

Abstract
Hyperkalemia increases the organization of ventricular fibrillation (VF) and may also terminate it by mechanisms that remain unclear. We previously showed that the left-to-right heterogeneity of excitation and wave fragmentation present in fibrillating guinea pig hearts is mediated by chamber-specific outward conductance differences in the inward rectifier potassium current (IK1). We hypothesized that hyperkalemia-mediated depolarization of the reversal potential of IK1 (EK1) would reduce excitability and thereby reduce VF excitation frequencies and left-to-right heterogeneity. We induced VF in Langendroff-perfused guinea pig hearts and increased the extracellular K+ concentration ([K+]o) from control (4 mM) to 7 mM (n = 5) or 10 mM (n = 7). Optical mapping enabled spatial characterization of excitation dominant frequencies (DFs) and wavebreaks, and identification of sustained rotors (>4 cycles). During VF, hyperkalemia reduced the maximum DF of the left ventricle (LV) from 31.5 ± 4.7 Hz (control) to 23.0 ± 4.7 Hz (7.0 mM) or 19.5 ± 3.6 Hz (10.0 mM; p < 0.006), the left-to-right DF gradient from 14.7 ± 3.6 Hz (control) to 4.4 ± 1.3 Hz (7 mM) and 3.2 ± 1.4 Hz (10 mM), the number of DF domains, and the incidence of wavebreak in the LV and interventricular regions. During 10 mM [K+]o, the rotation period and core area of sustained rotors in the LV increased, and VF often terminated. Two-dimensional computer simulations mimicking experimental VF predicted that clamping EK1 to normokalemic values during simulated hyperkalemia prevented all of the hyperkalemia-induced VF changes. During hyperkalemia, despite the shortening of the action potential duration, depolarization of EK1 increased refractoriness, leading to a slowing of VF, which effectively superseded the influence of IK1 conductance differences on VF organization. This reduced the left-to-right excitation gradients and heterogeneous wavebreak formation. Overall, these results provide, to our knowledge, the first direct mechanistic insight into the organization and/or termination of VF by hyperkalemia.
Abbreviations used: [K+]o, extracellular K+ concentration; VF, ventricular fibrillation; VT, ventricular tachycardia; SR, sinus rhythm; LV, left ventricle; RV, right ventricle; TSP, time space plot; DF, dominant frequency; DFmax, maximum dominant frequency; 2D, two dimensional; SP, singularity point; MRP, mean rotation period; MCA, mean core area; GK1, maximum conductance of IK1 current; GKr, maximum conductance of IKr current; EK, reversal potential for K+ current; h, fast inactivation gate of sodium current; j, slow inactivation gate of sodium current; hj, product of h and j and a measure of excitability/refractoriness; APD, action potential duration; DI, diastolic interval
Introduction
Changes in extracellular K+ ([K+]o) are commonly observed in cardiac disease (1). Recent studies suggest that treatment with inhibitors of the renin-angiotensin-aldosterone system leads to [K+]o elevation (hyperkalemia) in patients with chronic heart failure (2). Hyperkalemia also occurs during tachycardia and/or ischemia and is a harbinger of cardiac arrhythmias, including ventricular fibrillation (VF) (3), a leading cause of sudden cardiac death (4). Paradoxically, hyperkalemia has also been suggested to dampen the proarrhythmic effect of sympathetic stimulation and thus be a protective factor during or after exercise (5). A recent publication (6) reported a case study of a patient, who underwent spontaneous defibrillation, which the authors suggested may have been related to increases in potassium levels that are expected to occur during clinical VF. These observations suggest that hyperkalemia also has important antifibrillatory effects. However, the exact mechanisms by which increases in [K+]o cause antiarrhythmic effects remain unclear, and are the main focus of this study.
In experimental models, the antifibrillatory effect of hyperkalemia on sustained VF has long been recognized (7–10). Koller et al. (8) proposed a mechanism linked to the inhibition of alternans mediated by hyperkalemia-induced reduction of the action potential duration (APD) restitution curve slope. Nevertheless, as the authors pointed out (8), their data did not explain how the VF frequency was altered by hyperkalemia, which may have been partially related to the altered dynamics of spiral waves.
We recently showed that VF in the guinea pig heart is sustained by high-frequency rotors of excitation located in the left ventricle (LV) (11,12). Fibrillatory conduction emanating from such sources underlies activation of the right ventricle (RV), with the formation of wavebreak and a sharp reduction in excitation frequency occurring predominantly at the dominant frequency (DF) domain boundaries, including that at the LV-RV junction (11). Therefore, we postulated that we could better understand the mechanism underlying [K+]o-induced VF changes by quantifying the properties of rotors/spiral waves, which are thought to underlie VF in normal and pathophysiological conditions (13). Accordingly, we used optical mapping and numerical approaches to study the effects of hyperkalemia on VF dynamics (organization/termination) induced in the structurally normal guinea pig heart. Finally, we evaluated the ionic mechanisms underlying these changes in VF complexity via computer simulations.
Our results indicate that the evolution in VF organization during an increase in [K+]o can be explained via the changes that occur in the mother rotor/fibrillatory conduction paradigm that is postulated to maintain VF in this model. Furthermore, simulations point to an important role for the inward rectifier K+ current IK1 in mediating these hyperkalemia-induced changes.
Methods
Experiments
Hearts from 12 male guinea pigs (500–700 g) were Langendorff-perfused with Tyrode's solution at physiological temperatures (37°C) and paced for VF induction as described previously (12). During stable VF episodes (>5 min), the control perfusion and superfusion solutions (4 mM [K+]o) were changed to solutions containing 7 mM [K+]o (n = 5) or 10 mM [K+]o (n = 7) for 5–10 min, followed by washout (4 mM [K+]o). Movies of fluorescence (di-4-ANEPPS) were obtained via charge-coupled device cameras (64 × 64 pixels, 600 frames/s) and analyzed using DF, phase maps, and time-space plots (TSPs) to quantify VF frequency and excitation dynamics (12). The data are presented as means ± standard deviation (SD). Further details regarding the data analyses and statistics are presented in the Supporting Material.
