

Abstract
An increasing amount of evidence suggests that in several amyloid diseases, the fibril formation in vivo and the mechanism of toxicity both involve membrane interactions. We have studied Alzheimer's disease related amyloid beta peptide (Aβ). Recombinant Aβ(M1–40) and Aβ(M1–42) produced in Escherichia coli, allows us to carry out large scale kinetics assays with good statistics. The amyloid formation process is followed in means of thioflavin T fluorescence at relatively low (down to 380 nM) peptide concentration approaching the physiological range. The lipid membranes are introduced in the system as large and small unilamellar vesicles. The aggregation lagtime increases in the presence of lipid vesicles for all situations investigated and the phase behavior of the membrane in the vesicles has a large effect on the aggregation kinetics. By comparing vesicles with different membrane phase behavior we see that the solid gel phase dipalmitoylphosphatidylcholine bilayers cause the largest retardation of Aβ fibril formation. The membrane-induced retardation reaches saturation and is present when the vesicles are added during the lag time up to the nucleation point. No significant difference is detected in lag time when increasing amount of negative charge is incorporated into the membrane.
Introduction
To date, ∼30 proteins are known to form amyloid fibrils and deposits in humans. Depending on protein and location this causes different diseases. For the majority of the diseases, the detailed biochemical mechanisms are not known, but lipid membranes are gaining interest as a potentially important factor in disease propagation. In the amyloid plaques associated with several amyloidogenic diseases, tightly associated lipids have been identified, especially lipids associated with so-called lipid rafts (1). The amyloid fibril formation proceeds via transient oligomers that eventually go on to more stable fibrils and the intermediate small oligomers, have been found to be the toxic species rather than the monomers or fully formed amyloid fibrils (2). It is not known if this is due to specific interactions caused by the structure or size of the oligomers, or if it is because the oligomers are potent intermediates in the amyloid formation process. The molecular details behind the involvement of membrane in the amyloid formation mechanism are much debated. In one model, the lipid membrane produces a catalytic surface for amyloid formation (3). In another model, oligomers form ion channels in the membrane leading to apoptosis (4). Further models propose that amyloid formation changes the fluidity of the membrane or that the membrane is disrupted in the amyloid formation process (5–7).
The amyloid beta peptide (Aβ) (Fig. 1 C) associated with Alzheimer's disease is produced from the amyloid precursor protein (APP) by specific proteases. The C-terminus of the peptide originates from the membrane incorporated part of APP and this gives Aβ an amphiphilic character. The cleaved peptide can vary in length but Aβ(1–40) and Aβ(1–42) are the most common products. Due to the extension in the more hydrophobic C terminus, Aβ(1–42) is more aggregation prone than Aβ(1–40) and an increased level of Aβ(1–42) relative to Aβ(1–40) is a risk factor for developing Alzheimer's disease. Aggregation of Aβ has been associated to, e.g., membrane barrier disruption and lipid regulation in cells, and it has been proposed that variations in lipid composition, and in particular the presence of raft domains, can affect the amyloid toxicity in cells (8,9).
Figure 1.
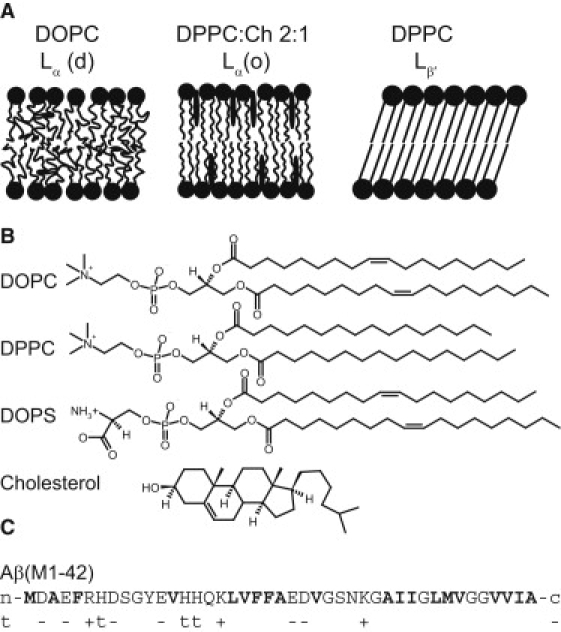
(A) Schematic illustration of the bilayer phases in the vesicles used in this study. By combining DOPC, DPPC, and cholesterol, three different phases were achieved with different degrees of translational diffusion and acyl chain order. (B) Chemical structure of used lipids. (C) Amino acid sequence of Aβ(M1–42) with hydrophobic residues in bold. Residues that are negatively (−) or positively (+) charged or titrating (t) at neutral pH are indicated under the sequence. The last two residues (IA) are missing in Aβ(M1–40).
A large number of model studies of Aβ–membrane interactions have been carried out with the membrane as a deposited bilayer, a monolayer, or in the form of vesicles dispersed in solution. Although the results are contradictory in some respects, well-founded conclusions are strong interactions between the Aβ peptide and lipid membranes and that membranes can be permeabilized by the Aβ peptide during the amyloid formation process. The composition of the lipid membrane is considered important for membrane interactions with Aβ. In particular, the liquid ordered domains known as lipid rafts have been related to amyloid formation. Common components of lipid rafts such as cholesterol, sphingomyelin, and gangliosides have all been reported as having increased interactions with Aβ peptide (10–12) and raft lipid components have been found tightly associated with amyloid plaques extracted from patients (1).
Lipid membranes have been reported to both accelerate and retard amyloid formation (reviewed in Murphy (13)). This may indicate that the precise conditions of the measurements will make one or the other effect dominating; however, high resolution kinetic data with good statistics of amyloid formation in the presence of lipid membranes have been lacking. In most cases, only a few or only endpoint measurements are reported but there are exceptions, e.g., Sabate et al. (14) show with good reproducibility that there is a small delay in Aβ(1–40) amyloid formation on soy bean phosphatidylcholine (PC) addition. The Aβ aggregation kinetics have been assumed to be stochastic on a macroscopic level (15), but recent developments show that this is not the case and Aβ aggregation kinetics are concentration dependent in a highly reproducible manner (16). These findings rely on the use of recombinant Aβ expressed in large scale in Escherichia coli (17) to overcome the high cost of synthetic peptide and allow for collection of enough data to obtain good statistics and refinement of the experimental setup. The collection from size exclusion chromatography of pure monomeric peptide in the degassed experimental buffer with no additives is especially essential.
