells could contribute to the chemokine milieu in the skin tumor microenvironment and thus to tumor formation. As seen in Figure 2A, introduction of oncogenic ras into keratinocytes upregulates mRNA expression of all three CXCR2 ligands (KC, MIP-2 and GCP-2). Keratinocytes also express the receptor CXCR2 itself, although oncogenic ras does not enhance the expression of the receptor CXCR2 mRNA (Figure 2A). Upregulation and secretion of these ligands by TPA has been shown to be mediated through PKCα and NF-κB (11;22). Oncogenic ras also increased the secretion of MIP-2 (Figure 2B) and KC (not shown) by keratinocytes. Blocking NF-κB transcriptional activity by introducing a dominant negative IκB adenovirus reduced the v-ras mediated secretion of MIP-2 (Figure 2B). PKCα overexpression also enhanced oncogenic ras-mediated MIP-2 secretion, and this was also regulated by NF-κB. Blocking NF-κB did not affect CXCR2 mRNA expression (data not shown).
Figure 2.

Oncogenic H-ras upregulates CXCR2 ligands in primary keratinocytes through EGFR, NF-κB and PKCα. A, real-time PCR analysis of KC/CXCL1, MIP-2/CXCL2/3, GCP-2/CXCL6 and CXCR2 mRNA expression in control and v-rasHa-transduced keratinocytes at indicated days after transduction. Bars represent mean value of triplicate determinations. B, MIP-2 secreted into culture supernatant collected from control or v-ras transduced K5-PKCα or WT keratinocytes infected with A-CMV (control) or degradation-resistant IκBα (IκBαSR) adenovirus to block NF-κB activity. Bars represent the mean ± SEM of triplicate determinations. Results are representative of three independent experiments. *P < 0.01 compared with respective non-ras transduced control; #, P < 0.05 compared with v-rasHa WT keratinocytes A-CMV transduced. C, Culture supernatants from EGFR WT or KO primary keratinocytes were collected after control or v-rasHa transduction. KC and MIP-2 concentrations were determined by ELISA. Bars represent the mean ± SEM of triplicate determinations.
Previous studies have shown that oncogenic ras establishes an autocrine loop through the EGFR in murine keratinocytes by upregulating EGFR ligands (23). To test whether EGFR autocrine signaling contributes to PKCα mediated induction of CXCR2 ligands in v-ras transformed keratinocytes, we used keratinocytes from mice genetically ablated for the EGFR (15). Disruption of autocrine EGFR signaling abolished the induction of MIP-2 and strongly reduced the induction of KC (Figure 2C). Collectively, these result suggest that ras-induced expression of CXCR2 ligands requires EGFR signaling and PKCα mediated activation of NF-κB.
CXCR2 deficient keratinocytes are defective in tumor formation
We have reported that the cutaneous inflammation in response to PKCα activation is ablated in the absence of CXCR2 (11). The functional significance of CXCR2 expression by keratinocyte during skin carcinogenesis was tested by transducing primary keratinocytes from CXCR2 null mice with the v-rasHa retrovirus and transplanting transduced tumorigenic cells to a prepared orthotopic graft site on nude mice that are otherwise CXCR2 intact. By this approach tumor formation is dependent solely on the grafted keratinocyte CXCR2 as the host is competent for CXCR2 expression. As shown in Figure 3A, tumor growth is retarded in the absence of keratinocyte CXCR2 relative to CXCR2 wildtype keratinocytes. Furthermore tumor latency was greater and fewer mice hosting grafts lacking CXCR2 formed tumors than intact controls (Table 1, panel B). The expression of rasHa was similar among the two keratinocyte genotypes as shown by western blot from an aliquot of grafted cells (Figure 3A, insert). Proliferation in tumors arising from CXCR2 deficient v-ras keratinocytes was reduced by 31% when assayed by BrdU staining (Figure 3B). The number of apoptotic cells detected by TUNEL staining was similar in tumors originating from either genotype (Figure 3C). ELR+ chemokines have been shown to have a direct pro-angiogenic activity on CXCR2 expressing endothelial cells. Because oncogenic ras induces CXCR2 ligand (MIP-2, KC and GCP-2) expression similarly in primary keratinocytes from both genotypes (Supplemental figure 2) and CD31 staining of tumors from both groups show a similar vascular pattern (Figure 3D) and microvasculature density was the same among the two groups (Figure 3D), therefore it is unlikely that the tumor growth difference was related to a defective paracrine angiogenic response by the host. Collectively these results suggest that an autocrine/paracrine activation of keratinocyte CXCR2 contributes to the tumorigenic properties of Ras transformed keratinocytes.
Figure 3.