Simulations
We used an ionic model of the guinea pig ventricular myocyte developed at physiological temperature (37°C (14)) to simulate spiral wave reentry in a two-dimensional (2D) sheet, as described in recent multicellular simulations (15). The 2D sheet (4 × 4 cm, 400 × 400 cells) was equally divided into two regions to represent the LV and RV, and the ionic properties were modified to simulate the control frequencies seen in experiments at 4 mM [K+]o. The details of the ionic modifications are presented in the Supporting Material. We initiated a rotor by cross-field stimulation and studied its behavior in hyperkalemia by increasing the concentrations of [K+]o from 4 to 10 mM in steps of 1 mM.
Results
Effect of 7 mM [K+]o on VF dynamics
To obtain direct information about the behavior of the driver/source of VF and the waves emanating from it (13,16) during hyperkalemia, we first quantified the spatial distribution of the DFs of excitation over the surface of the heart. In a previous study (11), we demonstrated that the DF distribution in the fibrillating guinea pig ventricles was stable for up to 20 min of perfusion with normal Tyrode's solution. Fig. 1 A shows a control DF map during stable VF at 4 mM [K+]o in a representative experiment. A relatively stable reentrant source (orange) within the blocked region on the LV resulted in a DF map in which the fastest and largest DF domains (domains 1 and 2, respectively) were 31.5 Hz and 12.0 Hz, respectively; i.e., the LV to RV DF difference (LV-RVDF) was 17.3 Hz. After [K+]o was increased to 7 mM, the fastest DF domain (domain 3) reduced to 23.0 Hz, whereas the largest DF domain in the RV (domain 4) increased to 16.0 Hz (Fig. 1 B). Consequently, the LV-RVDF (same areas) was reduced to 5.8 Hz. Table S1 of the Supporting Material summarizes the overall effects of 7 mM [K+]o on the measured properties of the DF.
Figure 1.
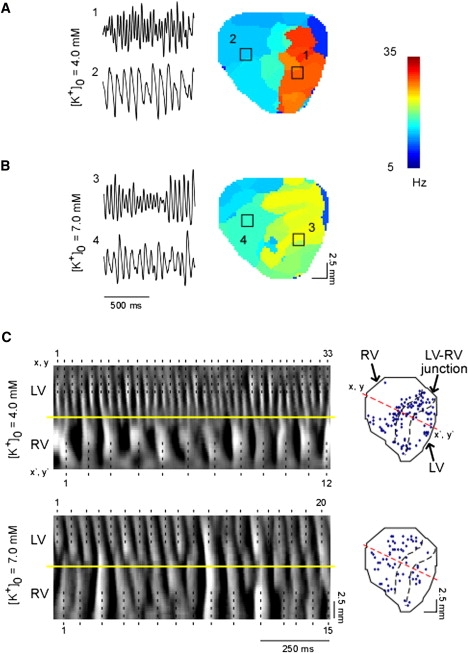
DF organization during 7 mM [K+]o. (A and B) Fluorescent signals (left) recorded from pixels located in the fastest (1 and 3) and largest (2 and 4) domains of DF maps (center) generated from control (A) and 7 mM [K+]o. (B) Movies of anterior wall excitation in a representative experiment. Squares indicate a 5 × 5 pixel area in the center of the LV and RV. (C) (Left) TSP of fluorescence along a line of pixels crossing the LV-RV junction (from (x,y) to (x′,y′)) during control (upper) and hyperkalemic (lower) VF in a similar experiment. (Right) Location of SP initiation in the same VF episodes. Black dotted lines and numeric labels on the left indicate activated sites. Yellow lines demarcate the LV-RV boundary. Black dotted lines on the right demarcate the LV-RV junction region from the two chambers. Red dotted line indicates the line of pixels used for TSP plots.
Time-space plots (TSPs) allow beat-to-beat quantification of the spatiotemporal patterns of wave propagation at specific locations on the surface of the ventricles (13). In addition, TSPs help determine the underlying basis of the formation of multiple DF domains and of the LV-RVDF gradient during VF. Fig. 1 C (upper panel, left) was taken from an experiment at 4 mM [K+]o. It shows a TSP along the line x,y-x′,y′ crossing both ventricles (see diagram on the right). High-frequency excitation on the anterior surface of the LV during VF resulted in a complex 33:12 (LV/RV) pattern of wave propagation, with intermittent blockade and wavebreaks at the interventricular septum. As shown by the diagram on the right, similar areas of wavebreak along multiple locations of the LV-RV junction resulted in abrupt transformations of DF, with formation of widely distributed DF domains (see Fig. 1 A). In Fig. 1 C (upper panel, right), quantification of the singularity points (SPs) at the sites of wavebreak (17) demonstrate that the largest SP density was at the LV-RV junction (1.16 SP mm−2 s−1), followed by LV regions around the highest DF domain (0.77 SP mm−2 s−1) and the RV (0.48 SP mm−2 s−1). In contrast, as illustrated at the bottom of Fig. 1 C, in TSPs obtained for 20 consecutive LV activations during hyperkalemia, the reduced LV activation rate enabled numerous instances of 1:1 LV-to-RV activation. As expected, the incidence of wavebreaks and the SP density was markedly reduced at the LV-RV junction and LV regions (0.17 SP mm−2 s−1 and 0.36 SP mm−2 s−1, respectively), but not on the RV (0.45 SP mm−2 s−1).
Overall, 7 mM [K+]o significantly reduced the total SP density from 0.90 ± 0.14 SP mm−2 s−1 to 0.48 ± 0.20 SP mm−2 s−1 (p = 0.021), which resulted from a significant reduction in SP density in the LV (from 1.19 ± 0.16 to 0.60 ± 0.4 SP mm−2 s−1, p = 0.039) and LV-RV junction (from 1.15 ± 0.15 to 0.45 ± 0.22 SP mm−2 s−1, p = 0.006), but not in the RV.
Effect of 10 mM [K+]o on VF dynamics
The effects of elevating [K+]o to 10 mM on the pattern of VF were more pronounced. In four out of seven hyperkalemic hearts, VF was converted to monomorphic ventricular tachycardia (VT) (≈11–16 Hz). In two of these hearts, VT spontaneously converted back to VF and then either remained in VF or converted to sinus rhythm (SR). In the third heart, VT was stable until washout ensued, and then spontaneously converted to VF. In the fourth heart, VT converted to SR. In the three other hearts, VF converted to SR after 6–9 min of 10 mM [K+]o without an apparent transitional episode of VT. Upon washout, VF was successfully reinduced in all nonfibrillating hearts. Fig. 2 A shows characteristic volume-conducted electrocardiograms recorded during control VF and during perfusion with 10 mM [K+]o in an episode of VF and an ensuing episode of VT in one experiment.