This study addresses the effect of phospholipid membranes on the aggregation kinetics of Aβ under conditions approaching the in vivo situation in terms of low Aβ concentration, low Aβ to membrane/surface ratio, and absence of cosolvents. We have studied the kinetics of Aβ aggregation in the absence and presence of model membranes in the form of large and small unilamellar vesicles of defined composition. Vesicles with different compositions of phospholipids and cholesterol were designed to elucidate different properties of lipid membranes such as phase behavior and charge. Aggregation of recombinant peptides Aβ(M1–40) and Aβ(M1–42) were monitored using thioflavin T (ThT) fluorescence. To study the role of membrane phase behavior (liquid disordered versus liquid ordered versus gel phase), we have compared the effect of vesicles containing phospholipids with the same PC headgroup but different acyl chains, with and without cholesterol. To study the role of membrane charge we have compared the effect of vesicles containing anionic phosphatidylserine (PS), and we relate the result to interaction studies with monolayer technique. Our data show that biological membranes may retard Aβ aggregation in a manner dependent on membrane phase behavior but independent of membrane charge.
Materials and Methods
Vesicle preparation
Dioleoyl-phosphatidylcholine (DOPC), dioleoyl-phosphatidylserine (DOPS), cholesterol were obtained from Avanti Polar Lipids (Alabaster, AL) and dipalmitoyl-phosphatidylcholine (DPPC) was obtained from Larodan (Malmö, Sweden) (Fig. 1 B). Unilamellar vesicles (200 nm) were prepared from 5 mM stock solutions of phospholipids and cholesterol in chloroform/methanol 9:1 v/v. Thin lipid films were deposited onto glass tubes under a slow flow of nitrogen and dried in vacuum over night at 50°C. The films were hydrated with 20 mM Tris/HCl pH 7.4 with 0.2 mM EDTA and 0.02% NaN3, unless otherwise stated, to a final concentration of 5 mM lipid. Large unilamellar vesicles (200 nm) were formed by 10 freeze-thaw cycles and 10 extrusions through a 200 nm cellulose filter. pH was readjusted before extrusion, keeping the ionic strength constant, with 20 mM Tris 0.2 mM EDTA 0.02% NaN3 to a final pH of 7.4. Sonicated small unilamellar vesicles were prepared by microtip sonication at 40% duty cycle for 20 min followed by centrifugation for 3 min at 13,000 × g.
Phospholipid concentrations were determined by phosphate analysis according to Rouser et al. (18). Dried samples were digested with 0.65 mL 70% perchloric acid at 180°C for 20 min and when cool, 3.3 mL ddH2O, 0.5 mL 25 g/L ammonium molybdate and 0.5 mL 100 g/L ascorbic acid were added. After heating for 5 min at 100°C, absorbance was read at 800 nm. A solution of KH2PO4 was used as standard in additions corresponding to 0–5 μg phosphor in each tube.
Aβ peptide
The peptides studied in this work, Aβ(M1–40) and Aβ(M1–42), comprise residues 671–711 and 671–713, respectively, of human APP (Fig. 1 C). The peptides were expressed in E. coli (strain BL21 star pLysS De3) from a synthetic gene with E. coli optimized codons and purified from inclusion bodies by ion exchange and size exclusion (17). Synthetic peptide Aβ(1–40) (≥95% pure) was purchased from Genscript (Piscataway, NJ). Both recombinant and synthetic peptide were further purified by gel filtration in the experimental buffer for the kinetic experiments to isolate monomer just before initiating the aggregation kinetics measurements (see below).
Kinetics experiments
Monomeric Aβ was collected from a 1 × 30 cm Superdex 75 column in 20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3 directly into a low bind Eppendorf tube on ice. The Aβ concentration was determined by integration of the absorbance peak in the FPLC chromatogram using an extinction coefficient of 1440 cm−1 M−1 at 280 nm. The monomer stock was then mixed with filtered buffer (20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3), ThT and vesicles to desired concentrations in low bind Eppendorf tubes on ice. One stock solution was made for each experimental condition and finally these stock solutions were distributed on a 96-well low bind half area plate (Corning 3881) on ice. Each well contained 100 μL solution with a ThT concentration of 20 μM. By using the maximum volume in a half area plate the air surface/volume ratio is minimized, which is important because Aβ is surface active. The plate was sealed with a plastic film, not in contact with the solution, to eliminate evaporation. The experiment was started by putting the plate in a plate reader, Fluostar Omega (BMG Labtech, Offenburg, Germany), thermostated at 37°C, and starting shaking at 100 rpm. ThT fluorescence was measured from the bottom of the plate every fifth minute through excitation and emission filters of 440 and 480 nm, respectively, with continuous orbital shaking between measurements. Vesicles incubated with ThT in absence of peptide showed no change in intensity over time and no change in vesicle size could be detected from dynamic light scattering when incubating DOPC vesicles for 10 h at 37°C and 100 rpm.
DSC measurements
DSC experiments were carried out using a vp DSC from MicroCal (Northampton, MA) with a cell volume of 0.5072 mL. Temperature was changed by 1°C/min during the measurements.
Surface balance
DPPC or 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine monolayer was applied on 20 mL 20 mM Tris, pH 7.4, 0.02% NaN3, 0.2 mM EDTA from 10 μL 1 mM lipid in chloroform/methanol 9:1 and was left for 10 min to evaporate the organic solvent. The monolayer was then compressed to 30 mN/m before relaxed to experimental surface pressure. Injection of Aβ(M1–40) or buffer was made by injecting 2 mL under one barrier and withdrawing 2 mL from the outside of the other barrier.