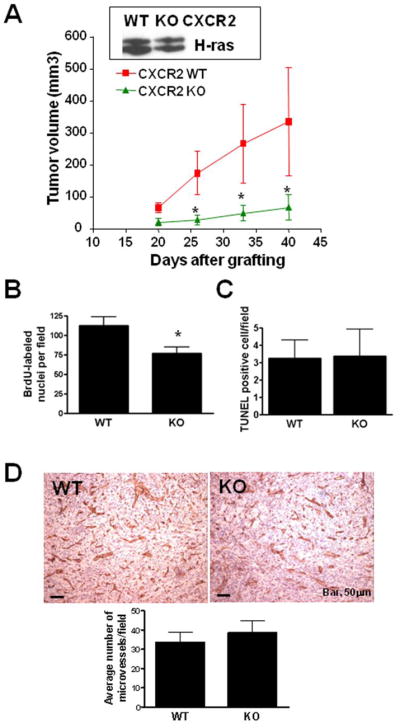
Targeted disruption of CXCR2 impairs growth of v-rasHa-induced squamous tumors. A, 2 × 106 CXCR2 WT or KO primary v-rasHa-transduced keratinocytes combined with CXCR2 WT fibroblasts (5 × 106) were grafted onto nude mice as a reconstituted skin and average tumor volume was calculated as described in methods. Data are reported as mean ± SE. *, P < 0.05 compared with CXCR2 WT for the same time-point. Each group contains 10–12 mice and results are representative of two independent experiments. Only mice bearing tumors were included in this figure. Insert shows H-ras expression in an aliquot of v-ras keratinocyte used for grafting. B, BrdU-labeled nuclei were counted in 5–7 randomly selected regions. Data are reported as mean ± SE. *, P < 0.05 compared with CXCR2 WT. Each group contains 7–9 tumors and graphed data are representative of two independent experiments. C, Apoptotic cells were identified using the Apoptag kit, which stains nuclei containing nicked DNA. Positive nuclei were counted in 5–7 randomly selected regions. Data are reported as mean ± SE. Each group contains 7–9 tumors and graphed data are representative of two independent experiments. D, immunostaining for CD31 antigen outlining blood vessels within tumors originating from CXCR2 WT (WT) or CXCR2 KO (KO) v-rasHa-transduced keratinocytes. Bottom graph, microvessels density based on CD31+ cells. The average number of positive cells in at least 5 fields per tumor originating from CXCR2 WT (WT) or CXCR2 KO (KO) v-rasHa-transduced keratinocytes is shown. Each group contains 7–9 tumors and graphed data are representative of two independent experiments. Data are reported as mean ± SE.
CXCR2 deficiency impairs the migratory properties of transformed keratinocytes
The small size of tumors from v-rasHa keratinocytes lacking CXCR2 strongly suggested a functional role for CXCR2 on keratinocytes and their transformed derivatives. To further explore this issue we used cultured genetically modified keratinocytes and recombinant CXCR2 ligands. Neither MIP-2 (Supplemental figure 3A) nor KC (data not shown) could stimulate 3H-thymidine incorporation in cultured ras transformed keratinocytes. Similarly, cell proliferation was identical in CXCR2 deficient v-rasHa transformed keratinocytes and their wildtype counterpart (Supplemental figure 3B) suggesting that the reduction of proliferation observed in the tumors in vivo might be a secondary event to CXCR2 deficiency. MTT assays revealed a similar TPA dose-dependent loss of cell viability in CXCR2 deficient v-rasHa transformed keratinocytes and their wildtype counterpart (Supp. Figure 3C). Since activation of CXCR2 on hematopoietic cells results in a migratory response, we considered that the receptor on keratinocytes might have a similar function. Using Transwell inserts we documented a chemotactic response of v-rasHa transformed wildtype keratinocytes toward a MIP-2 gradient and that response was absent in CXCR2 deficient v-ras keratinocytes (Figure 4A). Treatment of CXCR2 wildtype v-ras keratinocytes with a CXCR2 antagonist prevented their migratory response to MIP-2 (Figure 4A). In contrast keratinocytes of both genotypes migrate similarly in response to TGFα (Figure 4A). Basal cell motility (measured by tracking cell movement) was identical in CXCR2 wildtype (0.42 ± 0.07 μm/min.) and CXCR2 deficient v-ras keratinocytes (0.49 ± 0.07 μm/min.). These observations suggest that the lack of chemotaxis in CXCR2 deficient v-ras keratinocytes was not due to defective cell motility. These data support a primary autocrine role for CXCR2 on the migration of transformed keratinocytes that in turn may be required for their tumorigenic outgrowth.
Figure 4.