Figure 2.
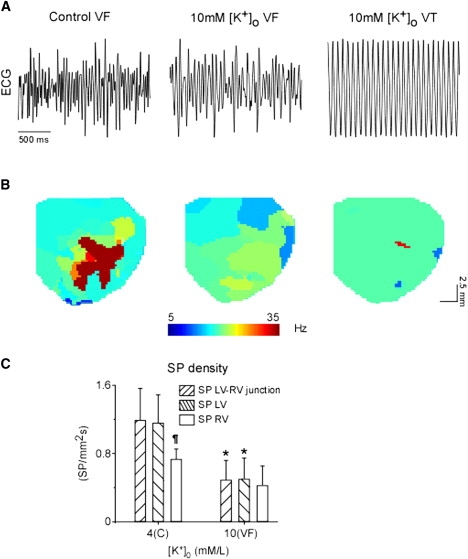
DF organization during 10 mM [K+]o. (A) Volume conductor electrocardiograms recorded during control (left) and 10 mM [K+]o VF (center) and VT (right). (B) Representative DF maps generated during normokalemic (4 mM [K+]o) and 10 mM [K+]o VF (center) and VT (right). (C) Mean rate of SP density in LV-RV junction, LV, and RV regions identified during control and 10 mM [K+]o VF. ∗p < 0.036 versus control. ¶p < 0.019 versus LV and LV-RV junction.
Fig. 2 B shows DF maps from another experiment in which the maximum DF (DFmax) was reduced from 35.5 Hz (domain 1) in control (left) to 18.5 Hz (center) during a 10 mM [K+]o episode of VF. The frequency of an ensuing episode of VT was 16 Hz (right). Concurrently, the LV-RVDF difference was reduced from 21.0 Hz in control to 4.5 Hz during 10 mM [K+]o VF, and further decreased to zero during VT. The overall DF parameter changes are summarized in Table S1. Of importance, a progressive decline in DFmax during hyperkalemic VF was associated with a reduction in the LV-RVDF and the number of DF domains, which respectively reached zero and one during VT episodes (Table S1).
As summarized in Fig. 2 C, changing [K+]o from 4 to 10 mM significantly reduced the SP density in the LV and LV-RV junction regions, but not in the RV. Altogether, at 10 mM [K+]o the total SP density decreased from 0.98 ± 0.21 to 0.47 ± 0.21 SP mm−2 s−1 (p = 0.038). As expected, the SP was zero throughout the ventricles during VT episodes at 10 mM [K+]o.
Effect of 10 mM [K+]o on wavelet organization
In addition to the frequency of excitation and SP density, we also quantified the rotation period and the tip meander of the reentrant sources (rotors) observed experimentally. These parameters give direct information about the stability of the rotors (13). The rotation period is likely to increase with reduced wavebreaks (or SPs), and an increase in tip meander indicates that the rotor is unstable and more likely to terminate as a result of its collision with a boundary (13).
During control, the majority of sustained rotors were located in the LV, but hyperkalemia increased their incidence in both chambers (Table S2). As summarized in Fig. 3 A, the mean rotation period (MRP) of sustained rotors was larger during hyperkalemic VF (56.1 ± 8.3 ms) than during control (27.6 ± 3.0 ms, p < 0.001). Additionally, there was a slight, albeit significant increase (p < 0.001) of the MRP of VT rotors compared to hyperkalemic VF rotors (66.7 ± 8.2 ms; Fig. 3 A, Table S2). As shown in Fig. 3 B, hyperkalemia also increased (p < 0.001) the tip meander, which was quantified as the mean core area (MCA) of sustained LV rotors from 0.90 ± 0.53 mm2 to 2.11 ± 0.88 mm2 during VF, and to 1.97 ± 0.89 mm2 during VT (see Table S2). However, the MCA of rotors identified during hyperkalemic VF and VT episodes was not significantly different. Of interest, the MCA of RV rotors was larger than that of LV rotors during both control and hyperkalemia (Table S2).
Figure 3.
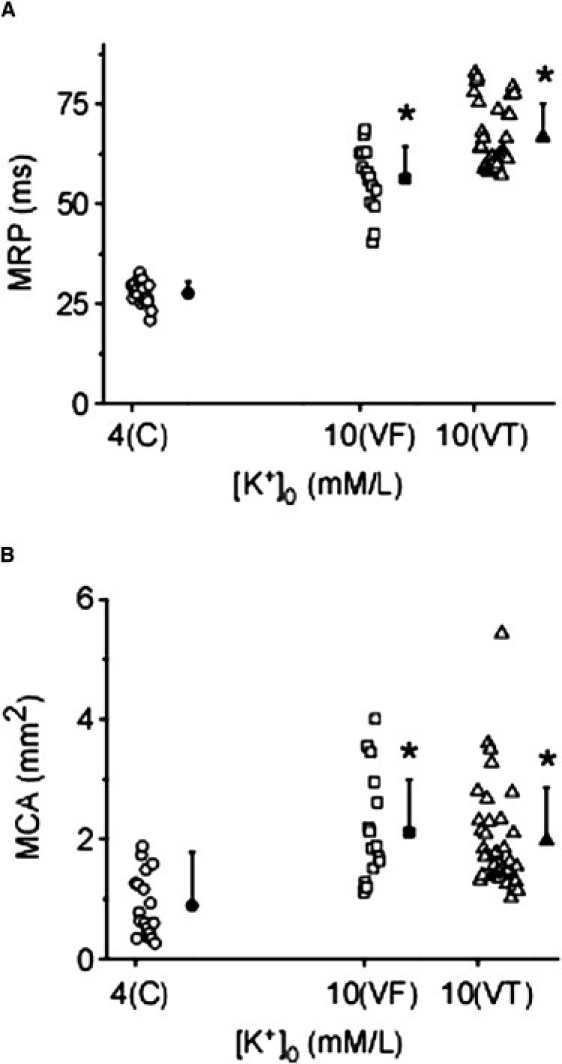
Quantification of rotor properties and SP. Individual (open symbols) and mean (solid symbols) values of MRP (A) and MCA (B) of LV rotors identified in control VF (●) and 10 mM [K+]o VF (□) and VT (Δ).