Dynamic light scattering
Dynamic light scattering measurements were carried out using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Dynamic light scattering experiments were carried out on vesicle suspensions at 22°C.
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were recorded in a 10-mm quartz cuvette between 300 and 190 nm using a Jasco J-815 CD spectrometer (Jasco, Easton, MD) at 1 nm bandwidth, 0.1 nm interval, and 1 s response time. Aβ(M1–40) monomer was isolated on a Superdex 75 column in 20 mM PO4, 0.05 mM EDTA buffer. After 3× dilution with ddH2O, one spectrum was recorded for 10 μM peptide. After addition of DOPC vesicles, extruded through a 100 nm filter, 17 spectra were recorded with 5-min intervals at ∼0.2 mM DOPC and 8.6 μM peptide.
Results
On binding to the cross-β structure in amyloid fibrils and oligomers, the fluorescence from the organic dye ThT experiences a red shift and gives an increased signal at 482 nm when excited at 442 nm. This feature allows for the use of ThT as a reporter on the propagation of amyloid fibril formation. The specificity and mechanism of ThT binding in the pathway of amyloid formation is still debated but in designed proteins with cross-β structures, ThT has been observed to bind cross-β ladders as small as four to five strands (19), corresponding to tetramer to pentamer of Aβ peptide. The ThT fluorescence trace from amyloid formation is characterized by a sigmoidal curve shape. During the initial lag phase, oligomers are built up to the critical size detectable by ThT and during the elongation phase the fluorescence intensity increases rapidly as fibrils elongate. After the elongation phase, the fluorescence intensity levels off to a plateau value when the monomer concentration approaches its equilibrium value in the presence of the formed fibrils. To allow good statistics in the kinetics studies, the amyloid formation was followed for multiple 100-μL replicates of each solution, in wells of a 96-well low bind half area plate. Pure monomer was collected on ice in the degassed experimental buffer from size exclusion chromatography just before the start of the experiment. Thus the experiment is carried out in a pure buffer system with no cosolvents. After dilution and addition of vesicle solutions to desired concentrations, the experiment was initiated by placing the sealed plate in a plate reader thermostated at 37°C. With 100 rpm shaking, the ThT fluorescence was measured from all wells every fifth minute.
The most common type of phospholipids in human cell membranes, PC, was chosen as the primary lipid component of the model membranes. The lipid was added as large unilamellar vesicles prepared by extrusion through a 200 nm filter or as small unilamellar vesicles prepared by sonication, ∼50 nm diameter as measured from dynamic light scattering. Depending on the length and degree of saturation of the phospholipid acyl chains (Fig. 1 B) and the amount of cholesterol dissolved in the membrane, the PC bilayer adopts different phase behavior as illustrated in Fig. 1 A. Fig. 2 shows kinetic traces from ThT fluorescence, reporting on amyloid formation, for Aβ(M1–40) and the more hydrophobic Aβ(M1–42) in the absence and presence of large unilamellar vesicles composed of DOPC. The presence of the vesicles cause a delay in the amyloid formation for both peptides investigated, indicating interference with the aggregation process. Incubation of ThT and vesicles in absence of peptide showed no change in signal over time (data not shown).
Figure 2.
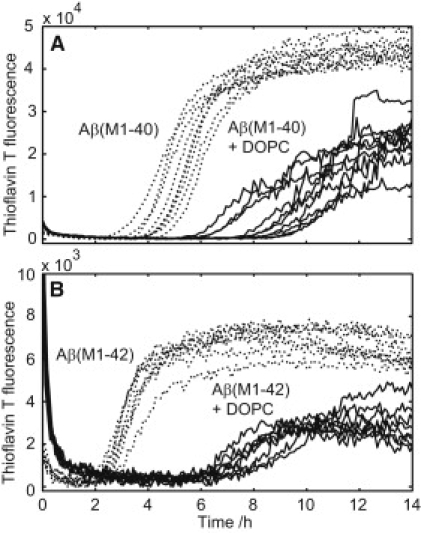
ThT fluorescence as a function of time in the absence (dashed line) and presence (solid line) of DOPC large unilamellar vesicles. Each line represents one replicate in a separate well in a 96-well plate. (A) Aβ(M1–40) (4.8 μM) with or without 1.8 mM phospholipid. (B) Aβ(M1–42) (0.38 μM) with or without 1.1 mM phospholipid. All experiments were carried out in 20 mM Tris/HCl, 0.2 mM EDTA, 0.02% NaN3, 20 μM ThT, pH 7.4. One replicate with peptide alone that never fibrillated was removed from A.
To investigate the influence of lipid phase behavior on the aggregation kinetics, the aggregation of Aβ(M1–40) was studied in the presence of DOPC vesicles as well as DPPC vesicles with and without cholesterol incorporation. DOPC has a melting temperature of ∼−20°C and at physiological temperature it forms liquid disordered lamellar phase, Lα(d), with high lateral diffusion and disordered acyl chains. DPPC, has a melting temperature of 42°C and forms a solid lamellar gel phase, Lβ′, at physiological temperature. The gel phase is characterized by low lateral diffusion and highly ordered acyl chains. Consequently, the gel phase has a tighter lipid packing with an average area per headgroup of 46 Å2 for DPPC in Lβ′ compared to 68 Å2 for DOPC in Lα(d) (20). When >20% cholesterol is introduced into the DPPC Lβ′ membrane, a liquid ordered bilayer phase, Lα(o), is formed (21), which has high order in the acyl chains but also a high lateral diffusion much closer to Lα(d) than Lβ′ (22,23). In contrast to the DPPC case, the introduction of cholesterol into the DOPC Lα(d) bilayer increases the acyl chain order, with just a small change in the translational diffusion, toward Lα(o) (23).
