

Abstract
The four-helix bundle protein Rd-apocyt b562, a redesigned stable variant of apocytochrome b562, exhibits two-state folding kinetics. Its transition-state ensemble has been characterized by Φ-value analysis. To elucidate the molecular basis of the transition-state ensemble, we have carried out high-temperature molecular dynamics simulations of the unfolding process. In six parallel simulations, unfolding started with the melting of helix I and the C-terminal half of helix IV, and followed by helix III, the N-terminal half of helix IV and helix II. This ordered melting of the helices is consistent with the conclusion from native-state hydrogen exchange, and can be rationalized by differences in intrinsic helix propensity. Guided by experimental Φ-values, a putative transition-state ensemble was extracted from the simulations. The residue helical probabilities of this transition-state ensemble show good correlation with the Φ-values. To further validate the putative transition-state ensemble, the effect of macromolecular crowding on the relative stability between the unfolded ensemble and the transition-state ensemble was calculated. The resulting effect of crowding on the folding kinetics agrees well with experimental observations. This study shows that molecular dynamics simulations combined with calculation of crowding effects provide an avenue for characterize the transition-state ensemble in atomic details.
Introduction
Many single-domain proteins fold without detectable intermediates in kinetic folding experiments. However, intermediate states that are more stable than the unfolded state may still exist after crossing the rate-limiting transition state of folding (1,2). In fact, such hidden intermediates have been detected in several small proteins by using the native-state hydrogen exchange method (3–12). One of these proteins is Rd-apocyt b562, a redesigned variant of the four-helix bundle protein apocytochrome b562 (13) (Fig. 1). Experimental studies on this protein have led to the identification of two partially unfolded forms (PUFs) after the rate-limiting transition state (8,14–17). PUF1 has the helix I and the C-terminal half of helix IV (or IV-C) unfolded but all other helical segments formed, whereas PUF2 has only helix I unfolded. A crude model for the folding pathway is thus (8,16)
| (1) |
where U, TS, and N denote the unfolded state, the transition state, and the native state, respectively; a pair of braces indicates a fast transition. NMR structures of mimetic models of the two PUFs have been solved (15,17). More recently PEG 20K has been used as a crowding agent to study the effect of macromolecular crowding on the folding kinetics (18). This multitude of experimental studies makes Rd-apocyt b562 a good model system for using molecular dynamics (MD) simulations to gain atomistic insight into the transition-state ensemble.
Figure 1.
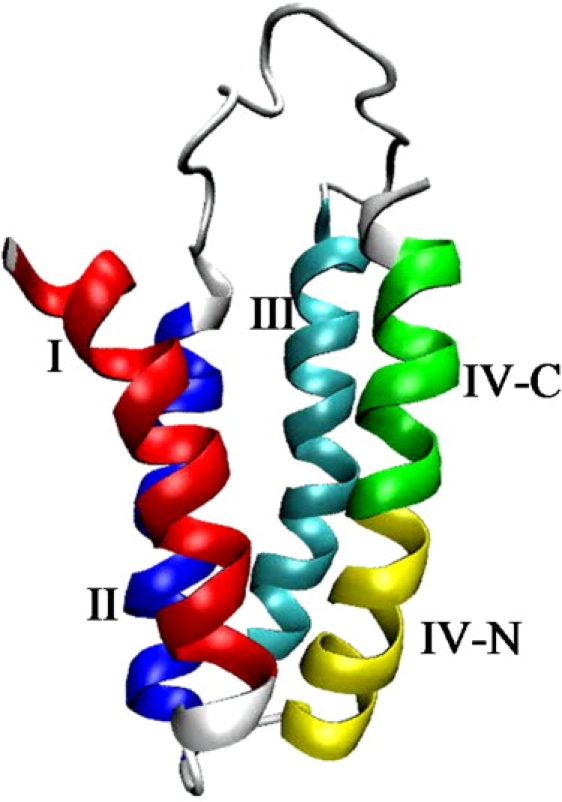
Structure of the four-helix bundle protein Rd-apocyt b562. In the color version of this figure, helices I, II, III, IV-N, and IV-C are displayed in red, blue, cyan, yellow, and green, respectively.
MD simulations have been used widely to characterize the transition-state ensembles in protein folding (19–27) and unfolding (28–45). In the latter simulations, high-temperature-induced unfolding has become a robust procedure since the pioneering work of Li and Daggett (28). Despite the successes and ongoing improvements of folding simulations, a number of advantages of unfolding simulations can be noted. Starting with the folded structure, a protein can be unfolded in a high-temperature simulation much faster than a simulation of the reverse process. Local-minimum traps are, by design, much easier to avoid in a high-temperature simulation. Folding simulations up to several microseconds are typically needed to fold small proteins (21–24,45), whereas simulations up to tens of nanoseconds are sufficient to unfold even medium-sized proteins. Beside the sampling problem, the inaccuracy of force fields is also a major obstacle to folding proteins correctly (46); a 10-μs simulation of a small ultrafast-folding protein in explicit solvent failed to reach any structure resembling the native state (27). The principle of microscopic reversibility asserts that the pathways of folding and unfolding are the reverse of each other; hence the information from unfolding simulations can be used to deduce the folding pathway. Because the unfolding simulations are carried out at high temperatures, an added assumption is that unfolding pathways are robust so as to remain valid at the lower temperatures accessible by experiments. The question of whether unfolding pathways depend on temperature has been explored to some extent (34,40); these studies suggest that unfolding pathways are generally independent of temperature. This assumption is made here.