Termination of SP during control and hyperkalemic VF
We conducted a detailed analysis of the events leading to the termination of SPs, which permitted us to classify the VF termination into three distinct categories depending on the progression of phase in the immediate vicinity of the termination site (Fig. 4). Consistent with the topological rules of formation and annihilation of wavelets (18), all SPs, with the exception of those colliding with the base of the heart or drifting out of the field of view, were terminated in pairs. Fig. 4 A depicts two counter-rotating waves (t = 0–12 ms), each around an SP near the apex. At 25 ms, the SPs drifted toward each other, substantially narrowing the isthmus between them (yellow-red). At 37 ms, the two counter-rotating wavefronts collided, and not having enough room to propagate down the exceedingly narrow isthmus, they stopped abruptly and VF terminated. Fig. 4 B shows at t = 0 ms two counter-rotating wavelets near the ventricular base, plus a third wavelet rotating clockwise around its own singularity. At t = 3 ms, the counter-rotating wavelets had fused, which effectively eliminated their respective SPs. The third wavelet continued to rotate in the clockwise direction, but eventually its SP also disappeared, which resulted in the consolidation of all wave fronts into a single large wave that traveled from right to left and downward. VF terminated shortly thereafter. Fig. 4 C shows two distinct forms of SP termination. The upper SP terminated upon collision with the base of the heart (upper limit), whereas the lower SP drifted outside the field of view to other nonmapped regions of the ventricle.
Figure 4.
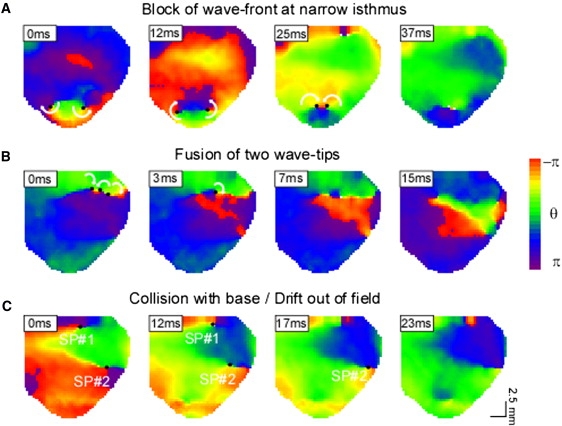
Mechanisms leading to the termination of SP during VF. (A) Termination after the collision of wavefronts (green) and inability to propagate through the narrow isthmus between their respective SPs. (B) Termination of SPs after the fusion of wavelets. (C) Termination of SP after collision of the SP with the base of the heart (SP No. 1) or after drift of SP out of the field of view (SP No. 2).
Table S3 summarizes the relative contribution of each of the three forms of VF termination in a representative experiment at 4 and 10 mM [K+]o just before conversion to VT. The data are normalized to account for the different time intervals used in the analysis. In addition to markedly reducing the number of SPs, hyperkalemia increased the likelihood of termination of VF by collision of drifting SPs with a boundary (from 1.7% to 5.8%).
Simulation of hyperkalemic VF
Frequency analyses of simulated rotors in 2D sheets (Fig. 5 A) demonstrate that during control ([K+]o = 4.0 mM), the DF distribution is clearly demarcated into a high-frequency domain in the LV (≈30 Hz) and a slower (≈18 Hz) domain in the RV, closely resembling the experimental values. Elevating [K+]o to 5.0 or 6.0 mM did not alter the LV-RVDF difference. However, at 7.0 mM [K+]o, the LV source frequency is reduced to ≈24 Hz. Of note, a substantial portion of the RV also now shows an identical DF of excitation. At 8.0 and 9.0 mM of [K+]o, the DFmax is further reduced, and the rotor terminates in the latter case (Movie S5). Fig. 5 B shows a comparison of experimental DFmax values and the DF of the simulated LV driving rotor. Note that the latter value was obtained from a location (∗, first panel of Fig. 5 A) that shows a 1:1 activation throughout hyperkalemia, and is away from the rotor core that displays harmonics of DF. Clearly, both simulations and experiments show a nonlinear reduction of the LV DF with increasing [K+]o. Of interest, in simulations the rotor terminated at 9.0 mM [K+]o, as compared with 10 mM [K+]o in experiments. This may be the result of inherent limitations in the simulations.
Figure 5.
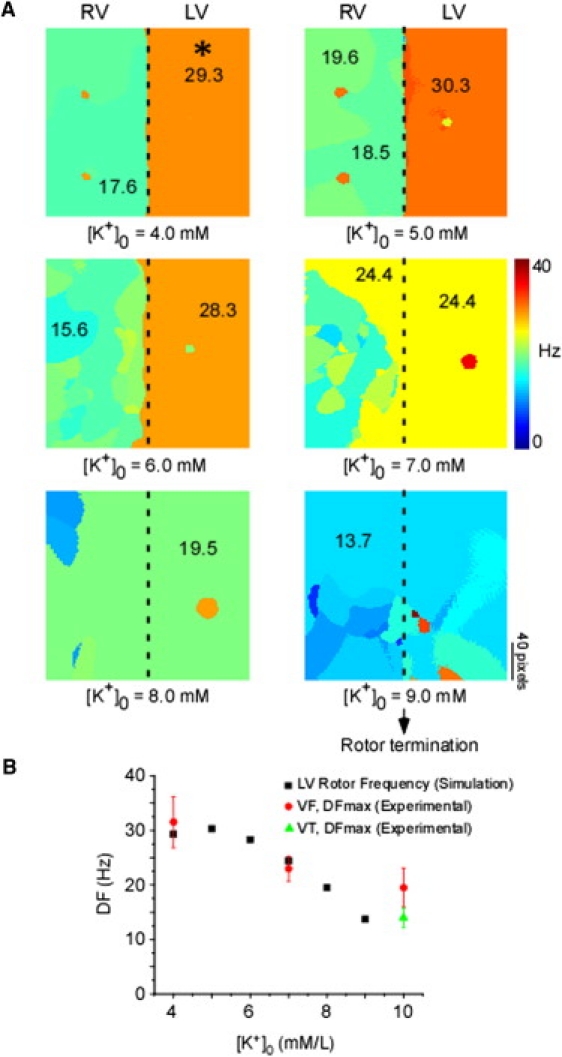
(A) DF maps of simulated spiral wave activity at [K+]o concentrations ranging from 4.0 to 9.0 mM, in 1.0 mM increments. Numbers on maps indicate DF values at the selected locations. Asterisk (∗) indicates pixel selected for plotting simulated values in panel B. (B) Simulated LV rotor frequency (■) and experimental values of DFmax during VF (•) or VT (▴) as a function of [K+]o.
