

Abstract
The atomic models of the Kv1.2 potassium channel in the active and resting state, originally presented elsewhere, are here refined using molecular dynamics simulations in an explicit membrane-solvent environment. With a minor adjustment of the orientation of the first arginine along the S4 segment, the total gating charge of the channel determined from >0.5 μs of molecular dynamics simulation is ∼12–12.7 e, in good accord with experimental estimates for the Shaker potassium channel, indicating that the final models offer a realistic depiction of voltage-gating. In the resting state of Kv1.2, the S4 segment in the voltage-sensing domain (VSD) spontaneously converts into a 310 helix over a stretch of 10 residues. The 310 helical conformation orients the gating arginines on S4 toward a water-filled crevice within the VSD and allows salt-bridge interactions with negatively charged residues along S2 and S3. Free energy calculations of the fractional transmembrane potential, acting upon key charged residues of the VSD, reveals that the applied field varies rapidly over a narrow region of 10–15 Å corresponding to the outer leaflet of the bilayer. The focused field allows the transfer of a large gating charge without translocation of S4 across the membrane.
Introduction
Voltage-gated potassium (Kv) channels are membrane proteins that respond to changes in the transmembrane potential by altering their conformation to allow the passage of K+ ions across the cell membrane. These channels are tetrameric proteins, in which each subunit comprises six transmembrane helical segments (S1–S6). The first four helical segments (S1–S4) in each subunit form a voltage-sensing domain (VSD) that is located in the lipid membrane at the periphery of the central pore domain. The ion conduction pore is located at the center of the tetrameric pore domain, formed by the S5 and S6 helices from the four subunits. Upon depolarization of the membrane, the VSD in each subunit undergoes a voltage-dependent transition from a resting to an active conformation, which then leads to the opening of the intracellular gate of the ion conduction pore (1–3).
The conformational changes associated with the activation of Kv channels result in the transfer of an electric charge ΔQ across the membrane, which can be measured experimentally as a small transient capacitive current (4,5). In the Shaker K+ channel, the gating charge corresponds to the transfer of 12–14 elementary charge (e) across the transmembrane electric field (6,7). Correspondingly, a change V in the membrane potential shifts the relative free energy of the closed and open conformations by VΔQ. The gating charge ΔQ is, thus, a key concept to explain how channel activation is coupled to the membrane potential. Initially postulated by Hodgkin and Huxley in 1952, the gating charge was first detected and measured more than 20 years later by Armstrong and Bezanilla (4).
Structural information about the different conformational states of K+ channels is a prerequisite to explain the mechanism of voltage-gating. Crystallographic studies have provided atomic resolution structures of KvAP (8), Kv1.2 (9), and a Kv1.2/Kv2.1 chimera (10) in their putative open conformation. The VSD, where the voltage function sensing lies, is a small bundle of four antiparallel helices (S1–S4) packed in a counterclockwise fashion (seen from the extracellular side). Four highly conserved arginine residues, located at every third position along the S4 segment, underlie the dominant contributions to the total gating charge (7,11). Negatively charged amino acids on S2 and S3 also affect voltage-sensing, but to a lesser degree. The discovery of similar domains in unrelated proteins lacking a conduction pore has established the concept of the VSD as an independent functional module (12–14).
Voltage-dependent changes in the conformation of the VSD have been detected in the Shaker K+ channel (15–18), as well as in the KvAP channel (19,20). The observations gave rise to three mechanistic models for the voltage-sensing motion in Kv channels, namely the paddle model (20,21), the helical-screw model and its variants (22–24), and the transporter model (17). The magnitude of the predicted motion for the key S4 segment varies between 2 Å and 20 Å among these models. However, as no atomic-resolution crystal structure of a Kv channel in the closed/resting state is currently available, experimental studies in combination with modeling have sought to complement the missing structural information (16,17,19,20,25–30).
Ultimately, explaining the voltage-gating mechanism of Kv channels in molecular terms requires gaining knowledge of the active and resting conformations, and then showing how those conformations are able to account for the experimentally observed gating charge ΔQ. One possible purely computational approach is to simulate, directly, the transition from the (putatively open) x-ray structure to the closed/resting state with all-atom molecular dynamics (MD) under the influence of an applied negative membrane potential. Unsurprisingly, even with microsecond trajectories, previous efforts were able to simulate only small Ångstrom-scale structural response of the channel to an applied potential (31–33). No spontaneous and complete transition from the open to the closed channel state has been observed in brute-force all-atom MD simulations with explicit solvent and membrane. The difficulties are further compounded by the fact that the x-ray structure probably represents a VSD-relaxed conformation of the channel, which returns very slowly to the resting state at a negative potential (34). Simulations of the transition from an active to a resting state conformation is essentially out of reach for all-atom MD due to the slow dynamics of Kv channels (on the order of ∼10–100 ms).
An alternative strategy adopted here is to start from carefully constructed atomic models of both active and resting states that are consistent with all available experimental information using protein structure prediction algorithms (26). Recently, Pathak et al. (30) have generated detailed atomic models of Kv1.2 in the open/active and closed/resting states using the Rosetta-Membrane structure prediction program (26). The model of the open/active state complements the information missing from the x-ray structure of the Kv1.2 channel (9) used as a template, while the interhelical loops of the VSD S1–S2 were modeled de novo (26). Confidence in the modeling methodology was strengthened by the subsequent x-ray structure of the Kv1.2/Kv2.1 chimera channel, which confirmed that the model of the open/active state was accurate, especially in the transmembrane region. The model of the closed/resting state is more uncertain, although it is broadly constrained by a wide range of experimental data (see discussion in (30)). Remarkably, the model was found to be consistent with two metal bridges engineered experimentally to stabilize the closed state, which were subsequently established in an independent study (27). The resulting models suggest a voltage-activated transition from the closed/resting to the open/active state in which the S4 segment moves outward by ∼6–8 Å relative to the membrane and rotates clockwise along its main axis as viewed from the extracellular side. The gating charge ΔQ for this transition was calculated to be 14e (30) using a continuum dielectric approximation based on a modified Poisson-Boltzmann theory (35).