We noted that the extruded Lβ′ DPPC vesicles sometimes were already significantly smaller than 200 nm after the freeze thaw cycles and therefore much smaller than the Lα(d) DOPC vesicles prepared according to the same protocol. To enable comparisons between systems with vesicles of similar size, we therefore used sonicated small unilamellar vesicles in the phase comparison. Fig. 3 shows that the lag time for the amyloid formation is considerably shorter when aggregation takes place in the presence of liquid ordered DPPC-cholesterol vesicles compared to solid gel phase DPPC vesicles. For the pure DOPC vesicles only a minor delay of the amyloid formation is observed, and introduction of cholesterol into the DOPC membrane only has a small accelerating effect. Because the headgroups of DOPC and DPPC are identical, this observed difference between the Lβ′ DPPC vesicles and the Lα(d) DOPC vesicles most likely comes from the difference in phase behavior. The traces from the peptide in absence of lipids are not included in Fig. 3 for clarity but overlapped with the curves for DPPC/cholesterol.
Figure 3.
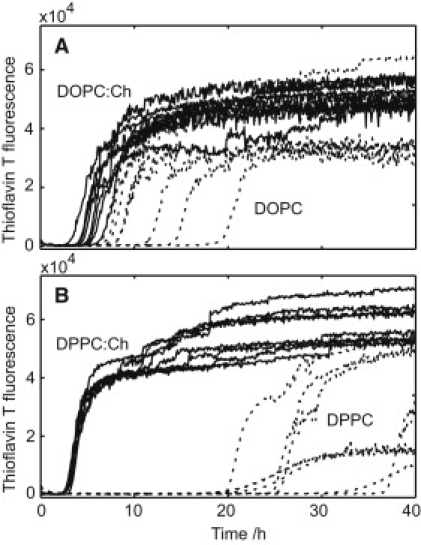
ThT fluorescence kinetic traces for 6.0 μM Aβ(M1–40) in the presence of (A) DOPC or (B) DPPC in small unilamellar vesicles with (solid lines) or without (dashed lines) membrane-incorporated cholesterol in 20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3 with 20 μM ThT. Cholesterol was added at a ratio of 2:1 PC/cholesterol and the total lipid concentration is in all cases 1.9 mM. Each line represents one replicate in a separate well.
The dependence of aggregation kinetics on the ratio between the vesicle and peptide concentration may contain further information about the mechanism of the interference in the amyloid formation process caused by the lipid vesicles. Fig. 4 shows how the time of half completion of the amyloid formation of Aβ(M1–40) varies with the concentration of DPPC present as large unilamellar vesicles. Although the concentration dependence has the appearance of a binding curve, it could not be fitted with any meaningful binding equation because both stoichiometry and binding affinity are unknown and interdependent parameters in a binding equation. However, the system seems to reach saturation around a DPPC/Aβ(M1–40) ratio of 1:15-30.
Figure 4.
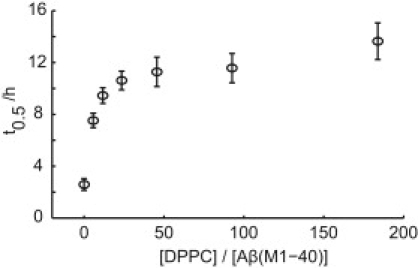
Lagtime dependence on DPPC concentration. 6.8 μM Aβ(M1–40) was incubated with different concentrations of DPPC present as large unilamellar vesicles in 20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3 with 20 μM ThT. The half-time of completion, t0.5, is defined as time at 50% of maximum intensity and the error bars represent the standard deviation from five wells.
To evaluate at which stage in the amyloid formation process the vesicles interfere, an experiment was started with multiple identical replicates of 50 μL 12 μM Aβ(M1–40) per well. At each time point (ranging from 0 to 254 min), two wells were complemented with 50 μL 2.7 mM DPPC and two wells were complemented with 50 μL buffer (Fig. 5). When the vesicles were added during the first hour of the experiment, the resulting lag time was prolonged from 2 to 9 h. When the addition was made later than 1 h after starting the experiment, the delay in lag time induced by the addition of vesicles diminished and after 2 h nearly no delay in lag time was observed on vesicle addition. When the vesicles were added in the elongation phase (steep part of the curve), the increase in ThT fluorescence was continued as in the undisturbed case. Any change in elongation rate on vesicle addition is hard to separate from the change in intensity caused by dilution of the peptide and light scattering from the vesicles.
Figure 5.
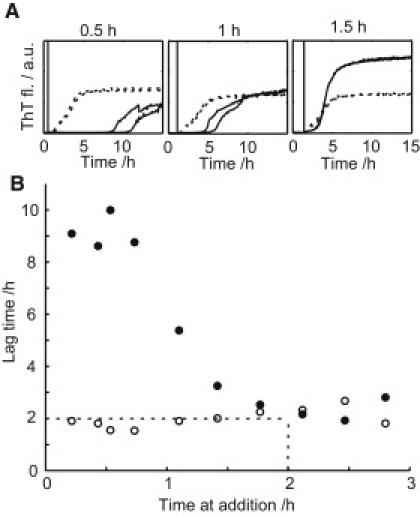
Addition of DPPC vesicles at different time points during the amyloid formation process. The experiment was started with 50 μL 12 μM Aβ(M1–40) in 20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3 with 20 μM ThT. At different time points, 50 μL 2.7 mM DPPC large unilamellar vesicles or 50 μL buffer was added. (A) ThT fluorescence versus time. Vertical lines indicate the time of vesicle or buffer addition as also given as numbers in each panel. Solid lines represent data from wells to which vesicles were added and dashed lines data from wells to which buffer was added. (B) Lag time as a function of time of vesicle (solid circles) or buffer (open circles) addition. Each point represents an average of two replica and the lag time is defined as the time at which 10% of the maximum fluorescence is reached. Dashed lines indicate the lag time in the absence of vesicles.