In this study, we carried out a set of high-temperature unfolding simulations of Rd-apocyt b562 and analyzed the unfolding pathway and the transition-state ensemble. The application of high-temperature unfolding simulations to Rd-apocyt b562 is particularly suitable because the PUFs identified by the experiments exist on the native side of the rate-limiting transition state, and hence are expected to occur early during unfolding. Indeed, as unfolding progressed in our simulations, helix I and then IV-C melted, producing PUF2 and PUF1, respectively. This unfolding order, on reversal, yields the same folding pathway as identified by the experimental studies (8,16). Furthermore, the higher stability of helices II, III, and IV-N (i.e., the N-terminal half of helix IV) over that of helices I and IV-C is correlated with the intrinsic helix propensities of the corresponding sequence segments.
Experimentally, Φ-value analysis yields valuable information about the transition-state ensemble (47). When complemented with MD simulations, an atomistic picture of the transition-state ensemble may be constructed (28–31,33,38,39). Here, based on the Φ-values (8,16), we empirically defined the transition-state ensemble in our simulations as comprised of all conformations in which helices I and IV-C have <3 residues remaining helical and at the same time helices II, III, and IV-N have at least 50% of their residues remaining helical. With this definition, the residue helical probabilities of the transition-state ensemble show good correlation with the Φ-values.
To further validate the putative transition-state ensemble, we calculated how its stability is affected by crowding. The resulting effects on the folding kinetics are consistent with the measurements with PEG 20K as a crowding agent by Ai et al. (18). The consistency confirms our placement of the transition state.
Simulation, Calculation, and Methods
Unfolding and native-state simulations
All MD simulations of Rd-apocyt b562 started from the NMR structure (15) (PDB entry 1yyj). In the native state, the helical segments of this 106-residue protein are: helix I, residues 2–18; helix II, residues 23–40; helix III, residues 59–81, helix IV-N, residues 84–94; and helix IV-C, residues 95–104) (Fig. 1). The simulations followed the procedure in our previous study (40). Six unfolding simulations were carried out at 500 K using the SANDER module in AMBER package (48) with ff99SB force field (20,49,50). These simulations are referred to as MD1 to MD6. To explore conformational sampling in the native state, a simulation was also carried out at 300 K. In each simulation, the protein was solvated in 4918 TIP3P water molecules (51) and neutralized by four sodium ions. Each simulation was prepared by energy minimization, with a harmonic restraint on the protein atoms gradually decreasing from 50 kcal/mol/Å2 to 0. For the native-state simulation, the energy-minimized structure was heated from 100 K to 300 K over 40 ps at constant volume. The simulation then continued at constant temperature and constant pressure to a total of 31 ns. For the high-temperature simulations, the energy-minimized structure was heated from 100 K to 500 K over 150 ps at constant volume, using different random number seeds. The simulations then continued at constant temperature and constant volume for a total of 15 ns each for MD1 to MD5 but for a total of 16 ns for MD6; the additional 1 ns of simulation is used to represent the unfolded ensemble (see below). The SHAKE algorithm (52) was used to constraint bonds involving hydrogen atoms. The timesteps were 2 fs for the 300-K simulation and 1 fs for the 500-K simulations. A cutoff of 10 Å for nonbonded interactions was used, and the particle mesh Ewald method (53–55) was used to treat the long range electrostatic interactions. After equilibration, the mass density was ∼1.0 g/cm3 and 0.76 g/cm3 in the 300-K and 500-K simulations, respectively. Protein conformations were sampled every 1 ps for analysis.
Specification of transition-state, unfolded, and native ensembles
The Φ-values indicate that helices I and IV-C are unfolded and helices II, III, and IV-N are partially formed in the rate-limiting transition state (8,16). Accordingly we specified the transition-state ensemble in our simulations as comprised of all conformations in which helices I and IV-C have less than three residues remaining helical whereas helices II, III, and IV-N have at least 50% of their residues remaining helical. Specifically, helices II, III, and IV-N must have at least 9, 12, and 6 residues, respectively, still helical. This empirical definition yielded a total of 256 conformations from the six unfolding simulations. The secondary structures of individual residues in the protein conformations were assigned according to the DSSP program (56). A residue was classified as helical if the DSSP output was H and nonhelical otherwise. In the DSSP program, a helix is considered to be comprised of at least three consecutive residues. Our intention of limiting the number of helical residues to less than three for helices I and IV-C was to make these two helices completely unfolded in the transition-state ensemble. Either helix could still have a small helical content (as in fact is the case) because the first or last one or two residues of the helix can be part of a longer helical stretch that goes beyond the N or C-terminal of the helix. Of course the residues beyond the N or C-terminal of the helix are not helical in the native state.