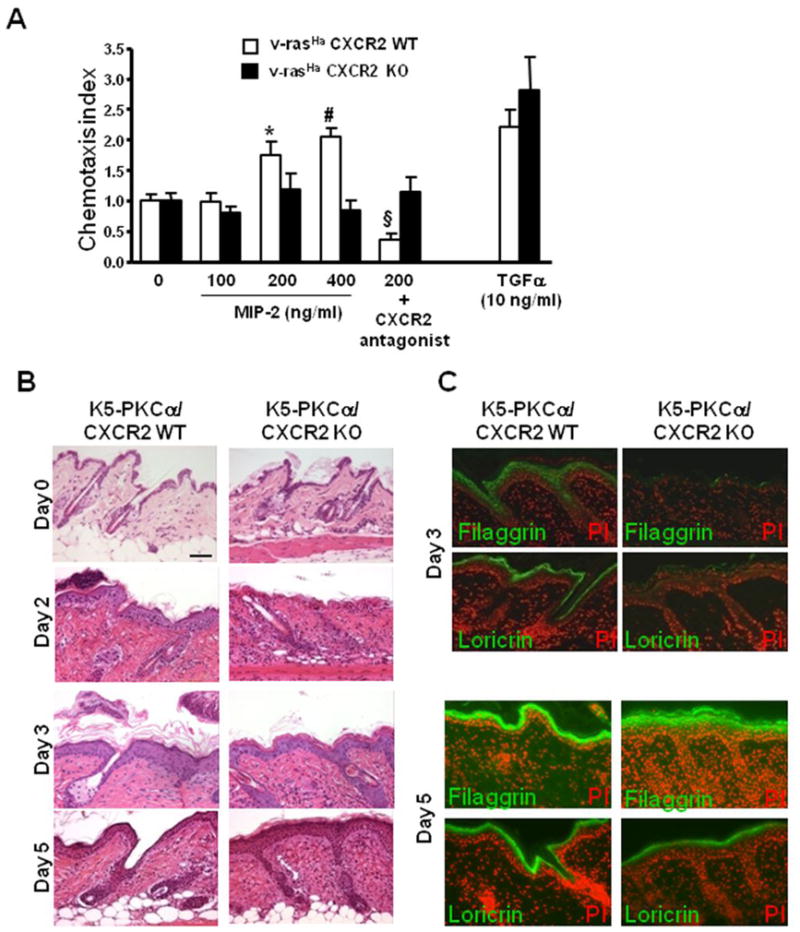
CXCR2 mediates migration of keratinocytes. A, Five days after v-rasHa transduction, migration of CXCR2 WT and KO keratinocytes was tested in Transwell assays as described in methods. MIP-2 or TGFα (10 ng/ml) diluted in low –serum containing media (0) was used as stimulus in the bottom chamber while cells were seeded in the top chamber in low –serum containing media. The CXCR2 antagonist was added to the cell suspension prior to the seeding in the top well. Fold increase (chemotactic index) of keratinocyte migration in response to treatment was compared to basal migration in the absence of stimulus. Bars represent the mean ± SEM of two triplicate determinations. *, P < 0.001; #, P < 0.0001 compared with untreated control, § P < 0.001 compared with untreated respective control. B, Delayed epidermal regeneration in K5-PKCα mice deficient for CXCR2 after a single topical application of TPA. Skin biopsies were collected at 2, 3 and 5 days after a single TPA application on K5-PKCα/CXCR2 WT and K5-PKCα/CXCR2 KO and H&E stained. Bar, 50μm. C, Immunohistochemistry for filaggrin and loricin was performed on similar biopsies as in B collected at day 3 and 5.
CXCR2 deficiency has previously been shown to delay skin wound healing after excisional punch biopsy owing to a migration defect of CXCR2 deficient keratinocytes (24). A single low dose TPA exposure on K5-PKCα skin causes sloughing of the epidermis followed by complete regeneration (9). We tested whether migration secondary to CXCR2 expression would contribute to the epidermal regenerative response. In crosses of K5-PKCα/CXCR2 KO mice the skin is normal (Figure 4B, Day 0). Two days after TPA treatment a nascent regenerative hyperplasia is observed in K5-PKCα/CXCR2 WT mice and a well organized stratification of the hyperplastic epidermis is evident at day 3. In contrast, regeneration and hyperplasia are delayed in K5-PKCα/CXCR2 KO mice, and the epidermis that forms is deficient in the late epidermal differentiation markers loricrin and filaggrin at day 3 (Figure 4C) in contrast to mice expressing CXCR2. By day 5 both differentiation markers are properly expressed in K5-PKCα/CXCR2 KO mice and hyperplasia is maximal. This delay in regenerative hyperplasia and expression of differentiation markers likely reflects the importance of CXCR2 in regulating keratinocyte migration both in vitro and in vivo.
To further demonstrate that CXCR2 is directly involved in keratinocytes migration, CXCR2 deficient v-ras keratinocytes were transduced with an adenovirus encoding for mouse CXCR2. As shown on Figure 5A, CXCR2 reconstitution restored the chemotactic response to MIP-2. The MAPK and PI3K pathways are two major signal transduction pathways activated by CXCR2 in response to its ligands (25;26). We tested the ligand-induced phosphorylation of ERK and Akt as indicators of MAPK and PI3K pathways activation in ras transformed keratinocytes. As shown in Figure 5B both MAPK and PI3K pathways are activated by MIP-2 when CXCR2 null ras-transformed keratinocyte are reconstituted with CXCR2 but not in the absence of reconstitution. TGFα activated similarly those pathways in both CXCR2 deficient and CXCR2 reconstituted ras-transformed keratinocytes. Whether those two pathways are functionally linked to the chemotactic response in ras-transformed keratinocytes require further pharmacological and genetic analysis. Thus two essential signaling pathways, linked to skin tumor formation are downstream from the interaction of keratinocyte CXCR2 and its ligands.
Figure 5.