To further investigate the effect of hyperkalemia on VF dynamics in the simulations, we analyzed the patterns of LV-to-RV wave propagation and the changes in the trajectory of the source wave tip. In Fig. 6 A, a snapshot of excitation and the TSP along a line of pixels crossing the simulated LV-RV border clearly show that during control, the LV source is unable to drive the RV in a 1:1 pattern, thus giving rise to the sharp DF transition seen in Fig. 5 (compare with experimental data in Fig. 1 C, upper panel). A similar representation of excitation during 7 mM [K+]o (Fig. 6 B) indicates that with a gradual passage of time (dashed yellow line), a larger area of the RV is being driven in a 1:1 fashion from the LV driving rotor (compare with Fig. 1 C, lower panel). This underlies the spatial DF distribution in 7.0 mM [K+]o (Fig. 5). Fig. 6 C shows snapshots of the spiral wave activity at 9.0 mM [K+]o. The left-most snapshots depict the previously described LV driving source terminating by collision with the left boundary (double red bars). Of interest, the remnant spiral wave activity in the RV now takes over as the primary rotor. It then meanders markedly as a single spiral wave across both the RV and LV (two right-most panels) before it terminates again due to a collision with the boundary (note that collision with a boundary is an SP termination mechanism enhanced by hyperkalemia). Fig. 6 D shows that the LV source tip meander, which is small during 4.0 mM [K+]o, is increased during 7.0 mM [K+]o. At 9 mM [K+]o, the tip meander of the rotor that drifts through the RV and sustains excitation is markedly increased just before termination of the arrhythmia.
Figure 6.
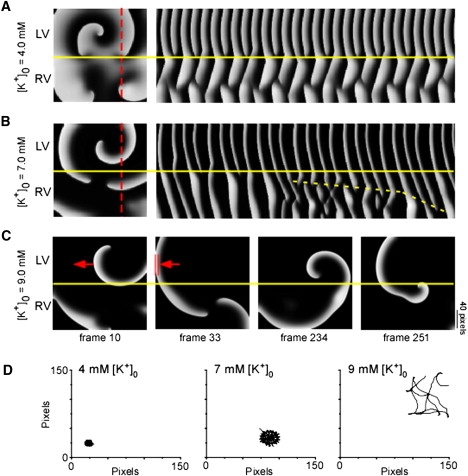
(A and B) Simulated spiral waves. Examples of spiral wave reentry during 4.0 mM [K+]o (A, left) and 7.0 mM [K+]o (B, left), with the corresponding TSP (A and B, right) along a selected line of pixels (red dashed line). The dashed yellow line indicates how RV gradually follows a 1:1 propagation from LV in 7.0 mM [K+]o. (C) Snapshots of spiral wave activity before its termination in 9.0 mM [K+]o. The red arrow denotes the direction of propagation, and double red bars indicate the site of collision with the boundary. Yellow lines demarcate the LV/RV boundary. (D) Trajectories followed by the LV spiral tip at 4.0 mM [K+]o (left), 7.0 mM [K+]o (center), and across the LV-RV border before termination (right). Note that the tip meander location is not same as in the snapshot, because of the way it was calculated. One pixel = 0.2 mm.
Ionic bases of hyperkalemic VF changes
The maximum conductance value of IK1 (GK1) is proportional to √[K+]o, and the reversal potential of IK1 (EK1) also varies with increasing [K+]o. The I/V relationship of IK1 at different values of [K+]o (see Fig. S2) depicts both of these changes. Similarly, the maximum conductance of IKr (GKr) and its reversal potential (EKr) are [K+]o dependent (I/V relationship not shown). We therefore investigated the relative importance of these [K+]o-induced changes in mediating spiral wave dynamics during hyperkalemia via numerical simulations. Fig. 7 A depicts DF maps when GK1 and GKr were allowed to change as [K+]o varied but the reversal potentials (EK1 and EKr, respectively) were not (i.e., they were clamped at their value corresponding to 4.0 mM [K+]o). Clearly, the DFmax in both LV and RV actually increased with hyperkalemia (7.0 and 9.0 mM) compared to the normal case (Fig. 5). Next, the reversal potentials, but not the maximum conductance values of IKr and IK1, were allowed to change with hyperkalemia. The resulting DF maps (Fig. 7 B) show a reduction of VF excitation frequencies, closely resembling the normal case (Fig. 5). Finally, we compared changes in IKr with those in IK1. When both the conductance and reversal potential of only IKr (but not IK1) were clamped to their respective constant values at [K+]o = 4.0 mM during simulated hyperkalemia, excitation frequencies were again decreased (Fig. 7 C), closely resembling the normal case. Fig. 7 D provides a quantitative comparison of the aforementioned simulations, which taken together provide evidence that the change in EK1 is the critical determinant leading to the reduced excitation frequency and transventricular homogenization of DF observed during hyperkalemic VF.
Figure 7.
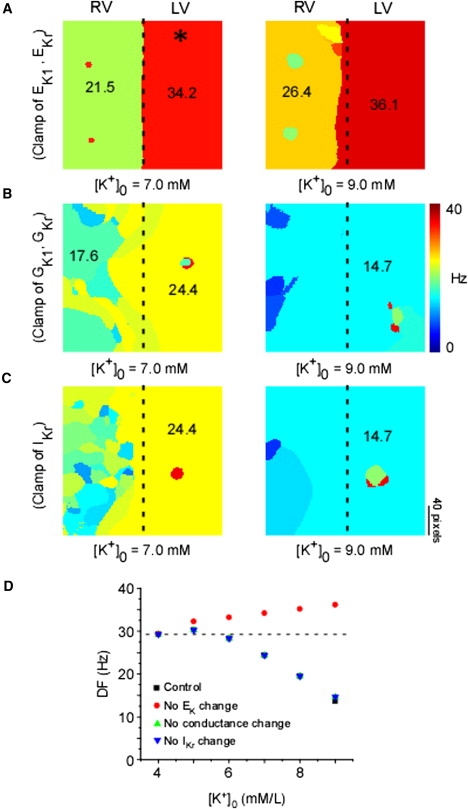
Ionic determinants of DF organization. DF maps of simulated hyperkalemic VF during artificial clamping to control values of EK1 and EKr (A), GK1 and GKr (B), and IKr (C). Asterisk (∗) indicates pixel location selected for plotting simulated values in panel D. (D) Plot of DF versus [K+]o for the control simulation in Fig. 6 (■) and simulations shown in panels A (•), B (▴), and C (▾).