The unfolded and native ensembles were already used in our previous study on the effects of crowding on the folding stability of Rd-apocyt b562 (57). The unfolded ensemble consisted of the 1000 conformations from the additional 1 ns of MD6 after 15 ns of simulation. The native ensemble consisted of 1000 conformations sampled at 10 ps intervals from 6 to 16 ns of the native-state simulation.
Calculation of crowding effects
We have developed a postprocessing approach for simulating crowding (57,58). In the conventional approach of direct simulations (59–63), the test protein and a number of crowders are mixed and their motions are followed simultaneously. In our approach, we simulate the motion of the test protein in a particular state (e.g., the native state or the unfolded state), and the effects of crowding on the free energy of this state are predicted by the transfer free energy when representative protein conformations are placed inside a box of crowders. The approach is similar in spirit to Widom's particle insertion method (64). The postprocessing approach holds a number of advantages over direct simulations, leading to gain in calculation speed and accuracy (58). In particular, in all direct simulations, test proteins have always been represented at a coarse-grained level. In contrast, in our approach the simulation of the test protein is done in the absence of crowders and is restricted to a particular state. Therefore we can afford to represent the protein at an atomic level and exhaustively sample the state of interest.
The postprocessing approach has been applied to the native state and unfolded state of Rd-apocyt b562 to predict the effects of crowding on the folding stability (57). Here we applied it to the transition-state ensemble as well, yielding predictions for the effects of crowding on the folding kinetics. As before (57), we calculated crowding effects for a range of crowder sizes and a range of crowder concentrations. Specifically, crowder radii were 15, 20, and 30 Å, and crowder volume fractions were 5%, 15%, 25%, and 35%. The crowding-induced change in chemical potential for each of the 256 conformations in the transition-state ensemble was Boltzmann-averaged to yield the change in the free energy of the transition-state ensemble by crowding. This change was finally combined with the counterparts of the unfolded state and native state to predict the effects of crowding on the folding and unfolding activation free energies.
Results and Discussion
The unfolding process
Fig. 2 displays the root mean-square deviations (RMSD) of the six high-temperature simulations and the native-state simulation from the starting NMR structure. The RMSD of the native-state simulation becomes steady in <1 ns and stays at relatively low values (<4 Å). In contrast, the RMSD of the high-temperature simulations show continued increase till the end of the 15 ns of simulation, reaching beyond 10 Å. At that point, the protein is mostly denatured (see below).
Figure 2.
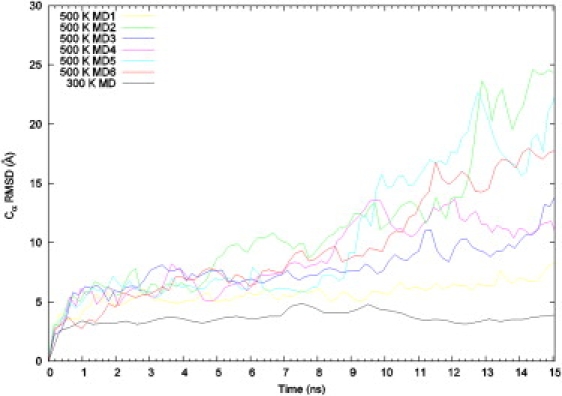
Root mean-square deviations of the simulations from the starting NMR structure, calculated over Cα atoms. The curves are smoothed using the bezier option in gnuplot. The RMSD of the native-state simulation remains at its low values all the way to the end of the 31-ns trajectory.
To gain further insight into the unfolding process, we used the DSSP program to calculate the secondary structures of individual residues along the six unfolding trajectories. The helical status of the residues as a function of the simulation time is displayed in Fig. 3. It is clear that, in all the six trajectories but MD1, the helices are melted by the end of the 15-ns simulations. Overall, helices I and IV-C melt relatively quickly, and helices II, III, and IV-N persist much longer before melting. In MD1, first helices I and IV-C and then III and IV-N melt, but helix II remains mostly intact.
Figure 3.
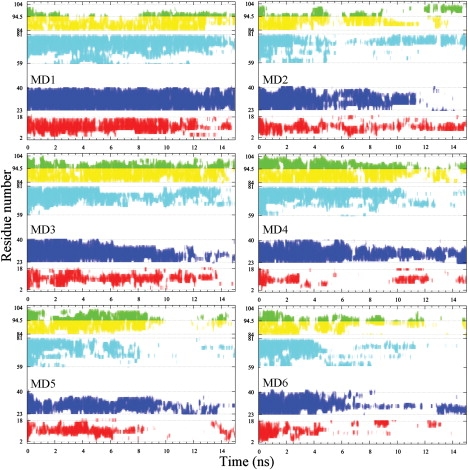
Helical status of the individual residues along the six high-temperature trajectories. Each helical residue is indicated by a vertical bar; the bars of the different helical segments have the same color code as in Fig. 1.