These results offer a promising starting point to expand our understanding of voltage gating in Kv channels. However, some critical issues must be addressed. In particular, the stability of the structural models in the complex dynamical environment of the lipid bilayer has not been ascertained. Furthermore, the gating charge associated with the models was only evaluated within a continuum approximation where water and membrane were treated as featureless dielectric media. There are strong suggestions that the hydration of the VSD is complex, with narrow aqueous crevices, where a representation of the water molecules as a featureless continuum may be questionable. In addition, it is known that lipid membranes are necessary for the stability and function of the protein, and several studies have suggested that direct salt-bridge interactions between the charged residues of the VSD and the lipid phosphate headgroups were implicated in the function of Kv channels (36,37). This raised fundamental questions regarding the possibility of a direct participation of the lipids molecules in the voltage-gating process. These issues cannot be addressed without using all-atom models with explicit solvent and membrane.
Our goal is to refine the atomic models of the closed/resting and open/activated states and test their ability to account for the experimentally observed gating charge using all-atom MD and an explicit representation of the lipid membrane environment. We have performed extensive computations on the Kv1.2 channel, for a total aggregate simulation time longer than 1.3 μs. The refined models are consistent with a wide range of experimental observations, and proved to be stable in the presence of external voltage biases (lasting >50 ns each). The models are validated by carrying out explicit all-atom MD calculations of the gating charge for the full-length and isolated VSD of Kv1.2. The contribution of individual VSD residues to the gating charge is determined via free energy perturbation (FEP). The results show that the hydration in the interior of the VSD causes a local focusing of the transmembrane electric field and that, for this reason, movement of the S4 segment over a distance of ∼7 Å is sufficient to account for the expected gating charge.
Methods
Calculations of the gating charge
The gating charge ΔQ represents the strength of the coupling between the applied membrane potential and the conformational changes of the channel. When the voltage changes by ΔV, the relative free energy of the closed and open conformations shifts by ΔQΔV. Three theoretical methods (Q-route, G-route, and W-route) have been formulated to characterize how a membrane protein is coupled to an applied voltage (38), the first two routes being employed here. The most straightforward estimate of ΔQ is obtained by considering averages of the linearly weighted displacement charge Qd within the simulation system (the Q-route),
| (1) |
where Lz is the length of the periodic box in the z direction, qi is the partial charge of atom i, N is the number of atoms in the system, and zui is the unwrapped coordinate of atom i along the z axis (i.e., not folded back within the interval [0, Lz] corresponding to the limits of the periodic system along z). The notation 〈⋯〉s, V stands for the ensemble average with channel state s and imposed voltage V, which is evaluated as the mean value over the time series from the corresponding equilibrium simulations. The sum over i in Eq. 1 runs over all atoms in the system, including the protein, the membrane lipids, the solvent, and the counterions. It can be shown that, as in experimental measurements of the gating charge (4,5), the shift in the average displacement charge as expressed in Eq. 1 tracks the net capacitive charge ΔQ flowing through the external circuit comprising the EMF and the electrodes when the conformation of the channel converts from the closed (c) to the open (o) state (38),
| (2) |
For a given state s, the average displacement charge varies linearly as 〈Qd〉c, 0 + CV, where C is the capacitance of the system. The gating charge ΔQ is the offset constant between the two linear relations.
Averages of the displacement charge were computed from 50-ns trajectories. The autocorrelation function of the displacement charge relaxes in <10 ns for the VSD and full-channel simulations, indicating that our trajectories are sufficiently long to obtain statistically converged averages. To avoid a residual effect of the previous runs on 〈Q〉s, V (after abrupt changes in V), snapshots from the first 10 ns of the simulations are not included in the calculation of 〈Q〉s, V.
A detailed decomposition of the contribution from individual residues to the total gating charge can be achieved by considering the charging free energies of a residue as a function of applied membrane potential and protein conformation (the G-route) (38). Accordingly, the contribution of a residue carrying a charge qi to the total gating charge ΔQ is expressed as qi[fc(i) − fo(i)], where the dimensionless quantities fs(i)
| (3) |
correspond to the fraction of the transmembrane potential acting upon residue i when the channel is in the open-active (s=o) and closed-resting (s=c) conformations, respectively. The expression ΔGs(V, qi) is the charging free energy of the residue i with charge qi with the channel in state s and applied voltage V. Details of the derivation are given in (38). Equation 3 is obtained perturbatively by recognizing that the total free energy of the system comprises an intrinsic contribution arising from the molecular environment affecting a residue even in the absence of an applied voltage, and a contribution reflecting the coupling to the applied membrane potential V. To lowest order in V, the latter is linear and can be estimated by calculating the charging free energy of a residue at two different voltages V1 and V2. Calculating fo(i) and fc(i) necessitates free energy perturbation molecular dynamics (FEP/MD) simulations at two different membrane voltages for each residue that is probed. Details of the FEP/MD calculations are given in the Supporting Material.