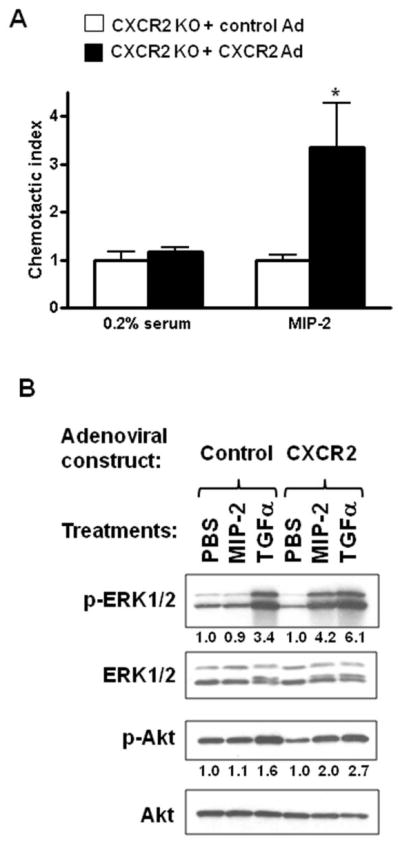
Reconstitution of CXCR2 KO keratinocytes with CXCR2 restores chemotaxis and signaling after MIP-2 treatment. On the fourth day after v-ras transduction, CXCR2 null primary keratinocytes were transduced with an empty adenovirus (Control Ad) or CXCR2 expressing adenovirus (CXCR2 Ad) for an additional 24 hours and assayed for their ability to migrate in response to MIP-2 using the Transwell assays (A) as described in figure 4 or (B) stimulated with 200ng/ml MIP-2 or 10ng/ml TGFα for 5 minutes. Western blots were performed for total and phospho-ERK 1/2 (p-ERK1/2) and total and phospho-Akt (p-Akt) as described under “materials and methods”. Relative levels of phospho-Akt and phospho-ERK are indicated in each figure normalized to the PBS treated cells. *, P < 0.05 compared with untreated control.
DISCUSSION
Cutaneous SCC is the second most common cancer in the United States after BCC. Activating mutations in ras genes are observed in human SCC (27) and in experimental cutaneous SCC induction in mice (28), and in the absence of mutations, human SCCs frequently show increased Ras activity and MAPK activation (29). While many downstream effectors of active ras have been delineated, those most critical for cutaneous SCC development remain obscure. Our current study demonstrates that CXCR2 ligands are transcriptional targets of oncogenic ras in primary keratinocytes. We further show that CXCR2 ligand induction by oncogenic ras requires intact NF-κB nuclear translocation, is potentiated by PKCα and is mediated through the EGFR. These findings connect three essential pathways in experimental skin carcinogenesis.
The skin carcinogenesis model has been particularly useful for illuminating the contribution of immune/inflammatory components for tumor development (4;5;7;30). These studies have revealed often opposing influences on tumor growth, immune surveillance and angiogenesis of both humoral and cellular factors that are present in the tumor microenvironment. They have also emphasized crosstalk between tumor cells and the stroma to create the proper microenvironment. Less focus has been directed at potential tumor-tumor autocrine consequences of factors in the microenvironment. The current studies suggest how potential autocrine interactions can influence tumor development. The development of K5-PKCα mice that exhibit cutaneous inflammation in response to TPA provided an inducible system for the recruitment of neutrophils to the skin that specifically depends on keratinocyte-derived CXCR2 ligands (9;11). TPA also causes epidermal desquamation on K5-PKCα mice that is independent of CXCR2 ligands (11). We now show that K5-PKCα mice exhibit increased sensitivity to tumor formation in the two-stage skin carcinogenesis model. Since recovery from epidermal desquamation occurs through migration and proliferation of hair follicles keratinocytes, we conclude that the initiated cells in this model must reside in the hair follicle compartment (31;32). Our results also link CXCR2 on keratinocytes to tumor formation in vivo. Specifically, when CXCR2 deficient v-ras transformed primary keratinocytes are grafted orthotopically to CXCR2 competent hosts, tumor growth is severely retarded.
CXCR2 is strongly expressed by leukocytes but it is also present on endothelial cells and embryonic and adult keratinocytes (24;33). Our study also shows that secreted CXCR2 ligands from ras transformed keratinocytes could not only recruit immune cells into the tumor microenvironment but could also promote angiogenesis and act directly on neoplastic keratinocytes in an autocrine/paracrine manner. It is also likely that transformed keratinocytes could respond to CXCR2 ligands produced by neutrophils and endothelial cells present in the stroma (34). CXCR2 ligands are pro-angiogenic in xenografts (21), grafted murine squamous cell carcinoma cells (35) and models of lung and pancreatic cancers (36). However, retarded tumor growth in our grafts of CXCR2 null transformed keratinocytes cannot be explained by defective angiogenesis. The CXCR2 null tumor cells express abundant CXCR2 ligands (resulting from intact NF-κB activity in those cells, see Supplemental figure 3E), the host endothelial cells were CXCR2 competent and blood vessel density was similar in tumors from CXCR2 null and intact keratinocytes.