As shown in Fig. 8, we determined the effects of hyperkalemia on the APD and excitability during VF. In panel A, we plot representative action potentials (APs) from a single point in the simulated LV for a duration of 200 ms at 4.0, 7.0, and 9.0 mM [K+]o. APD shortened as [K+]o increased. In addition, the resting membrane potential depolarized and the duration of the diastolic interval increased as the DFmax decreased progressively (see Fig. 5). We then quantified the APD at 80% repolarization (APD80) at different values of [K+]o (over a period of 1 s) from a single point in the LV and RV (Fig. 8 B), and both were seen to decrease monotonically. Further, the availability of the sodium current, which is quantified as the product of its inactivation gating variables (hj) and is a direct measure of tissue refractoriness, was quantified at the same points where the APD80 was measured, and was also seen to decrease monotonically as [K+]o increased (Fig. 8 C). Thus, although APD80 decreases with hyperkalemia, a concomitant increase in refractoriness (or decreasing hj) results in a slower rotor activity.
Figure 8.
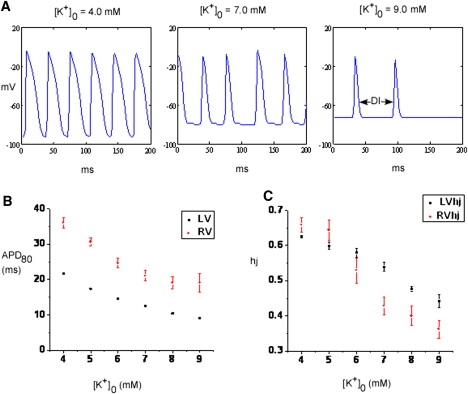
(A) Representative APs from a point located in the LV during spiral wave simulation at 4.0, 7.0, and 9.0 mM [K+]o. (B) Quantification of APD80 and (C) the availability of sodium current assessed by the product of its inactivation gating variables hj at a single point from LV and RV as a variation of [K+]o from 4.0 to 9.0 mM.
Discussion
The most important results of our study may be summarized as follows: 1) Elevation of [K+]o during VF in structurally normal guinea pig hearts reduces the DFmax of excitation, the LV-to-RV DF gradient, the number of DF domains, and the SP density on the LV and at the interventricular septum. 2) Hyperkalemia (10 mM [K+]o) markedly increases the MCA and the MRP of sustained rotors, and often converts VF to VT (driven by one or two counter-rotating rotors) or SR. 3) As demonstrated in both experiments and simulations, the increased organization of VF dynamics during hyperkalemia is a direct consequence of the slowing of the DFmax and is manifested by a reduction in wavebreak and SP incidence, fusion of wavelets, and increased rotor drift and collision with a boundary. 4) The numerical results demonstrate that a key mechanism underlying rotor drift and VF termination in hyperkalemia is the shift in EK1 and the resting membrane potential to less negative levels, with a consequent reduction in sodium current availability, excitability, and post-repolarization refractoriness.
Organization of excitation and wavebreak formation during hyperkalemia
Our data closely agree with previous studies that showed a reduction of ventricular excitation frequencies and VF termination during hyperkalemia (7–10). In similarity to results obtained in fibrillating rabbit hearts (7), we showed that hyperkalemia reduced the LV DFmax and the LV-RVDF gradient. Notably, however, our results demonstrate that the response of the VF frequency and dynamics to external K+ is highly nonlinear. As shown previously in the guinea pig heart, at 4 mM [K+]o, high-frequency rotors have a strong tendency to stabilize on the anterior surface of the LV, and the spiral waves emanating from them undergo numerous spatially distributed, intermittent block processes at the interventricular septum, which results in marked differences in LV versus RV activation rates. Increasing [K+]o to 7 mM reduced the LV activation frequency and the APD, resulting in more rapid recovery of excitability after each AP and a consequent reduction in wavebreak formation and increased propagation of waves toward the RV. At 10 mM [K+]o, the LV DFmax and the LV-RVDF gradient were further reduced, which occasionally led to VT that was associated with an additional reduction in excitation frequency, as well as a homogeneous distribution of DF that effectively eliminated all DF gradients. Of importance, in all such episodes of VT, ventricular activation was sustained by one or two counter-rotating reentrant sources. In the majority of episodes (35/40), the rotors were located in the LV, which emphasizes the key role of such periodic sources in determining the ventricular organization of excitation. In contrast, in severe hyperkalemia (9–10 mM [K+]o), excitability was substantially reduced. This led to rotor meandering or even drifting with termination by fusion with another wavefront propagating in the same direction, or by collision with an anatomical or functional obstacle (i.e., an incoming wavefront). Thus, the antifibrillatory action of hyperkalemia can be readily explained via the changes in spatiotemporal dynamics of the mother rotor/fibrillatory conduction schema that has been postulated to sustain VF (11,12,18).