Molecular dynamics simulations
All MD simulations were carried out using the program NAMD (39) and the CHARMM27 force field for proteins (40,41), ions (42), and phospholipids (43,44), with the TIP3P water model (45). All simulations were performed with periodic boundary conditions at constant temperature and constant pressure, with a fixed cross-sectional area. The computation of long-range electrostatic forces was done with the particle-mesh Ewald technique (46). Langevin dynamics with a very weak friction coefficient was used to keep the temperature constant. The Langevin Nosé-Hoover method (47) was used to maintain the pressure at 1 atm. The membrane potential was emulated by applying a constant electric field (normal to the plane of membrane) to every atom carrying a partial or a full charge in the system (38). The simulations of the full channel involved ∼350,000 atoms and covered altogether 805 ns (including FEP simulations); simulations of individual VSDs involved ∼94,000 atoms and covered altogether 515 ns (including FEP simulations).
Results
Refined Kv models in the resting and active states
The active and resting state models of Kv1.2 are refined using MD simulations of the full tetrameric channel and the isolated VSD in an explicit membrane-solvent environment. During equilibration simulations (50–100 ns each), water molecules hydrate the VSD and new contacts are formed between charged residues of the VSD to stabilize these highly charged protein domains inside the lipid bilayer. Snapshots of the full tetrameric channel and the isolated VSD are shown in Fig. 1 A and Fig. S1 in the Supporting Material, respectively.
Figure 1.
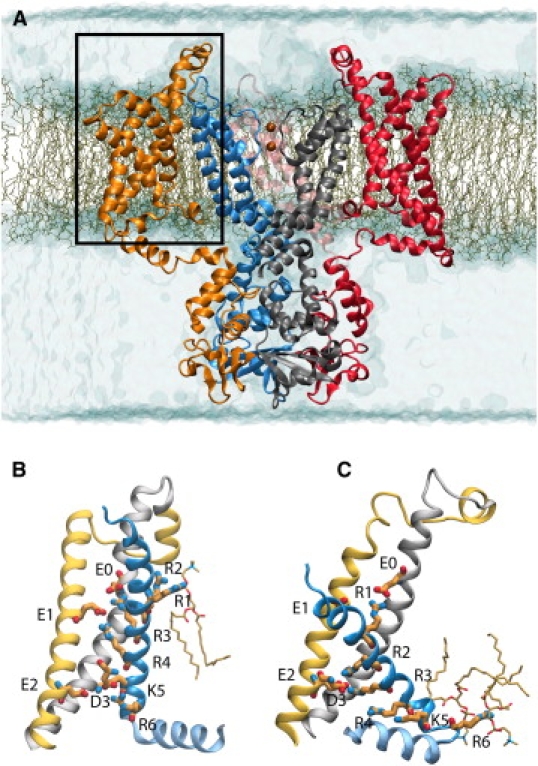
Kv1.2 in the open state embedded in a patch of DPPC lipid bilayer. The pore domain and the voltage sensor domains (VSD) of the tetrameric structure are shown for two of the four subunits. Each subunit is colored differently. Two K+ ions in the selectivity filter are shown. Water molecules are shown in transparent blue surface representation; lipid molecules are represented by lines. An individual VSD highlighted in panel A is shown in the active (B) and resting (C) state conformation. The S1, S2, and S4 segments are shown in silver, yellow, and blue, respectively, while the S3 segment is omitted for clarity. The S4–S5 linker connecting the VSD to the pore is colored in light blue. R1, R2, R3, and R4 (on S4), D3 (on S3), E1 and E2 (on S2), and E0 (on S1) are shown in licorice representation.
During equilibration simulations, the Cα root mean-square deviation with reference to the initial structures rapidly increases, but levels off during the last 10–20 ns of each trajectory (Fig. S3 A and Fig. S4 A). To assess the stability of the final conformations obtained from equilibration simulations, the Cα root mean-square fluctuations (RMSF) of each protein residue is calculated for four 50 ns-trajectories that followed the equilibration simulations. The RMSF profiles (Fig. S3 B and Fig. S4 B and C) show that the transmembrane helical region of the VSDs remain stable (RMSF < 3 Å). The peaks in the profile correspond to the S1–S2 loop, the N-terminal segment of S3, and the S3–S4 loop. The S1–S2 loop on the extracellular side of the membrane is more flexible in the active state of the VSD in comparison to the resting state. Previous simulations of an open state model of Kv1.2 have also identified the S1–S2 loop to be highly flexible (31). However, as shown in Fig. S3 and Fig. S4, the stability of S1 and S2 segments is not affected by the high mobility of the S1–S2 loop connecting these two transmembrane segments. The refined models of the VSD are shown in Fig. 1, B and C. Details of the structural models, in particular the gating charges, are given below.
Aqueous crevice at the center of the VSD
In all the simulations, water molecules diffuse from the bulk solution into the empty crevice at the center of the S1–S4 helical bundle, forming a narrow water-filled pore pinched near the center of the membrane. This is observed in the simulations of the active and resting conformations for both the full tetrameric channel and the isolated VSD. As a result, all the charged residues of the VSD become rapidly hydrated. The hourglass-shaped aqueous crevice running through the VSD is a recurring feature that has been observed in all previous MD simulations of Kv1.2 (31–33,48), as well as of isolated VSDs (49,50). The existence of an aqueous porelike region at the center of the S1–S4 helical bundle has experimentally been inferred by mutations enabling the rapid conduction of H+ and cations through the VSD (51,52).