To be quantitative about the timing of helix melting, we calculated the mean first melting time. In each simulation, the first melting time of a particular helix was defined as the first time when 70% of its residues become melted. To prevent the calculation from being biased by the briefest excursions below the 70% threshold, the calculation of the first melting time was done after replacing the helical content at each time point by the running average over a 51-ps window. The calculation was then averaged over MD2 through MD6 (MD1 was excluded because helix II remains not completely melted by the end of the 15-ns simulation). The final results for the mean first melting times of helices I, II, III, IV-N, and IV-C are 1.4 ± 0.3, 7.9 ± 3.7, 5.7 ± 2.0, 7.2 ± 3.0, and 1.5 ± 1.1 ns. The conclusion is again that helices I and IV-C melt early, and after an extended period helices III, IV-N and II then start to melt in that order. Our simulations seem to suggest that there are two distinct unfolding events, one is the melting of the apparently weaker helices I and IV-C, the other is the melting of the apparently stronger helices II, III, and IV-C. The conformations sampled between these two events can be identified as constituting PUF1 (see below).
Unfolding pathway and helix propensities
To further quantify the strengths of the helices to withstand high temperature, we calculated the fraction of time that each residue is in the helical state during the 15-ns simulation. The profile of the helical fractions of the individual residues captures the relative stability of the difference helices. Similar profiles are obtained from the separate simulations, but the magnitudes are somewhat different, reflecting the different paces of unfolding of the separate simulations. We scaled these profiles to the same maximum and averaged the results over MD2 through MD6 (again MD1 was excluded because of the incomplete melting of helix II). The composite profile of helical fractions is shown in Fig. 4. The peaks of helical fractions within the helices follow the order I < IV-C < III < IV-N < II. Not surprisingly, this order is the same as that of the mean first melting times.
Figure 4.
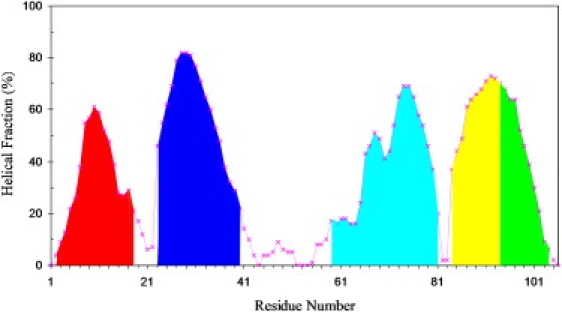
Composite profile of helical fractions calculated over the course of five high-temperature simulations. The areas above the individual helices of the native structure are separately shaded.
Based on the above analysis, we may conclude that the unfolding pathway in our simulations is
| (2) |
where MI denotes intermediate state in which helix I is melted and other helices are still intact; the symbols for the other intermediates can be similarly interpreted. On reversal, this pathway corresponds well with folding scheme 1, proposed by Chu et al. (8) and Zhou et al. (16) based on their experimental studies. In particular, MI and MI;IV-C correspond to PUF2 and PUF1, respectively. The transition state is passed when the other three helices are melted. We will return to the specification of the transition-state ensemble below.
What determines the relative stability of the helices to withstand high temperature and hence their order of melting? Our first instinct was that the intrinsic helix propensities dictated by the sequences play a role. To test this idea, we used the AGADIR program (http://agadir.crg.es/) to predict the helical contents from the corresponding sequences of the individual helical segments. The predictions are displayed in Fig. 5. They show surprisingly good match with the profile in Fig. 4. In particular, helices I and IV-C show lower peaks than helices II, III, and IV-C. Within each helix, the peaks occur at roughly the same residue positions in the two figures. For helix III, both figures show lower helical content in the N-terminal half than in the C-terminal half. Therefore intrinsic helix propensities seem to be a major determinant of the order of helix melting during the unfolding simulations. We caution, however, that a direct read-out of folding signals from protein sequence is challenging and no simple answer is expected. In this context, it is noted that the helix propensities of the sequence segments are all relatively low (<25%), and potentially can be overridden by other factors such as tertiary interactions (see below).
Figure 5.
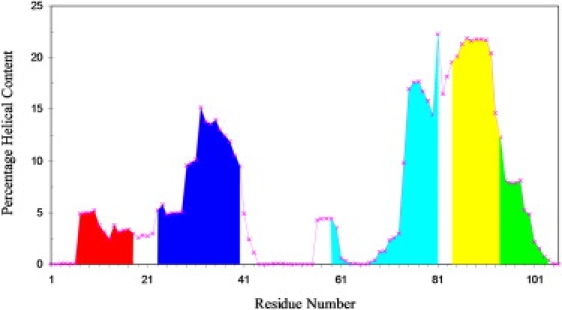
Intrinsic helix propensities predicted from the Rd-apocyt b562 sequence by the AGADIR program. Parameters used in the predictions were: 274 K; 0 ion strength; and pH 7. The areas above the individual helices of the native structure are separately shaded.
The initiation of the unfolding process is of interest. In most of the simulations, the hydrophobic core formed by W7, P45, P46, I98, Y101, and Y105 is broken early. The side chain of Y101 rotates outward, toward helix III, followed by a similar rotation of the side chain of Y105. These rotations break their interactions with W7, P45, and P46. Soon after, the side chain of W7 interacts with that of I98 for sometime, whereas the N-terminal half of helix I and the C-terminal half of helix IV become frayed, further exposing the hydrophobic core to the solvent. The unfolding then proceeds to form the MI and MI;IV-C intermediates.