Mechanisms leading to reduction of excitation frequency
Our data showed that the core area and the rotation period of sustained rotors were larger in the hyperkalemic LV than in the normokalemic LV. This was not unexpected, since it is well known that excitability determines the core area (19). Our simulations also showed that hyperkalemia depressed excitability by reducing Na+ current availability (Fig. 8), and that this was a consequence of the EK1 shift in the depolarized direction. As shown in Fig. S2, in addition to depolarizing EK1 progressively, when [K+]o was changed from 4 to 7 and then 10 mM, the slope conductance and the outward component of IK1 increased. Notably, in the simulations in which we clamped the EK1 to prevent depolarization (see Fig. 7), the increase in IK1 conductance with hyperkalemia increased the rotor frequency from ≈30 to 36 Hz. On the other hand, when GK1 was artificially forced to remain constant in hyperkalemia and EK1 was allowed to change as a function of [K+]o, the membrane depolarization (and the reduced sodium current availability) predominated, and thus both excitability and rotor frequency were reduced. Lastly, our simulations confirmed the dominant role of IK1 over IKr, since even though the hyperkalemia-induced changes in IKr were clamped, the K+-induced changes in VF frequency and organization persevered. This suggests that during high-frequency excitation in normal cardiac tissue, the contribution of IKr current is negligible compared to IK1. Note, however, that this does not exclude a role for IKr in inscribing high-frequency rotors when IKr conductance is increased and primarily causes AP shortening, as in short QT syndrome (20). Finally, provided that both Kir2.1 and Kir2.3 protein isoforms largely determine the IK1 profile in guinea pig ventricles (21), we suggest that the higher sensitivity of Kir2.1 to elevated [K+]o (21) gives this channel a dominant role in rotor stabilization during hyperkalemic VF.
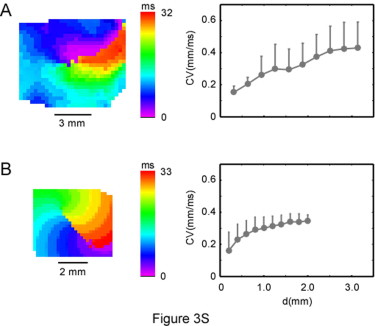
In previous studies, Buchanan et al. (22) also documented changes in APD and the conduction velocity (CV) as a function of changes in [K+]o. Their results suggested that although APD decreased monotonically as [K+]o was increased, there was a biphasic change in the longitudinal CV: CV initially increased at ≈7–8 mM of [K+]o and then decreased. The paradoxical increase in CV was attributed to the fact that the change in resting potential was now closer to the threshold for the AP. Simulations with the model used in this study (22) were also able to account for this phenomenon (23). However, both our simulations and experiments showed a monotonic decrease in the frequency of fibrillation, and thus no anomalous changes. There are two possible explanations for this discrepancy: 1), the measurements obtained by Buchanan et al. apply to pacing at relatively slow frequencies (1–2 Hz), not at the 20–30 Hz observed during VF (11,12); and 2), the CV that is relevant here is the CV measured as a function of the distance from the core, as described in our previous work (24). We observed a similar behavior when we plotted the CV versus core distance relation for experiments and simulations at 4 mM (see Fig. S3). Unfortunately, we were unable to characterize this same relationship at higher values of [K+]o because of the large meander of the reentrant wave tip.
Termination of VF
An earlier study in rabbits reported the occurrence of transitions to VT and/or SR during hyperkalemia (7.5 mM [K+]o) (9). Our data confirm that result, although we found that a higher [K+]o concentration (10 mM) was necessary to obtain a similar outcome. Of interest, high [K+]o (12.5 mM) in pig hearts induced transitions from VF to VT, but not to SR (10). In isolated canine preparations, Koller et al. (8) observed the conversion of VF to a single periodic rhythm at 10 mM [K+]o and termination of VF at 12 mM [K+]o. A comparison of the results suggests that ventricular mass and species may determine the outcome of hyperkalemic VF, with the largest hearts being less prone to VF termination. However, the apparent greater sensitivity of rabbit hearts to K+ changes compared to guinea pig hearts suggests the involvement of other mechanisms, such as species-dependent ion channel sensitivity to hyperkalemia.
Although the reduction of SP and DF gradients most likely favors VF termination during hyperkalemia, the mechanism involved in the transition remains elusive due to the uncertainty in the timing of this event. During stable VF, the formation of SP must on average be equal to the annihilation of SP. Thus, for a transition to occur, a critical event favoring the termination of SP must happen. Our analysis determined three different mechanisms leading to SP termination, any of which could be potentially relevant in determining VF transition to VT or SR. Our analysis showed that termination of SP as a result of conduction failure across a narrow isthmus between two closely apposed SPs, as well as drift and subsequent interaction with one of the ventricular boundaries, was enhanced during hyperkalemia. However, a detailed understanding of the event that ultimately leads to VF termination will require further studies designed to track the fate of SP during conversion of the arrhythmia. Our simulations did confirm the role of a reduction in SP and meander in transition from mother rotor/fibrillatory conduction during VF to a large meandering rotor during hyperkalemia.
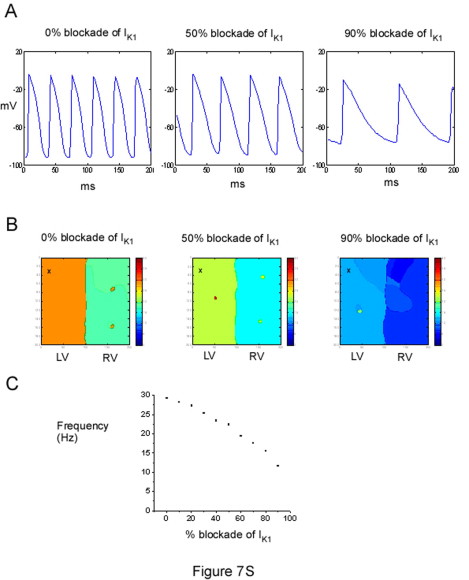
It is also important to note that the termination of VF by hyperkalemia is similar to the antiarrhythmic effect seen during IK1 blockade by relatively low concentrations (1–50 μM) of BaCl2 in previous experiments (12). Our simulations are able to account for this phenomenon (Fig. S7). They show that although hyperkalemia increases IK1 conductance, and barium decreases it, both manipulations lead to a depolarization of the resting membrane potential, resulting in a reduction of excitability and eventual termination of the rotor. However, we expect that the combined dual BaCl2 effect of reducing outward IK1 conductance and depolarizing EK1 should be more efficacious in terminating VF than hyperkalemia alone (which only depolarizes EK1, but increases the conductance).