Salt-bridge interactions within the VSD
Several salt-bridge interactions form spontaneously within the VSD during the simulations of the active and resting state conformations. Fig. 1, B and C, show snapshots of the resting and the active states of the VSD after equilibration and highlight the key basic and acidic residues: R294 (R1), R297 (R2), R300 (R3), and R303 (R4), K306 (K5), R309 (R6) along S4, E183 (E0) along S1, E226 (E1) and E236 (E2) along S2, and D259 (D3) along S3. Salt-bridge interactions within the VSD and between charged residues of the VSD and phospholipid headgroups are summarized in Table S4. In the active state conformation (Fig. 1 B), a salt-bridge is formed between R4 and E1. Transient interactions between R3 and E1 or E0 are also seen in all subunits. A network of salt-bridge interactions between E2 on S2, D3 on S3, and K5 is seen to be present in all the trajectories of the active state, shaping the interior structure and gluing together three helical segments of the VSD. Similar interactions were previously identified experimentally in the open state conformation of the homologous Shaker K+ channel (53,54), where they correspond to E283 (in S2) with R368 and R371 (in S4), and K374 (in S4) with E293 (in S2) and D316 (in S3).
In the resting state conformation (Fig. 1 C), R1, the first arginine along S4, interacts with E1 and E0. The latter is located near the extracellular end of S1 and corresponds to E247 in the Shaker K+ channel. Deeper inside the membrane, R3 forms a salt-bridge with E2 in two of the four subunits, and D3 in a third subunit. The forth gating arginine, R4, interacts with E2 in one of the four subunits. Most likely, the structural variations between the different VSDs and the absence of fourfold symmetry in the tetrameric channel configuration reflect an incomplete averaging in these simulations.
Phospholipid headgroups interact with the first two gating arginines, R2 and R1, in the active state conformation. In the resting state conformation, R6 forms salt-bridge interactions with the headgroups.
S4 converts to a 310 helix in the closed state simulation
In the resting conformation of the channel, the S4 segment, originally modeled as an α-helix by ROSETTA (26,30), converted spontaneously to a 310-helical conformation during MD. As shown in Fig. 2, a stretch of 10 residues on S4 fluctuate as a stable 310-helix during the resting state simulation of the full tetrameric channel. By virtue of the 310-helical configuration, the gating arginines (R1–R4) along S4 are aligned on the same helical face. Fig. 2 shows snapshots of the VSD in the active and resting state highlighting several charged residues of the VSD. In the resting state conformation, the R1–R4 side chains favorably point toward the aqueous crevice at the center of the VSD. Furthermore, as illustrated in Fig. 1, B and C, this configuration allows salt-bridge interactions with negatively charged residues in S2 (E1 and E2) and S3 (D3). It is worth noting that the CHARMM force field for a simple poly-alanine peptide in vacuum energetically favors the α-helix over the 310 helix. Therefore, the observed shift in the secondary structure of S4 is directly caused by the arginines seeking a more favorable local environment. The high propensity of the S4 segment to adopt a 310-helical conformation appears to be supported by two recent x-ray structures of other Kv channels (10,55) in which the inner-half of the S4 segment (∼11 residues) appears as a 310 helix, in the open state conformation.
Figure 2.

Extracellular view of the voltage-sensor domains in the active (A) and resting (B) state conformations, after equilibration. R1–R4 (S4), E0 (S1), E1 and E2 (S2), and D3 (S3) are shown. In the resting state conformation (B), the S4 segment adopts a 310 helical conformation, while in the active state (A), the S4 segment is α-helical. As a result, the gating arginines, R1–R4, located at every third position on S4, face the same side of the helix in the resting state, extending toward the central crevice of the VSD. In the active state conformation, only two of the four arginines, R3 and R4, extend to the central crevice of the VSD. The 310-helical propensity of S4 residues is shown for the active (C) and resting (D) state conformations. For comparison, the propensity of S1 residues is also shown for the active (E) and resting (F) state conformations. The 310-helical propensity for residue i is calculated as the probability of hydrogen-bond formation between residue i and residue i + 3.
Gating charge
Two independent theoretical methods were employed to characterize, quantitatively, the gating charge of the channel from all-atom MD simulations (38). In the first method (the Q-route based on Eqs. 1 and 2), the total gating charge is extracted from all-atom MD simulations by calculating the average displacement charge Qd as a function of the applied membrane voltage and the protein conformation. Changes in the displacement charge of the system between active and resting state conformations correspond to the gating charge of the channel. In addition, the individual contribution of key residues of the VSD to the gating charge was calculated by performing FEP/MD simulations (the G-route based on Eq. 3).
Fig. 3 shows the time-averaged displacement charge of the isolated VSD and the full-channel at different transmembrane potentials V. For the conformational state s, 〈Qd〉s,V varies as a function of the external voltage applied. For a sufficiently small membrane potential, the capacitive charge is a linear function of the applied voltage, CV, and thus, can be eliminated (38). The gating charge ΔQ is then calculated as the offset between the voltage-dependent displacement charge of the two channel states (active and resting). The magnitude of the gating charge calculated for the isolated VSD, and the full tetrameric channel, are 2.38 ± 0.07 and 10.25 ± 0.35e, respectively. The calculated gating charge for the refined models of the full channel is in good accord with the experimental estimates of 12–14e measured for the Shaker K+ channel (6,7,11). The reason why ΔQ might be slightly underestimated is discussed below.
Figure 3.

Displacement charge of the full tetrameric channel (top) and voltage-sensor domain (bottom) calculated from the active and resting state simulations. Each data point corresponds to the average displacement charge over 40 ns (40,000 snapshots) of simulation based on Eq. 1. The error bars at each point correspond to standard errors in the calculations.