It is worth noting that Feng et al. (10) have detected an additional state, denoted by N∗, in equilibrium unfolding studies using urea denaturation. The N∗ state has the native fold but nonnative hydrophobic interactions. Along the folding pathway, N∗ is placed between PUF2 and N. The main difference between N and N∗ is in hydrophobic core packing: in N, Y101 and Y105 interact with W7; but in N∗, Y101 and Y105 face outward and become closer to helix III, and W7 interacts with I98. These signatures of N∗ and its placement between N and PUF2 are just as observed during the initiation of the unfolding process, as outlined in the preceding paragraph. Feng et al. (15) have further detected nonnative hydrophobic interactions in PUF2. We observe very similar nonnative interactions in our MI intermediate, e.g., between F65 and F69, and between L68 and I98.
Transition-state ensemble
As described above, the unfolding of Rd-apocyt b562 consists of two distinct events: the early melting of helices I and IV-C and the melting of the remaining helices after a relatively long delay. Kinetically it seems logical to associate the crossing of the rate-limiting transition state with the second event. (This association is consistent with a suggestion made in our previous study using high-temperature simulations to characterize folding and unfolding pathways (40): the last remaining secondary structures should be part of the transition state for folding.) The Φ-values also indicate that helices I and IV-C are unfolded and helices II, III, and IV-N are partially formed in the transition state (8,16) (Fig. 6). These observations led us to define the transition-state ensemble as comprised of all conformations in which helices I and IV-C have less than three helical residues whereas helices II, III, and IV-N have at least 50% of helical residues. A total of 256 such conformations are collected from the six unfolding simulations. On average, the percentages of helical residues in the transition-state ensemble are (0.6 ± 2.5)%, (78.6 ± 16.0)%, (67.5 ± 13.0)%, (78.1 ± 16.8)%, and (6.6 ± 8.2)% for helices I, II, III, IV-N, and IV-C, respectively. The significant variances of the percent helicities give an indication of the diversity of the transition-state ensemble.
Figure 6.
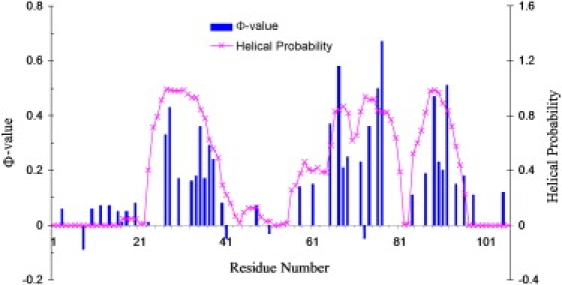
Comparison residue helix probabilities of the transition-state ensemble extracted from the simulations and Φ-values (8,16).
To assess this putative transition-state ensemble, we calculated the helical probabilities of individual residues (i.e., the percentage of times that a given residue is in the helical state in the 256-conformaiton ensemble). In Fig. 6 we compare the helical probabilities with the experimental Φ-values. A strong correlation is observed. As expected, the very low Φ-values of helices I and IV-C and the relatively high Φ-values of helices II, III, and IV-N are mapped by the corresponding disparities in helical probability among the different helices. Furthermore, the peak positions of Φ-values within helices II, III, and IV-N are well reproduced by the helical probabilities. Note that the correlation between the helical probabilities and the Φ-values is not intended as showing agreement between calculation and experiment; rather, it is meant to confirm that our putative transition-state ensemble, with atomic details not accessible to experiments, is quantitatively validated by Φ-value analysis.
To find a conformation that may serve as a representative of the transition-state ensemble, we carried out a cluster analysis according to RMSD of Cα atoms (65). Using a 8-Å RMSD cutoff, the 256 conformations are found to form a major cluster, including 207 conformations (81% of the total), and several much smaller clusters. The representative of this cluster is shown in Fig. 7. In this structure, helices II and III are still mostly intact, but they are no longer parallel as they are in the native state (see Fig. 1). As a result, these helices lose some of their native interactions. Our structural interpretation of the experimental observation that even the highest Φ-values, occurring in helices II, III, and IV-C, are significantly less than one is that it does not necessarily mean these helices are partially or largely unfolded in the transition-state ensemble. Rather, these helices may still be mostly intact, but their relative orientations are altered, thereby losing some of their native interactions, as illustrated by the representative transition-state structure of Fig. 7.
Figure 7.
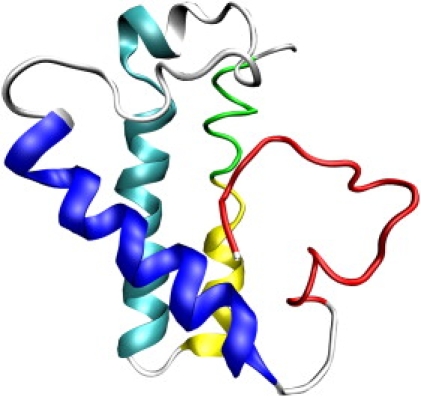
Structure of a representative of the transition-state ensemble.
From the transition-state ensemble, further unfolding is fast. Additional native interactions are lost as helices II and III further change their relative orientation and separation. Thereafter helix III starts to melt from the N-terminal half.