Amyloid formation of Aβ(M1–40) was finally investigated in the presence of vesicles composed of DOPS and DOPC at varying molar ratios (Fig. 6). DOPS is an anionic phospholipid, and the mixed DOPC/DOPS vesicles form a Lα(d) phase. Fig. 6 shows that aggregation lag time is approximately the same for all DOPS/DOPC compositions investigated, and thus no significant effect of increasing the membrane negative charge can be seen. To further analyze the electrostatic component of the Aβ peptide-membrane interaction, the influence of salt concentration was studied. The aggregation kinetics for both Aβ(M1–40) and Aβ(M1–42) are found to be strongly dependent on salt concentration, with a shorter lag time at higher salt concentration, and the lag time in the presence of DOPC vesicles decreased with increasing ionic strength. However, a longer lag time is observed in the presence of DOPC vesicles compared to Aβ alone at all NaCl concentrations studied ranging from 0 to 160 mM (in 20 mM Tris/HCl pH 7.4, 0.2 mM EDTA, 0.02% NaN3 with 20 μM ThT; data not shown).
Figure 6.
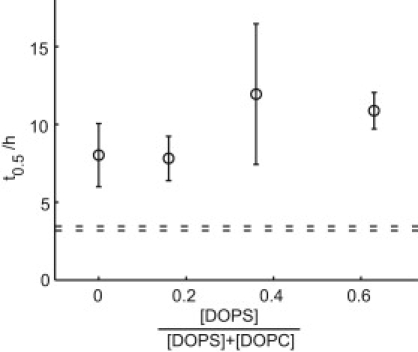
Lagtime dependence on membrane charge. Aβ(M1–40) (6.7 μM) was incubated with large unilamellar vesicles of different DOPS/DOPC ratios in 20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3 with 20 μM ThT. The total phospholipid concentration was kept constant at 1.2 mM. Each point represents the mean incubation time until 50% of maximum intensity is reached ± standard deviation from five wells. Dashed lines represent standard deviation error limits for five samples of peptide in absence of lipids.
The recombinant Aβ peptides used in this study have methionine preceding position 1 (that is an aspartate), whereas synthetic Aβ peptides start at position 1 like the majority of in vivo generated Aβ. Control experiments with monomeric synthetic peptide Aβ(1–40) purified from size exclusion were therefore carried out. A higher concentration of synthetic peptide was needed compared to recombinant Aβ(M1–40) peptide to produce equal lag times. This could be explained by the methods of preparation. The recombinant Aβ(M1–40) has no variation in length whereas the synthetic Aβ(1–40) preparation may be less homogeneous, containing shorter fragments affecting the kinetics. The aggregation kinetics were however slowed down for both synthetic and recombinant peptide when adding DOPC, DPPC, or DOPC/DOPS large unilamellar vesicles.
So far, the presented data all concern the influence of lipid vesicles on the aggregation kinetics. Although the relation between interference caused by the vesicles and peptide-vesicles association is not considered trivial, we believe it makes sense to relate the kinetic data to a study of peptide adsorption to lipid membranes. We carried out monolayer experiments to elucidate if there is any difference in membrane adsorption of monomeric Aβ peptide depending on lipid charge and lipid acyl-chain packing. Fig. 7 shows the change in surface pressure (ΔΠ) when monomeric Aβ(M1–40) was injected under PC and PS monolayers at 6 mN/m and 25 mN/m initial surface pressure. At the low initial surface pressure, the monolayer is in a liquid expanded (LE) phase, and at the high initial surface pressure, the monolayer is in a liquid condensed (LC) phase. The LE monolayer can be considered a fluid phase where the hydrocarbon chains have a high conformational disorder, whereas the LC phase have small cross-sectional areas per lipid and highly ordered hydrocarbon chains. Comparisons of the chain order and the headgroup areas with those seen in bulk lamellar phases suggest that the molecular properties of the LE and LC phases are similar to those found in the liquid crystalline and gel bilayers, respectively (24). From the data in Fig. 7 we conclude that surface pressure increases over time for both types of lipids both in the LE and the LC phases. At both high and low surface pressure the peptide adsorption to the monolayer is slightly faster for PC headgroups compared to PS. A deeper analysis of these data is complicated by the fact that the Aβ peptide is highly surface active in itself (25), and it is not evident what are that the driving forces for peptide adsorption to the monolayer. However, the data clearly show that there is no significant difference in peptide adsorption as measured as the total increase in ΔΠ between the zwitterionic and anionic monolayers. Monomeric Aβ peptide was used and we do not predict aggregation in the still bulk solution within the period of the experiment. We are therefore not able to draw any conclusion on association of oligomeric or fibrillar aggregates to the lipid monolayer.
Figure 7.
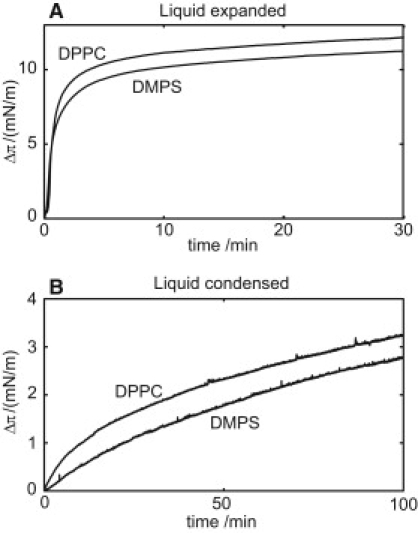
Langmuir monolayer experiment. 2 ml 30 μM Aβ(M1–40) was injected under a monolayer of DPPC or 2,3-dimercapto-1-propanesulfonic acid on a subphase of 20 mL 20 mM Tris/HCl, pH 7.4, 0.2 mM EDTA, 0.02% NaN3. (A) Liquid expanded phase (π0 = 6 mN/m) as a model for liquid disordered bilayer phase. (B) Liquid condensed phase (π0 = 25 mN/m) as a model for gel bilayer phase. The small pressure drop during injection due to mechanical disturbance is corrected for and Δπ is thus defined as pressure increase after injection.
We also measured the CD spectra of Aβ(M1–40) but saw no change in the presence of DOPC-vesicles. No helix structure could be detected in the absence or presence of lipids (data not shown).