The fraction of the transmembrane potential felt by specific key residues carrying a charge was calculated according to Eq. 3 using FEP/MD simulations. The calculations were performed for nine key residues, including six positively charged residues (R1, R2, R3, R4, K5, and R6), and three negatively charged residues (E1, E2, and D3) of the VSD. These residues are highly conserved in Kv channels, and their mutation affects voltage-sensing of the channel (7,11). The location of these residues is highlighted in Fig. 1, B and C. In the case of the full tetrameric channel, the FEP/MD simulations were performed on five snapshots, taken at 8-ns intervals from four 50-ns trajectories at +500 mV and −500 mV voltages that followed the equilibration simulations. For a given channel state, the electrostatic charging free energies, ΔGs(V, qi), were averaged over the five snapshots for residue i of charge qi at each voltage bias V. The standard deviation in the FEP/MD calculation represents local fluctuations of the residue side chain while the channel is in state s. The results are shown in Fig. 4; in all these figures, the potential on the extracellular side is kept grounded at V = 0.
Figure 4.
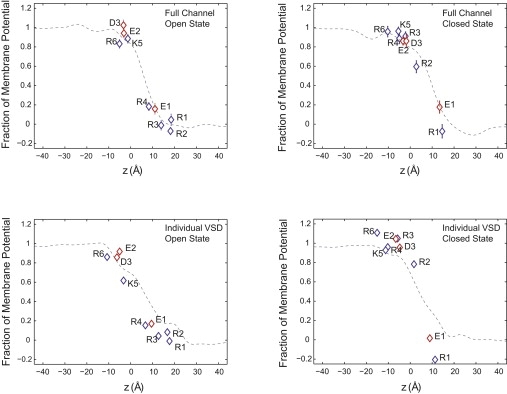
Fraction of the transmembrane potential at the position of key charged residues within the VSD, calculated for the full tetrameric channel (top) and the isolated VSD (bottom), using the G-route (38) based on Eq. 3. The position z corresponds to the average geometric center of the atomic partial charges of the residue normal to the membrane. The data for the basic residues (R1–R4, K5, and R6) are plotted in blue and for the acidic residues (E1, E2, and D3) in red. Thin dotted lines represent the transmembrane potential along a vertical line passing through the VSD, extracted from time-averaged electrostatic maps, using the PMEPOT plug-in (62) of VMD (63).
In the active state of the channel the three outermost arginines (R1, R2, and R3) are positioned near the extracellular solution at ∼0.1 of the transmembrane potential. Deeper inside the VSDs, E1 on S2 and R4 on S4 are located at ∼0.2 of the transmembrane potential. Further along S4, K5, is located at ∼0.6 of the potential in the isolated VSD, and at ∼0.9 of the potential in the full tetrameric channel. Near the intracellular membrane-solution interface are E2 on S2, D3 on S3, and R6 on S4, which are positioned within 0.8 of the transmembrane potential.
In the resting state of the channel, R1 is located at the membrane-solution interface, at the same transmembrane potential as the extracellular solution. E1 on the S2 segment is located near R1 at ∼0.2 of the transmembrane potential. R2 is positioned near the center of the bilayer at ∼0.8 of the potential in the isolated VSD, and at ∼0.4 of the potential in the full tetrameric channel. R3 and R4 are positioned near the (intracellular) membrane-solution interface, and are located at the same potential as the intracellular solution. E2 on S2, D3 on S3, and K5 and R6 on S4 are also positioned within 0.9 of the potential.
The contribution to the total gating charge ΔQ from residues carrying a charge qi can be calculated from the state-dependent fraction of transmembrane potential shown in Fig. 4 as qi[fo(i) − fc(i)]. Table 1 shows the contribution to the gating charge from nine key residues in the VSD. The calculations were performed for the isolated VSD and the full tetrameric channel. The results indicate that the main contributions to the gating charge are from the three basic residues of S4 (R2, R3, and R4). R1, the outermost arginine along S4, makes only a negligible contribution to the gating charge of the active and resting conformations tested here. Of the three acidic residues examined (E1, E2, and D3), none contributes significantly to the gating charge.
Table 1.
Gating charge contribution from key charged residues
| Residue | Δq (Full-channel) | Δq (Isolated VSD) |
|---|---|---|
| R1 (S4) | −0.12 ± 0.13 | −0.20 |
| R2 (S4) | 0.67 ± 0.10 | 0.70 |
| R3 (S4) | 0.92 ± 0.09 | 1.01 |
| R4 (S4) | 0.70 ± 0.10 | 0.81 |
| K5 (S4) | 0.08 ± 0.09 | 0.31 |
| R6 (S4) | 0.13 ± 0.10 | 0.25 |
| E1 (S2) | −0.02 ± 0.12 | 0.15 |
| E2 (S2) | 0.08 ± 0.09 | −0.13 |
| D3 (S3) | 0.16 ± 0.12 | 0.10 |
As mentioned above, the calculated gating charge for the entire channel appears to be slightly smaller (by 2–4e) than the experimental estimates for the Shaker K+ channel (6,7). The individual contribution of the VSD residues to the gating charge (Table 1) indicates that the outermost arginine R1 makes a negligible contribution, whereas R2 makes only a contribution of 0.7e. This is not in accord with experiments, which show that these two arginines contribute significantly to the total gating charge (6,7,56). As shown in Fig. 4, in the resting state R1 and R2 experience ∼0.0 and 0.6 of the membrane potential, respectively. To make a larger contribution to the total gating charge, these two arginines need to move deeper in the membrane field toward the intracellular solution.