What then are the consequences of ligand activation of keratinocyte CXCR2? BrdU labeling shows a modest reduction in the number of proliferative cells in tumors derived from CXCR2 null keratinocytes, but, in vitro, CXCR2 ligands did not stimulate keratinocyte proliferation nor did oncogenic ras have a differential effect on proliferation of the two genotypes. Our studies indicate that CXCR2 activation mediates the migration of transformed keratinocytes in vitro, and migration is delayed in CXCR2 null epidermis after TPA imposed epidermal desquamation. This result is consistent with the slow migration seen in CXCR2 null epidermis during cutaneous wound repair (24) and could perhaps impede cell turnover in tumors. Keratinocyte migration in response to autocrine or paracrine CXCR2 activation could provide the initiated cells with an advantage particularly during the early steps of tumor development when forming a critical clone size is necessary to expand in the context of a tightly regulated epithelial compartment (37). This assumption is consistent with the finding that CXCR2 deficiency did not alter the malignant conversion rate of grafted tumor cells which occurs as a late event in tumor progression. Previously, CXCR2 ligands have been shown to enhance proliferation of human cancer cell lines (38;39) but in the transformation of lining epithelial cells, migration may be particularly important to expand the zone of early transformants(37). Crosstalk between CXCR1/2 with EGFR has also been reported in human ovarian cancer cells (40), and we considered that the smaller tumor size could be related to changes in EGFR responses. However, the migratory response (Figure 4) and proliferative response (data not shown) to exogenous TGFα in both CXCR2 wildtype and KO keratinocytes were similar. MAPK activity was increased in response to TGFα in CXCR2 deficient and CXCR2 reconstituted transformed keratinocytes (Figure 5) although MAPK was not activated in CXCR2 deficient cells by CXCR2 ligands. Our reconstitution studies show that activation of both MAPK and Akt are downstream of CXCR2 activation in transformed keratinocytes. These two pathways have been linked to CXCR2 mediated cell migration in several cell lines (25;26), but recent studies suggest the association is more complex and involves additional pathways (41). Further studies will be required to understand how signaling through CXCR2 in transformed keratinocytes regulates migration and how this migratory pathway contributes to tumor formation.
In human keratinocytes, EGFR activation causes a strong upregulation of IL-8 (42;43). Similarly, oncogenic ras induces transcripts of the EGF ligand family (namely TGFα, betacellulin, amphiregulin and HB-EGF) and therefore contributes to the establishment of an autocrine loop (23;44;45). We have shown previously that papillomas expressing v-ras but deficient for EGFR are growth impaired but the proliferative pool of cells is not reduced. Instead, the tumor cells show altered migration and premature differentiation (46). Our current study reveals that autocrine stimulation of EGFR is necessary for oncogenic ras mediated induction of CXCR2 ligands. This result suggests that upregulation of CXCR2 ligands cannot be directly compensated by the constitutively active viral ras protein, and autocrine-activated EGFR is an essential mediator of this pathway in keratinocytes.
The K5-PKCα mouse model has proven useful for addressing other consequences of inflammation in skin tumor development. For example it has been proposed that the inflammatory infiltrate contributes to the sustained hyperplasia associated with tumor promotion. However neutrophil depletion in K5-PKCα mice using an anti-Gr-1 antibody had no overt consequence on skin hyperplasia following TPA application. Because an anti-Gr1 based approach allows only for a temporary depletion of neutrophil as it is followed by a rebound in granulocytes counts (47), it therefore does not allow for the study of the contribution of neutrophils during a complete promotion stage. Interestingly, the pioneer work of the Schreiber’ group showed that elimination of Gr-1+ leukocytes in athymic nude mice slowed the growth of a variant of a UV light-induced tumor (48). Activation of CXCR2 on Gr-1+CD11b+ myeloid cells (also called myeloid-derived suppressor cells) was recently shown to mediate their recruitment to mammary carcinoma (49). Gr1+CD11b+ cells can contribute to tumor immune evasion (for review (50)), angiogenesis but also tumor invasion and metastasis (49). Therefore blocking CXCR2 appears as an attractive target to prevent the recruitment of Gr-1+ leukocytes to the tumor site. That therapeutic approach would also benefit from the blockade of keratinocyte CXCR2 as our results suggest that it contributes important autocrine/paracrine function during skin carcinogenesis.
Supplementary Material
Supplemental figure 1: Ly-6G treatment of K5-PKCα mice prevented systemic neutrophilia and neutrophil infiltration and microabscess formation in K5-PKCα mice after topical TPA treatment. A single dose of TPA (1 μg) in acetone was applied to the shaved backs of K5-PKCα transgenic mice previously inoculated with control IgG or Ly-6G antibodies by IP injection. Blood was drawn at 12h, and differential white blood cell counts were performed. Bars represent the mean number of neutrophils per microliter of blood ± SEM from 5–7 mice per group. The baseline values for neutrophil count is represented by the dotted line (415±85 neutrophil/μl of blood) in non TPA treated K5-PKCα mice. B, Skin sections from similarly treated mice were immunostained using myeloperoxidase (MPO) antibody that recognizes granulocytes. Inflammatory cells infiltrating the hair follicles and epidermis and forming intraepidermal microabscesses are seen in K5-PKCα mice treated with control IgG but not in Ly-6G treated animals.
Supplemental figure 2: CXCR2 ligands are induced by oncogenic ras (v-rasHa) in primary keratinocytes independent of CXCR2 genotype. A, culture supernatants from mock or v-rasHa-transduced CXCR2 WT or KO primary keratinocytes were collected over 24 hours for cells in culture for 5 days. MIP-2 concentrations were determined by ELISA. Bars represent the mean ± SEM of triplicate determinations. B, C and D, real-time PCR analysis of MIP-2 (panel B), KC (panel C) or GCP-2 (panel D) mRNA expression in CXCR2 WT or CXCR2 KO keratinocytes 3 days after mock or v-rasHa-transduction. Bars represent mean value of triplicate determinations ± SEM. E) Three days after control or v-rasHa transduction, CXCR2 WT and KO primary keratinocytes were transiently transfected with the NF-kB reporter activity plasmid construct, and 48h later cells were harvested for reporter activity. Values are expressed as relative light units (RLU) per 100ug total protein as determined by Bradford. Bars represent the mean ± SEM of six culture wells. Results are representative of two independent experiments.