Limitations
The limitations of optical mapping experiments have been described in previous studies (11,12). Our computer simulations represent a simplified model of the myocardium and do not incorporate ionic heterogeneities or structural components (e.g., fiber orientation and the 3D nature of the heart), which are important determinants of VF organization. As in previous studies (11), we had to alter the guinea pig ventricular myocyte ionic mathematical model to better emulate experimental VF. This suggests that further improvements in the collection of ion channel data, especially at high pacing rates from both the LV and RV, are required. Lastly, in our simulations the rotor terminated at 9 mM [K+]o. In our experiments, VF often converted to VT or terminated at 10 mM [K+]o. One possible explanation for this discrepancy may reflect the fact that we decreased the maximum sodium conductance in our computer model (from 16.0 to 12.8; see the Supporting Material) to better simulate the rotor frequencies seen experimentally. This may have resulted in a slightly faster decrease in the excitability as a function of hyperkalemia compared to that observed in experiments. Additionally, it may also be related to the difficulty of accurately modeling the Na+ current and related parameters. Because of the fast kinetics involved, measurements are carried out at room temperature and then extrapolated to physiological temperatures for modeling purposes.
Supporting Material
Methods, results, references, three tables, seven figures, and six movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00262-6.
Supporting Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Drs. Alexey Zaitsev, Javier Moreno, Samie Noujaim, Sharon Zlochiver, Prabal Guha, and Justus Anumonwo for useful discussions and suggestions.
This work was supported by National Heart, Blood and Lung Institute grants PO1-HL039707, PO1-HL070074, and RO1-HL080159 (J.J.); a Heart Rhythm Society Michael Mirowski International Fellowship (M.W.); and an American Heart Association postdoctoral fellowship and Scientist Development Grant (S.V.P.).
References
- 1.Gettes L.S. Electrolyte abnormalities underlying lethal and ventricular arrhythmias. Circulation. 1992;85(1, Suppl):I70–I76. [PubMed] [Google Scholar]
- 2.Pitt B., Bakris G., EPHESUS Investigators Serum potassium and clinical outcomes in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) Circulation. 2008;118:1643–1650. doi: 10.1161/CIRCULATIONAHA.108.778811. [DOI] [PubMed] [Google Scholar]
- 3.Janse M.J., Wit A.L. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- 4.Zipes D.P., Wellens H.J. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 5.Paterson D.J. Antiarrhythmic mechanisms during exercise. J. Appl. Physiol. 1996;80:1853–1862. doi: 10.1152/jappl.1996.80.6.1853. [DOI] [PubMed] [Google Scholar]
- 6.Nair K., Umapathy K., Nanthakumar K. Aborted sudden death from sustained ventricular fibrillation. Heart Rhythm. 2008;5:1198–1200. doi: 10.1016/j.hrthm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Caldwell J., Burton F.L., Cobbe S.M. Heterogeneity of ventricular fibrillation dominant frequency during global ischemia in isolated rabbit hearts. J. Cardiovasc. Electrophysiol. 2007;18:854–861. doi: 10.1111/j.1540-8167.2007.00867.x. [DOI] [PubMed] [Google Scholar]
- 8.Koller M.L., Riccio M.L., Gilmour R.F. Effects of [K(+)](o) on electrical restitution and activation dynamics during ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H2665–H2672. doi: 10.1152/ajpheart.2000.279.6.H2665. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe Y., Toda H., Uchida H. Electrophysiological mechanisms for the initiation and maintenance of ventricular fibrillation in nonischemic rabbit hearts. Heart Vessels Suppl. 1987;2:69–87. [PubMed] [Google Scholar]
- 10.Zaitsev A.V. Mechanisms of ischemic ventricular fibrillation: who's the killer? In: Zipes D.P., Jalife J., editors. Cardiac Electrophysiology: From Cell to Bedside. W. B. Saunders; Philadelphia: 2004. 399–406. [Google Scholar]
- 11.Samie F.H., Berenfeld O., Jalife J. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ. Res. 2001;89:1216–1223. doi: 10.1161/hh2401.100818. [DOI] [PubMed] [Google Scholar]
- 12.Warren M., Guha P.K., Jalife J. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J. Cardiovasc. Electrophysiol. 2003;14:621–631. doi: 10.1046/j.1540-8167.2003.03006.x. [DOI] [PubMed] [Google Scholar]
- 13.Jalife J. Ventricular fibrillation: mechanisms of initiation and maintenance. Annu. Rev. Physiol. 2000;62:25–50. doi: 10.1146/annurev.physiol.62.1.25. [DOI] [PubMed] [Google Scholar]
- 14.Faber G.M., Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys. J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pandit S.V., Berenfeld O., Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys. J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berenfeld O., Mandapati R., Jalife J. Spatially distributed dominant excitation frequencies reveal hidden organization in atrial fibrillation in the Langendorff-perfused sheep heart. J. Cardiovasc. Electrophysiol. 2000;11:869–879. doi: 10.1111/j.1540-8167.2000.tb00066.x. [DOI] [PubMed] [Google Scholar]
- 17.Gray R.A., Pertsov A.M., Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature. 1998;392:75–78. doi: 10.1038/32164. [DOI] [PubMed] [Google Scholar]
- 18.Davidenko J.M., Salomonsz R., Jalife J. Effects of pacing on stationary reentrant activity. Theoretical and experimental study. Circ. Res. 1995;77:1166–1179. doi: 10.1161/01.res.77.6.1166. [DOI] [PubMed] [Google Scholar]
- 19.Mandapati R., Asano Y., Jalife J. Quantification of effects of global ischemia on dynamics of ventricular fibrillation in isolated rabbit heart. Circulation. 1998;98:1688–1696. doi: 10.1161/01.cir.98.16.1688. [DOI] [PubMed] [Google Scholar]
- 20.Brugada R., Hong K., Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 21.Dhamoon A.S., Pandit S.V., Anumonwo J.M. Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ. Res. 2004;94:1332–1339. doi: 10.1161/01.RES.0000128408.66946.67. [DOI] [PubMed] [Google Scholar]
- 22.Buchanan J.W., Gettes L. Ionic environment and propagation. In: Zipes D.P., Jalife J., editors. Cardiac Electrophysiology: From Cell to Bedside. W.B. Saunders; Philadelphia: 1990. 149–156. [Google Scholar]
- 23.Shaw R.M., Rudy Y. Electrophysiologic effects of acute myocardial ischemia. A mechanistic investigation of action potential conduction and conduction failure. Circ. Res. 1997;80:124–138. doi: 10.1161/01.res.80.1.124. [DOI] [PubMed] [Google Scholar]
- 24.Noujaim S.F., Pandit S.V., Jalife J. Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J. Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.