Macromolecular crowding
Although our proposed unfolding pathway and transition-state ensemble seem to rationalize a host of experimental observations, it remains important to find independent validation of the putative transition-state ensemble. To that end, we calculated the effects of macromolecular crowding on the free energy of the transition-state ensemble, following our previous work presenting similar calculations on the unfolded and native states (57).
In Fig. 8, we display the predicted effects of crowding on the folding and unfolding activation free energies, for a range of crowder radii (15–30 Å) and a range of crowder volume fractions (5–35%). Crowding is seen to exert modest stabilization of the transition-state ensemble relative to the unfolded state, and vanishingly small destabilization of the transition-state ensemble relative to the native state. These results are in qualitative agreement with the measurements by Ai et al. (18), who found that the addition of 85 g/l of PEG 20K results in a modest increase in the folding rate of Rd-apocyt b562 but essentially no effect on the unfolding rate. Specifically, an increase of 45% in folding rate and a decrease of 6% in unfolding rate are observed at 25°C. Such effects are predicted by our calculation for crowders with a 20-Å and occupying 15% of volume. Atomistic representation of the crowding agent in future work will make the comparison more quantitative.
Figure 8.
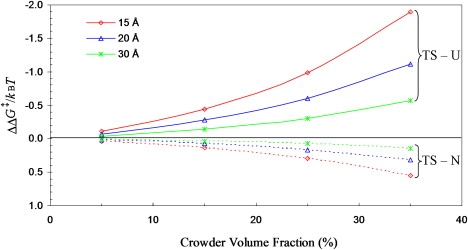
Effects of crowding on the folding and unfolding activation free energies, ΔΔG‡TS-U and ΔΔG‡TS-N, in units of kBT where kB is Boltzmann constant and T is the absolute temperature. Results for three crowder radii (with values shown) are displayed.
In our calculation, the crowding-induced decrease in folding activation free energy originates from the fact that the transition-state conformations are more compact than the unfolded conformations. In general, compact conformations are less destabilized by crowding than open conformations. The modest increase in folding rate by the addition of PEG 20K is thus an indication that the conformations of Rd-apocyt b562 experience a compaction when it moves from the unfolded state to the rate-limiting transition state. Similarly, the negligible effect of PEG 20K on the folding rate is an indication that the transition state is not significantly less compact than the native state. The experimental results on crowding effects thus provide both qualitative and quantitative support of our putative transition-state ensemble. Use of different crowding agents and their mixtures (66) will further increase the discriminatory power of macromolecular crowding as a probe of transition-state ensembles.
Conclusions
By high-temperature simulations, we observed the following unfolding pathway of Rd-apocyt b562: helices I and IV-C melt quickly, and after an extended delay, helices III, IV-N, and II start to melt; the melting of the latter helices finally unfold the protein completely. The reversal of the unfolding pathway can be proposed as the folding pathway. It seems that the folding/unfolding pathway is governed mainly by intrinsic helix propensity, whereby helices with higher helix propensities form first in the folding process. Our simulation results reproduce well experimental observations (8,16,17) and provide a rationalization. In addition to the folding/unfolding pathway, the simulations yield an atomic representation of the transition-state ensemble. The putative transition-state ensemble can be further validated, as we do here by calculation of crowding effects. We show that a modest increase in folding rate and null effect on the unfolding rate by the addition of a crowding agent (18) both qualitatively and quantitatively support the transition-state ensemble extracted from the simulations. MD simulations combined with calculation of crowding effects thus provide an avenue for characterizing the transition-state ensemble in atomic details.
Acknowledgments
This work was supported in part by the National Institutes of Health (grant No. GM058187).
Footnotes
Harianto Tjong's present address is Department of Biological Sciences, University of Southern California, Los Angeles, CA.