Supplemental figure 3: CXCR2 status does not affect the growth of v-rasHa-transduced keratinocytes. A, three days after ras transduction primary keratinocytes were starved overnight in 0.2% serum containing media, then treated with indicated concentration of MIP-2 or complete media (serum) for 24 hours. Cells were pulsed with 3H-thymidine for the last 4 hours. Bars represent the mean ± SEM of quadruplicate determinations. Results are representative of two independent experiments. B, growth of CXCR2 WT and KO keratinocytes following transduction by the v-rasHa oncogene as described in methods. Primary keratinocytes were grown in 24-well tissue culture plates and cell counts were performed on the indicated days. Points represent value based on counts from four wells, ± SEM. C, primary keratinocytes from CXCR2 WT and KO mice were cultured in 0.05 mM Ca2+ medium and treated overnight with TPA on the fourth day after ras transduction. Cell viability was assayed by MTT assay. Bars represent the mean ± SEM of quadruplicate determinations. Results are representative of two independent experiments.
Table 2.
| B | |||||
|---|---|---|---|---|---|
| Experiment | CXCR2 status | Latencya (days) | Tumor incidenceb | Histology | Graft age at sacrifice (days) |
| 1 | WT | 14 | 9/12 | Papilloma (1/9) Carcinoma (8/9) |
40 |
| KO | 21 | 7/12 | Papilloma (1/7) Carcinoma (6/7) |
40 | |
| 2 | WT | 7 | 10/10 | Papilloma (1/10) Carcinoma (9/10) |
48 |
| KO | 14 | 5/10 | Carcinoma (5/5) | 48 | |
time interval after grafting when tumor(s) first reported.
maximum number of tumor-bearing animals/total animals.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The authors thank Dr. Victor E. Marquez and Dr. Krishnan Malolanarasimhan (Laboratory of Medicinal Chemistry, NCI) for synthetizing the CXCR2 small-molecule antagonist, Dr. Ji-Liang Gao for providing the mouse CXCR2 construct, Dr. Mary-Ann Stepp for time-lapse-microscopy studies, Marta Custer and Susana Walters for care of the mouse colonies and the Pathology/Histotechnology Laboratory (SAIC Frederick) for excellent technical assistance.
References
- 1.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 2.Robinson SC, Coussens LM. Soluble mediators of inflammation during tumor development. Adv Cancer Res. 2005;93:159–87. doi: 10.1016/S0065-230X(05)93005-4. [DOI] [PubMed] [Google Scholar]
- 3.Mueller MM. Inflammation in epithelial skin tumours: old stories and new ideas. Eur J Cancer. 2006;42:735–44. doi: 10.1016/j.ejca.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Moore RJ, Owens DM, Stamp G, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 5.Arnott CH, Scott KA, Moore RJ, Robinson SC, Thompson RG, Balkwill FR. Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene. 2004;23:1902–10. doi: 10.1038/sj.onc.1207317. [DOI] [PubMed] [Google Scholar]
- 6.Girardi M, Glusac E, Filler RB, et al. The distinct contributions of murine T cell receptor (TCR)gammadelta+ and TCRalphabeta+ T cells to different stages of chemically induced skin cancer. J Exp Med. 2003;198:747–55. doi: 10.1084/jem.20021282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–23. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Obermueller E, Vosseler S, Fusenig NE, Mueller MM. Cooperative autocrine and paracrine functions of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the progression of skin carcinoma cells. Cancer Res. 2004;64:7801–12. doi: 10.1158/0008-5472.CAN-03-3301. [DOI] [PubMed] [Google Scholar]
- 9.Cataisson C, Joseloff E, Murillas R, et al. Activation of cutaneous protein kinase C alpha induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J Immunol. 2003;171:2703–13. doi: 10.4049/jimmunol.171.5.2703. [DOI] [PubMed] [Google Scholar]
- 10.Wang HQ, Smart RC. Overexpression of protein kinase C-alpha in the epidermis of transgenic mice results in striking alterations in phorbol ester-induced inflammation and COX-2, MIP-2 and TNF-α expression but not tumor promotion. J Cell Sci. 1999;112:3497–506. doi: 10.1242/jcs.112.20.3497. [DOI] [PubMed] [Google Scholar]
- 11.Cataisson C, Pearson AJ, Tsien MZ, et al. CXCR2 ligands and G-CSF mediate PKCalpha-induced intraepidermal inflammation. J Clin Invest. 2006;116:2757–66. doi: 10.1172/JCI27514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schon M, Denzer D, Kubitza RC, Ruzicka T, Schon MP. Critical role of neutrophils for the generation of psoriasiform skin lesions in flaky skin mice. J Invest Dermatol. 2000;114:976–83. doi: 10.1046/j.1523-1747.2000.00953.x. [DOI] [PubMed] [Google Scholar]
- 13.Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109:1207–17. doi: 10.1083/jcb.109.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Threadgill DW, Dlugosz AA, Hansen LA, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–4. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 16.Roop DR, Lowy DR, Tambourin PE, et al. An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature. 1986;323:822–4. doi: 10.1038/323822a0. [DOI] [PubMed] [Google Scholar]