Discussion
We believe the present data shows clearly that lipid vesicles can interfere with the aggregation of the Aβ peptide, and that this interference can be modulated by varying the properties of the lipid system. The lipid vesicles provide an interface that can potentially act as a catalytic or inhibiting surface, and can thereby influence the aggregation kinetics in similar manner as has been shown for e.g., nanoparticles (26,27). Moreover, the lipid vesicles are dynamic entities and the self-assembled structure can be altered due to the lipid-peptide interactions. In all experiments carried out in this study, the presence of unilamellar vesicles either retards or has no effect on Aβ amyloid formation. Aβ(M1–42) is intrinsically more aggregation-prone than Aβ(M1–40) and Aβ(M1–42) was therefore studied at lower peptide concentration than Aβ(M1–40). However, the addition of lipid vesicles produces qualitatively the same effects on the aggregation kinetics for both peptides, and in no case do we see an acceleration of fibril formation due to the membranes. Thus, in the pure buffer system used in this study, there is no indication that the membrane acts as a general nucleating surface for amyloid formation. Rather, the lipid membrane systems studied here interact with the peptide monomers or oligomers in a way that hinders the kinetics of the process under the conditions of this study (4.8–12 μM Aβ(M1–40) or 0.38–0.59 μM Aβ(M1–42) with 0.85–2.7 mM phospholipid in 20 mM Tris/HCl, 0.2 mM EDTA, 0.02% NaN3, pH 7.4 with no cosolvents or other additives such as hexafluoroisopropanol or DMSO). This does not exclude the possibility that lipid membranes have accelerating effects at other combinations of peptide and lipid concentration, or in the presence of other solution components. A notable difference between this and other studies of the effect of membranes on Aβ aggregation is that we use lower peptide concentration. The low peptide concentrations used here (380 nM–12 μM) approaches typical physiological concentrations (1.5 nM in healthy individuals and higher in patients (28,29)) and in a pure buffer system with no additives, such low concentrations are needed to obtain kinetic data with a distinct lag phase. The low peptide concentration also assures for a low peptide to lipid ratio as is the case in vivo. Other studies have reported distinct effects of agents (surfactants or nanoparticles) on aggregation kinetics depending on the ratio between protein and agent with an acceleration of aggregation in the regime of high protein/agent ratio and retardation in the low protein/agent regime (30,31). This further underscores the importance of the experimental conditions used. Thus, with low Aβ concentration, low Aβ/vesicle ratio and solutions free from cosolvents, we find either a retardation or no effect depending on the membrane composition.
The ThT assay used is primary in the respect that it allows us to directly monitor the propagation of amyloid formation in real time, but the effect from lipid bilayers is seen as a secondary kinetic effect. We believe the present data are consistent with adsorption (association) of Aβ peptide to the lipid bilayers, and provides quantitative measures of the aggregation kinetics at given conditions for the lipid/peptide ratio in solution. However, the data cannot be quantitatively correlated to the amount of peptide associated with the vesicles, and should therefore be looked on in relation to the extensive literature on this matter (3,10,27,28).
The model systems are chosen so that the phase behavior is varied by using lipids with different acyl-chains or by introducing cholesterol, whereas the lipid headgroup and the PC concentrations are the same. The observation that the delay in aggregation in the presence of DPPC vesicles is greatly reduced when the vesicles are instead in liquid ordered or liquid disordered state either by cholesterol incorporation or exchange of DPPC for DOPC, imply that the aggregation kinetics is in some way related to the lipid phase behavior.
Two scenarios for how lipid membranes can affect the aggregation kinetics are consistent with the prolonged lag-time: i), the amyloid formation can take place in bulk with part of the peptide partitioning to the membrane, causing an effectively lower bulk concentration and thus slower aggregation (16); or ii), the amyloid formation can take place at the membrane surface with the peptide membrane associated. The observed differences in aggregation kinetics may arise from higher partitioning of the peptide to gel state bilayers compared to the liquid crystalline bilayers, which would then support the first scenario described above. Indeed, high Aβ binding specificity for gel phase lipid bilayers finds some support in the literature (32,33). Still, this mechanism alone cannot explain that the addition of vesicles of DOPC or DPPC with cholesterol causes only very minor delays in the aggregation kinetics, as there are accumulating evidences pointing to higher affinity of Aβ for bilayers and monolayers that contain cholesterol (10,11,34,35).
The aggregation kinetics may be influenced by the mode of Aβ peptide association with the membrane, although it is not trivial to predict how a specific interaction will affect the aggregation process. The peptide may be located in the bilayer interfacial headgroup region, or it may penetrate into the hydrophobic layer of the bilayer. Several report show that Aβ fragments can bury in the acyl-chain regions in zwitterionic and anionic phospholipid Lα(d) bilayers (5,34,36,37), whereas less or no penetration into the hydrophobic core of the bilayer has been detected at high cholesterol concentrations (Lα(o) bilayers) or for gel phase bilayers. Corresponding results have been obtained for expanded and condensed phospholipid monolayers (37,38). It is also possible that there is accumulation of peptides in domain boundaries or defects, as reported for line-active proteins (39,40). It is well-known that small unilamellar gel phase vesicles can exhibit faceted structure with areas of planar bilayer structures (41,42). In the edges between the facets, the bilayer is highly curved and the packing of the acyl-chain is naturally different from the planar areas of the bilayer. It is possible that the peptides accumulate in such areas with packing defects.
For a diffusion-controlled process, such as the aggregation, the influence of membrane association on the diffusion might influence the aggregation. This is particularly relevant if there is a tight membrane association, where the peptide spends a large fraction of time in a membrane associated state. The different model membrane systems used in this study show clearly different lateral diffusion characteristics (23). This might provide an explanation for the results obtained for the PC systems showing a large delay in aggregation for the solid gel phase vesicles compared to the liquid crystalline vesicles, and it is in agreement with the second scenario of membrane associated aggregation suggested above. Assuming equal partitioning of Aβ to the PC membrane regardless of phase behavior, Aβ would be considerably more hindered in the translational diffusion in the membrane surface in the DPPC solid bilayer compared to the liquid crystalline bilayer. In such case, the half time, t0.5, seen at high DPPC concentration in Fig. 4 would represent t0.5 when all peptide is membrane bound and hindered by the low translational diffusion in the DPPC solid gel phase.