To explore this possibility, steered molecular dynamics (SMD) simulations (57,58) were performed starting from the resting state models, described above. SMD was performed on the full tetrameric channel and on the individual VSD, in which R1 residue side chains were pulled toward the intracellular side within 30 ns. The displacement charge of the system, Qd, was monitored over the course of the SMD simulation, the results being shown in Fig. 5. The variation in Qd is equal to the increase in the gating charge that is transferred across the membrane as the R1 side chains are pulled down. After 20 ns of simulation, Qd reaches a plateau corresponding to a gating charge of 12.7e. A similar plateau is observed in the case of the individual VSD, where this time the plateau corresponds to a gating charge of 3.0e. Most importantly, the salt-bridge interactions between R1 and E0 in S1 are disrupted in all four subunits during the first 10 ns of the SMD simulation. As the arginine side chains are pulled further down, a strong salt-bridge interaction is formed between R1 and E1 on S2. These interactions remain present for the rest of the simulation in two of the four subunits, and appear to block the entry of water molecules from the extracellular side. Otherwise, the conformation reached during the SMD simulations does not differ considerably from the refined resting state. The z direction displacement of Cα atoms of S4 (after 15 ns) is ∼2.5 Å for R1, whereas the average displacement is 0.5 Å near the intracellular end of S4, indicating that the change in the gating charge arises mainly from the orientation of residue side chains of the VSD. When considering this alternate resting conformation obtained by SMD, the total gating charge is 12.7e, which is in excellent agreement with experimental estimates.
Figure 5.

Snapshots of the voltage-sensor domain (A–D) taken from SMD simulations of the full tetrameric channel. During the simulation, the salt-bridge between E0 (S1) and R1 (S4) is broken, and a new salt-bridge is formed between E1 (S2) and R1 on S4. Time-dependent variations of the displacement charge Qd of the isolated VSD (E) and the full tetrameric channel (F) during SMD simulations. Blue lines represent Qd values calculated at every 10 ps in each trajectory. The thick black lines represent the Qd values block-averaged over every 300 ps.
Conclusion and Discussion
Active and resting state models of Kv1.2 (30) were refined through MD simulations of the channel in an explicit membrane-solvent environment. Each model was equilibrated for 50 or 100 ns in the presence of a transmembrane potential to obtain stable conformations of each channel state. An hourglass-shaped aqueous pore running across the VSD appears in all the simulations, as observed in previous simulations (31–33,48–50). Furthermore, several salt-bridges within the VSD and between charged residues of the VSD and lipid headgroups stabilize the configuration of the VSD. The salt-bridge interactions observed in the closed and active conformations reflect an overall movement of the S4 segment in which this segment undergoes a sliding motion with respect to a set of countercharges of the VSD-membrane complex. Those countercharges, which include the polar lipid headgroups at the membrane solution interfaces as well as some highly conserved acidic residues in S2 (E1 and E2) and S3 (D3), provide a set of favorable electrostatic interactions complementing the positively charged residues along S4.
During the simulation of the resting state model, the extracellular end of S4 spontaneously converted into a 310-helix over a stretch of 10 residues. The 310 helical conformation, which places the gating charges, located at every third residue, on the same face of S4, is energetically advantageous because it orients the positively charged side chains of S4 toward the water-filled crevice within the VSD where they can also form favorable salt-bridges with the negatively charged residue along S2 and S3. Thus, it appears that the 310 helical conformation of S4 in the resting state is dictated by the highly heterogeneous and anisotropic environment experienced by the charged residues of the VSD. Partial conversion of the S4 segment to a 310-helix has also been observed in MD simulations of the open state of Kv1.2 (31), and in recent crystal structure of the related MlotiK1 channel (55). These results, together with the present simulations, support the notion that S4 adopts a 310-helical conformation in the resting state of the channel. Interestingly, kinetic and thermodynamic analyses of the voltage-sensitive phosphatase Ci-VSP have suggested that the existence of additional inactive substates is a generic property of all VSD proteins (34). On a speculative note, it is possible that the interconversion of S4 from an α to a 310 helix underlies some of the substates of VSDs.
The fraction of the membrane potential felt by key charged residues in the VSD was calculated using FEP/MD simulations. Across a perfectly homogeneous, planar membrane, the potential drops linearly from the intracellular side to the extracellular side. However, the irregular shape and dielectric inhomogeneity of the VSD with its complex water-filled crevices modulates the spatial variations of the membrane potential. As shown in Fig. 4, the calculations reveal that the transmembrane potential drops rapidly over a distance of ∼10–15 Å within the VSD. In the active state (left), the potential acting on the gating residues along S4 drops sharply between the center of the bilayer and the extracellular solution. The membrane potential roughly varies from its full value to zero over the extracellular half of the membrane bilayer. This trend is also seen for the resting state. The sharp drop in the transmembrane potential is due to the presence of a water-filled crevice located within the VSD, which provides a high-dielectric medium in contrast to the low-dielectric medium of the surrounding protein and lipid molecules. The field is even more markedly focused in the case of the tetrameric channel (Fig. 4, top), due to the high dielectric vestibular cavity at the intracellular entrance of the open channel (48).