- 17.White JR, Lee JM, Young PR, et al. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J Biol Chem. 1998;273:10095–8. doi: 10.1074/jbc.273.17.10095. [DOI] [PubMed] [Google Scholar]
- 18.Stepp MA, Liu Y, Pal-Ghosh S, et al. Reduced migration, altered matrix and enhanced TGFbeta1 signaling are signatures of mouse keratinocytes lacking Sdc1. J Cell Sci. 2007;120:2851–63. doi: 10.1242/jcs.03480. [DOI] [PubMed] [Google Scholar]
- 19.Hennings H, Glick AB, Lowry DT, Krsmanovic LS, Sly LM, Yuspa SH. FVB/N mice: an inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis. 1993;14:2353–8. doi: 10.1093/carcin/14.11.2353. [DOI] [PubMed] [Google Scholar]
- 20.Jansen AP, Dreckschmidt NE, Verwiebe EG, Wheeler DL, Oberley TD, Verma AK. Relation of the induction of epidermal ornithine decarboxylase and hyperplasia to the different skin tumor-promotion susceptibilities of protein kinase C alpha, -delta and -epsilon transgenic mice. Int J Cancer. 2001;93:635–43. doi: 10.1002/ijc.1395. [DOI] [PubMed] [Google Scholar]
- 21.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–58. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 22.Cataisson C, Pearson AJ, Torgerson S, Nedospasov SA, Yuspa SH. Protein kinase C alpha -mediated chemotaxis of neutrophils requires NF-kappa B activity but is independent of TNF alpha signaling in mouse skin In vivo. J Immunol. 2005;174:1686–92. doi: 10.4049/jimmunol.174.3.1686. [DOI] [PubMed] [Google Scholar]
- 23.Dlugosz AA, Cheng C, Williams EK, et al. Autocrine transforming growth factor α is dispensible for v-rasHa-induced epidermal neoplasia: potential involvement of alternate epidermal growth factor receptor ligands. Cancer Res. 1995;55:1883–93. [PubMed] [Google Scholar]
- 24.Devalaraja RM, Nanney LB, Du J, et al. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–44. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sai J, Walker G, Wikswo J, Richmond A. The IL sequence in the LLKIL motif in CXCR2 is required for full ligand-induced activation of Erk, Akt, and chemotaxis in HL60 cells. J Biol Chem. 2006;281:35931–41. doi: 10.1074/jbc.M605883200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sai J, Fan GH, Wang D, Richmond A. The C-terminal domain LLKIL motif of CXCR2 is required for ligand-mediated polarization of early signals during chemotaxis. J Cell Sci. 2004;117:5489–96. doi: 10.1242/jcs.01398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog. 1991;4:196–202. doi: 10.1002/mc.2940040306. [DOI] [PubMed] [Google Scholar]
- 28.Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17:1–7. doi: 10.1016/s0923-1811(97)00071-6. [DOI] [PubMed] [Google Scholar]
- 29.Dajee M, Lazarov M, Zhang JY, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 30.Coussens LM, Raymond WW, Bergers G, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–97. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Binder RL, Gallagher PM, Johnson GR, et al. Evidence that initiated keratinocytes clonally expand into multiple existing hair follicles during papilloma histogenesis in SENCAR mouse skin. Mol Carcinog. 1997;20:151–8. doi: 10.1002/(sici)1098-2744(199709)20:1<151::aid-mc17>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 32.Morris RJ, Tryson KA, Wu KQ. Evidence that the epidermal targets of carcinogen action are found in the interfollicular epidermis of infundibulum as well as in the hair follicles. Cancer Res. 2000;60:226–9. [PubMed] [Google Scholar]
- 33.Luan J, Furuta Y, Du J, Richmond A. Developmental expression of two CXC chemokines, MIP-2 and KC, and their receptors. Cytokine. 2001;14:253–63. doi: 10.1006/cyto.2001.0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warner KA, Miyazawa M, Cordeiro MM, et al. Endothelial cells enhance tumor cell invasion through a crosstalk mediated by CXC chemokine signaling. Neoplasia. 2008;10:131–9. doi: 10.1593/neo.07815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loukinova E, Dong G, Enamorado-Ayalya I, et al. Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene. 2000;19:3477–86. doi: 10.1038/sj.onc.1203687. [DOI] [PubMed] [Google Scholar]
- 36.Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2004;172:2853–60. doi: 10.4049/jimmunol.172.5.2853. [DOI] [PubMed] [Google Scholar]
- 37.Glick AB, Yuspa SH. Tissue homeostasis and the control of the neoplastic phenotype in epithelial cancers. Semin Cancer Biol. 2005;15:75–83. doi: 10.1016/j.semcancer.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Metzner B, Hofmann C, Heinemann C, et al. Overexpression of CXC-chemokines and CXC-chemokine receptor type II constitute an autocrine growth mechanism in the epidermoid carcinoma cells KB and A431. Oncol Rep. 1999;6:1405–10. doi: 10.3892/or.6.6.1405. [DOI] [PubMed] [Google Scholar]
- 39.Miyazaki H, Patel V, Wang H, Edmunds RK, Gutkind JS, Yeudall WA. Down-regulation of CXCL5 inhibits squamous carcinogenesis. Cancer Res. 2006;66:4279–84. doi: 10.1158/0008-5472.CAN-05-4398. [DOI] [PubMed] [Google Scholar]