The concentration dependence of DPPC vesicles on t0.5 of Aβ(M1–40) aggregation reaches a plateau value at a stoichiometry of 15–30 lipids/peptide (Fig. 4). Because the membrane is a double layer and the majority of the vesicles are unilamellar, this corresponds to a presented phospholipid to peptide ratio of 1:7.5–15. The t0.5 value at the plateau may represent membrane bound peptide that is diffusion limited by the solid gel phase DPPC. Given a DPPC headgroup area in the gel phase of 46 Å2 (20), a stoichiometry of 15–30 corresponds to 345–690 Å2 or 19 × 19 to 26 × 26 Å membrane surface area presented per peptide. These numbers should be considered as maximum surface area per peptide, in case not all vesicles are unilamellar, and imply a rather dense incorporation of Aβ in the membrane. If globular, the Aβ peptide would have a diameter of ∼20 Å, whereas a cross section with smaller or larger diameter may be achieved for an extended peptide depending on orientation.
An important aspect of physiological membranes is their negative charge, and interactions of Aβ with anionic phospholipids are somewhat controversial (12). We observe approximately the same lengthening of the lag phase for Aβ fibrillation regardless of the DOPS/DOPC ratio in the range 0:1–2:1 (Fig. 6).
The Aβ peptide is highly surface active and it unspecifically adsorbs to many surfaces. This complicates the design of adsorption experiments. Comparative experiments between supported bilayers are difficult to evaluate as the surface coverage for bilayers of different composition is unknown and the peptide often binds to the underlying supporting material. Lipid monolayers have the benefit of always having 100% coverage and were therefore chosen for interaction studies to compare binding to monolayers of different composition and surface pressure. The monolayer experiments show no significant difference in the adsorption of monomeric Aβ(M1–40) to PS and PC monolayers at the air-water interface.
At pH 7.4, Aβ has a net charge of −3 (see Fig. 1 C), whereas PC is zwitterionic and PS has a net charge of −1. It is somewhat surprising that neutral or highly negatively charged large unilamellar vesicles affect the rate of amyloid formation equally. This suggests that the electrostatic repulsion between Aβ and the negatively charged phospholipid headgroups is overcome by other highly favorable interactions, e.g., between the hydrophobic tail of Aβ and the interior of the bilayer. The aggregation kinetics for both Aβ(M1–40) and Aβ(M1–42) were found to be strongly dependent on salt concentration, with a shorter lag time observed at higher salt in agreement with earlier studies (43). This implies that the net negative charge (−3) of Aβ counteracts aggregation and that the electrostatic repulsion between peptides is screened by salt. A reduced inhibition of aggregation due to reduced electrostatic repulsion may also explain the faster aggregation of familial Aβ mutants with −1 or −2 net negative charge (44). The lag time in the presence of DOPC vesicles decreased with increasing ionic strength but was always prolonged compared to the peptide in absence of vesicles. This shows that the retarding effect of DOPC membranes on Aβ fibril formation persists at physiological salt, and that the screening of inter-Aβ repulsion by salt may speed up fibril formation also for the membrane associated peptide. Based on this, it seems unlikely that the interaction between the peptide and the membrane should be mainly electrostatic in its nature. We believe, however, that any quantification here is impossible because there is no trivial function describing the relation between the peptide-membrane interaction and the lag time.
No clear trend could be detected in the elongation rates. This indicates that the mechanism behind the increased lag time must originate in interferences taking place during the build up of small oligomers from monomers. Once nucleated, the aggregation process proceeds with the same rate in the absence and presence of membranes.
Conclusion
Phospholipid membranes delay the onset of Aβ fibril formation. By comparing vesicles with different membrane phase behavior we conclude that the solid gel phase DPPC retards the fibril formation. The most possible explanation for this retardation is due to bilayer defects in faceted vesicles and low lateral diffusion in the bilayer. The retardation of Aβ fibril formation kinetics in the presence of vesicles reaches saturation and is present when the vesicles are added during the lag time up to the nucleation point. Both free and membrane-bound peptide is screened by salt leading to a shortening of the lag phase. Neutral and negatively charged PC and PS model membranes retard fibril formation to the same extent implying that the electrostatic repulsion between Aβ and the negatively charged phospholipid headgroups is overcome by other interactions that allow for the same retardation effect as with uncharged membranes. This is supported by monolayer experiments.
Acknowledgments
This work was supported by the Swedish Research Council and its Linneaus programme OMM (to S.L., E.S.), The Swedish Foundation for Strategic Research (to E.S.), Per-Eric and Ulla Schyberg's Foundation (to E.S.), the Crafoord Foundation (to S.L.), and the Royal Physiographic Society (to E.H.).