The present results show that, rather than being spread over the width of the entire bilayer, the transmembrane field is focused over a fairly narrow region corresponding roughly to the outer leaflet of the bilayer. A striking example is provided by R4 and K5 for the active state. These two basic residues experience 0.16 and 0.89 of the potential, respectively, even though they are separated by less than one helix turn along S4. The concept of a focused field across the VSD is supported by experimental evidence for a proton-conducting pore in the R1H mutant of the Shaker K+ channel (51). Dynamic fluorometric measurements of the electrostatic field inside the VSD (59) and tethered measurements of the movement of S4 residues (56) also show that the transmembrane electric potential falls within a distance of 4–10 Å around S4. Most importantly, a strong salt-bridge interaction formed during the SMD simulation between R1 and E2 on S2 appears to seal, dielectrically, the extracellular side of the VSD, preventing the penetration of water molecules. Presumably, this salt-bridge locks the VSD in the fully resting state and helps further focus the membrane field. From a functional and mechanistic point of view, the focused field is a critical feature that makes it possible to achieve the transfer of a large effective gating charge without requiring a full translocation of S4 across the membrane.
The magnitude of the gating charge ΔQ that is effectively transferred across the membrane upon the activation of the tetrameric channel is critical for explaining the strong coupling between the conformation of the channel and the applied membrane potential. It was calculated from the displacement charge Qd as defined in Eq. 1. The calculated gating charge obtained for the refined models of the active and resting states of the full tetrameric Kv1.2 channel is 10.25 ± 0.35e. Consistent with this value, the calculated gating charge obtained for the refined models of the active and resting state of the isolated VSD is approximately one-fourth of the ΔQ for the tetramer (2.38 ± 0.07e). The calculated gating charge is a little bit smaller than the experimental estimates of 12–14e measured for the Shaker K+ channel (6,7,11). The difference can be accounted for by considering the individual contribution of key residues of the VSD to the gating charge (Table 1), indicating that the contribution from the two outermost arginines, R1 and R2, is underestimated compared to experiments (6,7,56). However, a relatively small motion of the side chains of R1 and R2 pulled down by SMD is sufficient to increase the gating charge by ∼2.45e (Fig. 5). The total gating charge is 12.7e for the alternate resting conformation obtained by SMD, in excellent agreement with experiment. Because of the focused field across the VSD, the increase in gating charge with SMD is achieved with a relatively small motion of the side chains of R1 and R2. The result suggests that, rather than being the true inactive/resting state that exists under hyperpolarizing conditions, the (stable) closed state model in the MD simulations corresponds to an intermediate substate appearing early during channel activation. The existence of such intermediate substates during the activation of Kv channels is supported by a variety of experiments (60,61). For instance, it has been noted that the activation of Kv channels is accompanied by a very fast (<10 μs) jump in the gating charge by ∼4e, corresponding to the transition from the resting state to an intermediate closed conformation (61). Of particular interest, the calculated contribution from the lipids to the total gating charge is only ∼0.28e. This shows that, despite the important interactions between some residues along S4 and the lipid headgroups, lipid charges do not participate actively in the voltage-gating mechanism.
Supporting Material
Details of FEP/MD calculations, four figures, and six tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00361-9.
Supporting Material
Acknowledgments
The authors gratefully acknowledge the help and support of Ray Loy at Argonne National Laboratory and Sadaf Alam at Oak Ridge National Laboratory.
This work is supported by the National Institutes of Health via grants Nos. P41-RR005969 and R01-GM06788716 (to F.K.-A., E.T., and K.S.) and grant No. R01-GM062342 (to V.J. and B.R.). This research benefited from an INCITE grant and used resources of the Argonne Leadership Computing Facility at Argonne National Laboratory and the National Center for Computational Sciences at Oak Ridge National Laboratory, both of which are supported by the Office of Science of the U.S. Department of Energy under contract No. DE-AC02-06CH11357.
Contributor Information
Benoît Roux, Email: roux@uchicago.edu.
Klaus Schulten, Email: kschulte@ks.uiuc.edu.
References
- 1.Bezanilla F., Perozo E., Stefani E. Gating of Shaker K+ channels: II. The components of gating currents and a model of channel activation. Biophys. J. 1994;66:1011–1021. doi: 10.1016/S0006-3495(94)80882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zagotta W.N., Hoshi T., Aldrich R.W. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ledwell J.L., Aldrich R.W. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong C.M., Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- 5.Keynes R.D., Rojas E. Kinetics and steady-state properties of the charged system controlling sodium conductance in the squid giant axon. J. Physiol. 1974;239:393–434. doi: 10.1113/jphysiol.1974.sp010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoppa N.E., McCormack K., Sigworth F.J. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255:1712–1715. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- 7.Seoh S.A., Sigg D., Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 8.Lee S.Y., Lee A., MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. USA. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 10.Long S., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;15:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 11.Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 12.Murata Y., Iwasaki H., Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki M., Takagi M., Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 14.Ramsey I.S., Moran M.M., Clapham D.E. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cha A., Snyder G.E., Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- 16.Posson D.J., Ge P., Selvin P.R. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 2005;436:848–851. doi: 10.1038/nature03819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chanda B., Asamoah O.K., Bezanilla F. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 2005;436:852–856. doi: 10.1038/nature03888. [DOI] [PubMed] [Google Scholar]