- 40.Venkatakrishnan G, Salgia R, Groopman JE. Chemokine receptors CXCR-1/2 activate mitogen-activated protein kinase via the epidermal growth factor receptor in ovarian cancer cells. J Biol Chem. 2000;275:6868–75. doi: 10.1074/jbc.275.10.6868. [DOI] [PubMed] [Google Scholar]
- 41.Sai J, Raman D, Liu Y, Wikswo J, Richmond A. Parallel Phosphatidylinositol 3-Kinase (PI3K)-dependent and Src-dependent Pathways Lead to CXCL8-mediated Rac2 Activation and Chemotaxis. J Biol Chem. 2008;283:26538–47. doi: 10.1074/jbc.M805611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303–12. doi: 10.1016/S0002-9440(10)63654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pastore S, Mascia F, Mariotti F, Dattilo C, Mariani V, Girolomoni G. ERK1/2 regulates epidermal chemokine expression and skin inflammation. J Immunol. 2005;174:5047–56. doi: 10.4049/jimmunol.174.8.5047. [DOI] [PubMed] [Google Scholar]
- 44.Glick AB, Sporn MB, Yuspa SH. Altered regulation of TGF-β 1 and TGF-α in primary keratinocytes and papillomas expressing v-Ha-ras. Mol Carcinog. 1991;4:210–9. doi: 10.1002/mc.2940040308. [DOI] [PubMed] [Google Scholar]
- 45.Dlugosz AA, Hansen L, Cheng C, et al. Targeted disruption of the epidermal growth factor receptor impairs growth of squamous papillomas expressing the v-rasHa oncogene but does not block in vitro keratinocyte responses to oncogenic ras. Cancer Res. 1997;57:3180–8. [PubMed] [Google Scholar]
- 46.Hansen LA, Woodson RL, II, Holbus S, Strain K, Lo Y-C, Yuspa SH. The epidermal growth factor receptor is required to maintain the proliferative population in the basal compartment of epidermal tumors. Cancer Res. 2000;60:3328–32. [PubMed] [Google Scholar]
- 47.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–98. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181:435–40. doi: 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–45. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1: Ly-6G treatment of K5-PKCα mice prevented systemic neutrophilia and neutrophil infiltration and microabscess formation in K5-PKCα mice after topical TPA treatment. A single dose of TPA (1 μg) in acetone was applied to the shaved backs of K5-PKCα transgenic mice previously inoculated with control IgG or Ly-6G antibodies by IP injection. Blood was drawn at 12h, and differential white blood cell counts were performed. Bars represent the mean number of neutrophils per microliter of blood ± SEM from 5–7 mice per group. The baseline values for neutrophil count is represented by the dotted line (415±85 neutrophil/μl of blood) in non TPA treated K5-PKCα mice. B, Skin sections from similarly treated mice were immunostained using myeloperoxidase (MPO) antibody that recognizes granulocytes. Inflammatory cells infiltrating the hair follicles and epidermis and forming intraepidermal microabscesses are seen in K5-PKCα mice treated with control IgG but not in Ly-6G treated animals.
Supplemental figure 2: CXCR2 ligands are induced by oncogenic ras (v-rasHa) in primary keratinocytes independent of CXCR2 genotype. A, culture supernatants from mock or v-rasHa-transduced CXCR2 WT or KO primary keratinocytes were collected over 24 hours for cells in culture for 5 days. MIP-2 concentrations were determined by ELISA. Bars represent the mean ± SEM of triplicate determinations. B, C and D, real-time PCR analysis of MIP-2 (panel B), KC (panel C) or GCP-2 (panel D) mRNA expression in CXCR2 WT or CXCR2 KO keratinocytes 3 days after mock or v-rasHa-transduction. Bars represent mean value of triplicate determinations ± SEM. E) Three days after control or v-rasHa transduction, CXCR2 WT and KO primary keratinocytes were transiently transfected with the NF-kB reporter activity plasmid construct, and 48h later cells were harvested for reporter activity. Values are expressed as relative light units (RLU) per 100ug total protein as determined by Bradford. Bars represent the mean ± SEM of six culture wells. Results are representative of two independent experiments.
Supplemental figure 3: CXCR2 status does not affect the growth of v-rasHa-transduced keratinocytes. A, three days after ras transduction primary keratinocytes were starved overnight in 0.2% serum containing media, then treated with indicated concentration of MIP-2 or complete media (serum) for 24 hours. Cells were pulsed with 3H-thymidine for the last 4 hours. Bars represent the mean ± SEM of quadruplicate determinations. Results are representative of two independent experiments. B, growth of CXCR2 WT and KO keratinocytes following transduction by the v-rasHa oncogene as described in methods. Primary keratinocytes were grown in 24-well tissue culture plates and cell counts were performed on the indicated days. Points represent value based on counts from four wells, ± SEM. C, primary keratinocytes from CXCR2 WT and KO mice were cultured in 0.05 mM Ca2+ medium and treated overnight with TPA on the fourth day after ras transduction. Cell viability was assayed by MTT assay. Bars represent the mean ± SEM of quadruplicate determinations. Results are representative of two independent experiments.