References
- 1.Bai Y., Englander S.W. Future directions in folding: the multi-state nature of protein structure. Proteins. 1996;24:145–151. doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 2.Bai Y. Energy barriers, cooperativity, and hidden intermediates in the folding of small proteins. Biochem. Biophys. Res. Commun. 2006;340:976–983. doi: 10.1016/j.bbrc.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 3.Bai Y., Sosnick T.R., Englander S.W. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chamberlain A.K., Handel T.M., Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Mol. Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 5.Fuentes E.J., Wand A.J. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry. 1998;37:9877–9883. doi: 10.1021/bi980894o. [DOI] [PubMed] [Google Scholar]
- 6.Llinas M., Gillespie B., Marqusee S. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Mol. Biol. 1999;6:1072–1078. doi: 10.1038/14956. [DOI] [PubMed] [Google Scholar]
- 7.Fersht A.R. A kinetically significant intermediate in the folding of barnase. Proc. Natl. Acad. Sci. USA. 2000;97:14121–14126. doi: 10.1073/pnas.260502597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu R., Pei W., Bai Y. Relationship between the native-state hydrogen exchange and folding pathways of a four-helix bundle protein. Biochemistry. 2002;41:7998–8003. doi: 10.1021/bi025872n. [DOI] [PubMed] [Google Scholar]
- 9.Yan S., Kennedy S.D., Koide S. Thermodynamic and kinetic exploration of the energy landscape of Borrelia burgdorferi OspA by native-state hydrogen exchange. J. Mol. Biol. 2002;323:363–375. doi: 10.1016/s0022-2836(02)00882-3. [DOI] [PubMed] [Google Scholar]
- 10.Feng H., Vu N.-D., Bai Y. Detection and structure determination of an equilibrium unfolding intermediate of Rd-apocytochrome b562: native fold with non-native hydrophobic interactions. J. Mol. Biol. 2004;343:1477–1485. doi: 10.1016/j.jmb.2004.08.099. [DOI] [PubMed] [Google Scholar]
- 11.Vu N.-D., Feng H., Bai Y. The folding pathway of barnase: the rate-limiting transition state and a hidden intermediate under native conditions. Biochemistry. 2004;43:3346–3356. doi: 10.1021/bi0362267. [DOI] [PubMed] [Google Scholar]
- 12.Feng H., Vu N.-D., Bai Y. Detection of a hidden folding intermediate of the third domain of PDZ. J. Mol. Biol. 2005;346:345–353. doi: 10.1016/j.jmb.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 13.Feng H., Bai Y. Repacking of hydrophobic residues in a stable mutant of apocytochrome b562 selected by phage-display and proteolysis. Proteins. 2004;56:426–429. doi: 10.1002/prot.20161. [DOI] [PubMed] [Google Scholar]
- 14.Takei J., Pei W., Bai Y. Populating partially unfolded forms by hydrogen exchange-directed protein engineering. Biochemistry. 2002;41:12308–12312. doi: 10.1021/bi026491c. [DOI] [PubMed] [Google Scholar]
- 15.Feng H., Takei J., Bai Y. Specific non-native hydrophobic interactions in a hidden folding intermediate: implications for protein folding. Biochemistry. 2003;42:12461–12465. doi: 10.1021/bi035561s. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z., Huang Y., Bai Y. An on-pathway hidden intermediate and the early rate-limiting transition state of Rd-apocytochrome b562 characterized by protein engineering. J. Mol. Biol. 2005;352:757–764. doi: 10.1016/j.jmb.2005.07.057. [DOI] [PubMed] [Google Scholar]
- 17.Feng H., Zhou Z., Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc. Natl. Acad. Sci. USA. 2005;102:5026–5031. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ai X., Zhou Z., Choy W.Y. 15N NMR spin relaxation dispersion study of the molecular crowding effects on protein folding under native conditions. J. Am. Chem. Soc. 2006;128:3916–3917. doi: 10.1021/ja057832n. [DOI] [PubMed] [Google Scholar]
- 19.Snow C.D., Nguyen H., Gruebele M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 2002;420:102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 20.Simmerling C., Strockbine B., Roitberg A.E. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 2002;124:11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 21.Chowdhury S., Lee M.C., Duan Y. Ab initio folding simulation of the Trp-cage mini-protein approaches NMR resolution. J. Mol. Biol. 2003;327:711–717. doi: 10.1016/s0022-2836(03)00177-3. [DOI] [PubMed] [Google Scholar]
- 22.Pitera J.W., Swope W. Understanding folding and design: replica-exchange simulations of “Trp-cage” miniproteins. Proc. Natl. Acad. Sci. USA. 2003;100:7587–7592. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei H., Duan Y. Two-stage folding of HP-35 from ab initio simulations. J. Mol. Biol. 2007;370:196–206. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L., Grubb M.P., Gao Y.Q. Application of the accelerated molecular dynamics simulations to the folding of a small protein. J. Chem. Phys. 2007;126:125102–125107. doi: 10.1063/1.2709639. [DOI] [PubMed] [Google Scholar]
- 25.Ensign D.L., Kasson P.M., Pande V.S. Heterogeneity even at the speed limit of folding: large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J. Mol. Biol. 2007;374:806–816. doi: 10.1016/j.jmb.2007.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maragakis P., Lindorff-Larsen K., Shaw D.E. Microsecond molecular dynamics simulation shows effect of slow loop dynamics on backbone amide order parameters of proteins. J. Phys. Chem. B. 2008;112:6155–6158. doi: 10.1021/jp077018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freddolino P.L., Liu F., Schulten K. Ten-microsecond molecular dynamics simulation of a fast-folding WW domain. Biophys. J. 2008;94:L75–L77. doi: 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li A., Daggett V. Characterization of the transition state of protein unfolding by use of molecular dynamics: chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li A., Daggett V. Identification and characterization of the unfolding transition state of chymotrypsin inhibitor 2 by molecular dynamics simulations. J. Mol. Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 30.Lazaridis T., Karplus M. “New view” of protein folding reconciled with the old through multiple unfolding simulations. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 31.Li A., Daggett V. Molecular dynamics simulation of the unfolding of barnase: characterization of the major intermediate. J. Mol. Biol. 1998;275:677–694. doi: 10.1006/jmbi.1997.1484. [DOI] [PubMed] [Google Scholar]