- 18.Glauner K.S., Mannuzzu L.M., Isacoff E.Y. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y., Ruta V., MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 20.Ruta V., Chen J., MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 21.Jiang Y., Lee A., MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 22.Guy H.R., Seetharamulu P. Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. USA. 1986;83:508–512. doi: 10.1073/pnas.83.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catterall W.A. Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 1986;55:953–985. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- 24.Keynes R.D., Elinder F. The screw-helical voltage gating of ion channels. Proc. Biol. Sci. 1999;266:843–852. doi: 10.1098/rspb.1999.0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lainé M., Lin M.-C.A., Papazian D.M. Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron. 2003;39:467–481. doi: 10.1016/s0896-6273(03)00468-9. [DOI] [PubMed] [Google Scholar]
- 26.Yarov-Yarovoy V., Baker D., Catterall W.A. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. USA. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campos F.V., Chanda B., Bezanilla F. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc. Natl. Acad. Sci. USA. 2007;104:7904–7909. doi: 10.1073/pnas.0702638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grabe M., Lai H.C., Jan L.Y. Structure prediction for the down state of a potassium channel voltage sensor. Nature. 2007;445:550–553. doi: 10.1038/nature05494. [DOI] [PubMed] [Google Scholar]
- 29.Lewis A., Jogini V., Roux B. Atomic constraints between the voltage sensor and the pore domain in a voltage-gated K+ channel of known structure. J. Gen. Physiol. 2008;131:549–561. doi: 10.1085/jgp.200809962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pathak M.M., Yarov-Yarovoy V., Isacoff E.Y. Closing in on the resting state of the Shaker K+ channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 31.Bjelkmar P., Niemelä P.S., Lindahl E. Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLOS Comput. Biol. 2009;5:e1000289. doi: 10.1371/journal.pcbi.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Treptow W., Tarek M., Klein M.L. Initial response of the potassium channel voltage sensor to a transmembrane potential. J. Am. Chem. Soc. 2009;131:2107–2109. doi: 10.1021/ja807330g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishizawa M., Nishizawa K. Molecular dynamics simulation of Kv channel voltage sensor helix in a lipid membrane with applied electric field. Biophys. J. 2008;95:1729–1744. doi: 10.1529/biophysj.108.130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villalba-Galea C.A., Sandtner W., Bezanilla F. S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. USA. 2008;105:17600–17607. doi: 10.1073/pnas.0807387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roux B. Influence of the membrane potential on the free energy of an intrinsic protein. Biophys. J. 1997;73:2980–2989. doi: 10.1016/S0006-3495(97)78327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmitz G., Liebisch G., Langmann T. Lipidomic strategies to study structural and functional defects of ABC-transporters in cellular lipid trafficking. FEBS Lett. 2006;580:5597–5610. doi: 10.1016/j.febslet.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt D., MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. USA. 2008;105:19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roux B. The membrane potential and its representation by a constant electric field in computer simulations. Biophys. J. 2008;95:4205–4216. doi: 10.1529/biophysj.108.136499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of protein s. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 41.Mackerell A.D., Feig M., Brooks C.L. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 42.Beglov D., Roux B. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J. Chem. Phys. 1994;100:9050–9063. [Google Scholar]
- 43.Schlenkrich M., Brickmann J., Karplus M. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In: Merz K.M., Roux B., editors. Biological Membranes: A Molecular Perspective from Computation and Experiment. Birkhauser; Boston, MA: 1996. pp. 31–81. [Google Scholar]
- 44.Feller S.E. Molecular dynamics simulations of lipid bilayers. Curr. Opin. Colloid Interface Sci. 2000;5:217–223. [Google Scholar]
- 45.Jorgensen W.L., Chandrasekhar J., and, Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 46.Darden T., York D., Pedersen L. Particle mesh Ewald. An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 47.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 48.Jogini V., Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J. 2007;93:3070–3082. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freites J.A., Tobias D.J., White S.H. A voltage-sensor water pore. Biophys. J. 2006;91:L90–L92. doi: 10.1529/biophysj.106.096065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sands Z.A., Sansom M.S. How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure. 2007;15:235–244. doi: 10.1016/j.str.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Starace D.M., Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 52.Tombola F., Pathak M.M., Isacoff E.Y. The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature. 2007;445:546–549. doi: 10.1038/nature05396. [DOI] [PubMed] [Google Scholar]
- 53.Tiwari-Woodruff S.K., Schulteis C.T., Papazian D.M. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiwari-Woodruff S.K., Lin M.A., Papazian D.M. Voltage-dependent structural interactions in the Shaker K+ channel. J. Gen. Physiol. 2000;115:123–138. doi: 10.1085/jgp.115.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clayton G., Altieri S., Morais-Cabral J. Structure of the transmembrane region of a bacterial cyclic nucleotide-regulated channel. Proc. Natl. Acad. Sci. USA. 2008;105:1151–1155. doi: 10.1073/pnas.0711533105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahern C.A., Horn R. Focused electric field across the voltage sensor of potassium channels. Neuron. 2005;48:25–29. doi: 10.1016/j.neuron.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 57.Isralewitz B., Baudry J., Schulten K. Steered molecular dynamics investigations of protein function. J. Mol. Graphics Mod. 2001;19:13–25. doi: 10.1016/s1093-3263(00)00133-9. [DOI] [PubMed] [Google Scholar]
- 58.Sotomayor M., Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316:1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 59.Asamoah O.K., Wuskell J.P., Bezanilla F. A fluorometric approach to local electric field measurements in a voltage-gated ion channel. Neuron. 2003;37:85–97. doi: 10.1016/s0896-6273(02)01126-1. [DOI] [PubMed] [Google Scholar]
- 60.Silverman W.R., Roux B., Papazian D.M. Structural basis of two-stage voltage-dependent activation in K+ channels. Proc. Natl. Acad. Sci. USA. 2003;100:2935–2940. doi: 10.1073/pnas.0636603100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sigg D., Bezanilla F., Stefani E. Fast gating in the Shaker K+ channel and the energy landscape of activation. Proc. Natl. Acad. Sci. USA. 2003;100:7611–7615. doi: 10.1073/pnas.1332409100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aksimentiev A., Schulten K. Imaging α-hemolysin with molecular dynamics: ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005;88:3745–3761. doi: 10.1529/biophysj.104.058727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.