- 32.Tsai J., Levitt M., Baker D. Hierarchy of structure loss in MD simulations of src SH3 domain unfolding. J. Mol. Biol. 1999;291:215–225. doi: 10.1006/jmbi.1999.2949. [DOI] [PubMed] [Google Scholar]
- 33.Gsponer J., Caflisch A. Role of native topology investigated by multiple unfolding simulations of four SH3 domains. J. Mol. Biol. 2001;309:285–298. doi: 10.1006/jmbi.2001.4552. [DOI] [PubMed] [Google Scholar]
- 34.Day R., Bennion B.J., Daggett V. Increasing temperature accelerates protein unfolding without changing the pathway of unfolding. J. Mol. Biol. 2002;322:189–203. doi: 10.1016/s0022-2836(02)00672-1. [DOI] [PubMed] [Google Scholar]
- 35.Daggett V. Molecular dynamics simulations of the protein unfolding/folding reaction. Acc. Chem. Res. 2002;35:422–429. doi: 10.1021/ar0100834. [DOI] [PubMed] [Google Scholar]
- 36.Day R., Daggett V. Ensemble versus single-molecule protein unfolding. Proc. Natl. Acad. Sci. USA. 2005;102:13445–13450. doi: 10.1073/pnas.0501773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snow C.D., Sorin E.J., Pande V.S. How well can simulation predict protein folding kinetics and thermodynamics? Annu. Rev. Biophys. Biomol. Struct. 2005;34:43–69. doi: 10.1146/annurev.biophys.34.040204.144447. [DOI] [PubMed] [Google Scholar]
- 38.White G.W.N., Gianni S., Daggett V. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation-condensation to framework mechanism of folding. J. Mol. Biol. 2005;350:757–775. doi: 10.1016/j.jmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Daggett V. Protein folding-simulation. Chem. Rev. 2006;106:1898–1916. doi: 10.1021/cr0404242. [DOI] [PubMed] [Google Scholar]
- 40.Huang X., Zhou H.-X. Similarity and difference in the unfolding of thermophilic and mesophilic cold shock proteins studied by molecular dynamics simulations. Biophys. J. 2006;91:2451–2463. doi: 10.1529/biophysj.106.082891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eleftheriou M., Germain R.S., Zhou R. Thermal denaturing of mutant lysozyme with both the OPLSAA and the CHARMM force fields. J. Am. Chem. Soc. 2006;128:13388–13395. doi: 10.1021/ja060972s. [DOI] [PubMed] [Google Scholar]
- 42.Das A., Mukhopadhyay C. Application of principal component analysis in protein unfolding: an all-atom molecular dynamics simulation study. J. Chem. Phys. 2007;127:165103. doi: 10.1063/1.2796165. [DOI] [PubMed] [Google Scholar]
- 43.Beck D.A.C., Daggett V. A one-dimensional reaction coordinate for identification of transition states from explicit solvent P(fold)-like calculations. Biophys. J. 2007;93:3382–3391. doi: 10.1529/biophysj.106.100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Settanni G., Fersht A.R. High temperature unfolding simulations of the TRPZ1 peptide. Biophys. J. 2008;94:4444–4453. doi: 10.1529/biophysj.107.122606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L., Shao Q., Gao Y.Q. Thermodynamics and folding pathways of trpzip2: an accelerated molecular dynamics simulation study. J. Phys. Chem. B. 2009;113:803–808. doi: 10.1021/jp803160f. [DOI] [PubMed] [Google Scholar]
- 46.Freddolino P.L., Park S., Schulten K. Force field bias in protein folding simulations. Biophys. J. 2009;96:3772–3780. doi: 10.1016/j.bpj.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fersht A.R., Matouschek A., Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 48.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco, CA: 2004. AMBER 8. [Google Scholar]
- 49.Wang J.M., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 50.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 52.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 53.Darden T., York D., Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 54.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 55.Sagui C., Darden T.A. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 57.Qin S., Zhou H.-X. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys. J. 2010;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qin S., Minh D.D., Zhou H.-X. A method to predict crowding effects by postprocessing molecular dynamics trajectories: application to the flap dynamics of HIV-1 protease. J Phys. Chem. Lett. 2009;1:107–110. doi: 10.1021/jz900023w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheung M.S., Klimov D., Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc. Natl. Acad. Sci. USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stagg L., Zhang S.Q., Wittung-Stafshede P. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc. Natl. Acad. Sci. USA. 2007;104:18976–18981. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mittal J., Best R.B. Theory and simulation of macromolecular crowding effects on protein folding stability and kinetics. Biophys. J. 2009 Accepted. [Google Scholar]
- 62.Pincus D.L., Thirumalai D. Crowding effects on the mechanical stability and unfolding pathways of ubiquitin. J. Phys. Chem. 2009;113:359–368. doi: 10.1021/jp807755b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Minh D.D., Chang C.E., McCammon J.A. The influence of macromolecular crowding on HIV-1 protease internal dynamics. J. Am. Chem. Soc. 2006;128:6006–6007. doi: 10.1021/ja060483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Widom B. Some topics in the theory of fluids. J. Chem. Phys. 1963;39:2802–2812. [Google Scholar]
- 65.de Vries S.J., van Dijk A.D.J., Bonvin A.M.J.J. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 66.Batra J., Xu K., Zhou H.X. Nonadditive effects of mixed crowding on protein stability. Proteins. 2009;77:133–138. doi: 10.1002/prot.22425. [DOI] [PMC free article] [PubMed] [Google Scholar]