References
- 1.Gellermann G.P., Appel T.R., Fändrich M. Raft lipids as common components of human extracellular amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:6297–6302. doi: 10.1073/pnas.0407035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh D.M., Klyubin I., Selkoe D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 3.Gorbenko G.P., Kinnunen P.K.J. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids. 2005;141:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Durell S.R., Guy H.R., Pollard H.B. Theoretical models of the ion channel structure of amyloid beta-protein. Biophys. J. 1994;67:2137–2145. doi: 10.1016/S0006-3495(94)80717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kremer J.J., Pallitto M.M., Murphy R.M. Correlation of beta-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry. 2000;39:10309–10318. doi: 10.1021/bi0001980. [DOI] [PubMed] [Google Scholar]
- 6.Sparr E., Engel M.F.M., Killian J.A. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577:117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 7.Yang A.J., Chandswangbhuvana D., Glabe C.G. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J. Neurosci. Res. 1998;52:691–698. doi: 10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Cecchi C., Nichino D., Relini A. A protective role for lipid raft cholesterol against amyloid-induced membrane damage in human neuroblastoma cells. Biochim. Biophys. Acta. 2009;1788:2204–2216. doi: 10.1016/j.bbamem.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 9.Subasinghe S., Unabia S., Small D.H. Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J. Neurochem. 2003;84:471–479. doi: 10.1046/j.1471-4159.2003.01552.x. [DOI] [PubMed] [Google Scholar]
- 10.Devanathan S., Salamon Z., Tollin G. Effects of sphingomyelin, cholesterol and zinc ions on the binding, insertion and aggregation of the amyloid Abeta(1-40) peptide in solid-supported lipid bilayers. FEBS J. 2006;273:1389–1402. doi: 10.1111/j.1742-4658.2006.05162.x. [DOI] [PubMed] [Google Scholar]
- 11.Ji S.R., Wu Y., Sui S.F. Cholesterol is an important factor affecting the membrane insertion of beta-amyloid peptide (A beta 1-40), which may potentially inhibit the fibril formation. J. Biol. Chem. 2002;277:6273–6279. doi: 10.1074/jbc.M104146200. [DOI] [PubMed] [Google Scholar]
- 12.Matsuzaki K. Physicochemical interactions of amyloid-peptide with lipid bilayers. Biochim. Biophys. Acta. 2007;1768:1935–1942. doi: 10.1016/j.bbamem.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Murphy R.M. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim. Biophys. Acta. 2007;1768:1923–1934. doi: 10.1016/j.bbamem.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Sabate R., Gallardo M., Estelrich J. Spontaneous incorporation of beta-amyloid peptide into neutral liposomes. Colloids Surf. A. Physicochem. Eng. Asp. 2005;270:13–17. [Google Scholar]
- 15.Hortschansky P., Schroeckh V., Fändrich M. The aggregation kinetics of Alzheimer's beta-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 2005;14:1753–1759. doi: 10.1110/ps.041266605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hellstrand E., Boland B., Linse S. An improved method for monitoring Aβ aggregation produces highly reproducible kinetic data and indicate a two-phase process. ACS. Chem. Neurosci. 2009;1:13–18. doi: 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walsh D.M., Thulin E., Linse S. A facile method for expression and purification of the Alzheimer's disease-associated amyloid beta-peptide. FEBS J. 2009;276:1266–1281. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rouser G., Fkeischer S., Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 19.Biancalana M., Makabe K., Koide S. Molecular mechanism of thioflavin-T binding to the surface of beta-rich peptide self-assemblies. J. Mol. Biol. 2009;385:1052–1063. doi: 10.1016/j.jmb.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cevc G. Marcel Dekker; New York, NY: 1993. Phospholipids Handbook. [Google Scholar]
- 21.Vist M.R., Davis J.H. Phase-equilibria of cholesterol dipalmitoylphosphatidylcholine mixtures: H-2 nuclear magnetic resonance and differential scanning calorimetry. Biochemistry. 1990;29:451–464. doi: 10.1021/bi00454a021. [DOI] [PubMed] [Google Scholar]
- 22.Karmakar S., Raghunathan V.A., Mayor S. Phase behavior of dipalmitoyl phosphatidylcholine (DPPC)-cholesterol membranes. J. Phys. Condens. Matter. 2004;17:1177–1182. [Google Scholar]
- 23.Lindblom G., Oradd G., Filippov A. Lipid lateral diffusion in bilayers with phosphatidylcholine, sphingomyelin and cholesterol—an NMR study of dynamics and lateral phase separation. Chem. Phys. Lipids. 2005;141:179–184. doi: 10.1016/j.chemphyslip.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Larsson K. Oily Press; Dundee, Scotland: 1994. Lipids—Molecular Organization, Physical Functions and Technical Applications. [Google Scholar]
- 25.Chi E.Y., Ege C., Lee K.Y. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer's disease amyloid-beta peptide. Proteins. 2008;72:1–24. doi: 10.1002/prot.21887. [DOI] [PubMed] [Google Scholar]
- 26.Cabaleiro-Lago C., Quinlan-Pluck F., Linse S. Inhibition of amyloid beta protein fibrillation by polymeric nanoparticles. J. Am. Chem. Soc. 2008;130:15437–15443. doi: 10.1021/ja8041806. [DOI] [PubMed] [Google Scholar]
- 27.Linse S., Cabaleiro-Lago C., Dawson K.A. Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA. 2007;104:8691–8696. doi: 10.1073/pnas.0701250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Höglund K., Hansson O., Wiltfang J. Prediction of Alzheimer's disease using a cerebrospinal fluid pattern of C-terminally truncated beta-amyloid peptides. Neurodegener. Dis. 2008;5:268–276. doi: 10.1159/000119457. [DOI] [PubMed] [Google Scholar]
- 29.Kuo Y.M., Emmerling M.R., Roher A.E. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 30.Cabaleiro-Lago C.,, Quinlan-Pluck F., Linse S. Dual effect of amino modified polystyrene particles on amyloid β protein fibrillation. ACS Chem. Neurosci. 2010 doi: 10.1021/cn900027u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giehm, L. O. C., G. Christiansen, …, D. Otzen. 2010. SDS-induced fibrillation of a-synuclein: an alternative fibrillation pathway. Submitted. [DOI] [PubMed]
- 32.Choucair A., Chakrapani M., Johnston L.J. Preferential accumulation of A beta(1–42) on gel phase domains of lipid bilayers: an AFM and fluorescence study. Biochim. Biophys. Acta. 2007;1768:146–154. doi: 10.1016/j.bbamem.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Yoda M., Miura T., Takeuchi H. Non-electrostatic binding and self-association of amyloid beta-peptide on the surface of tightly packed phosphatidylcholine membranes. Biochem. Biophys. Res. Commun. 2008;376:56–59. doi: 10.1016/j.bbrc.2008.08.093. [DOI] [PubMed] [Google Scholar]
- 34.D'Errico G., Vitiello G., D'Ursi A.M. Interaction between Alzheimer's A beta(25–35) peptide and phospholipid bilayers: the role of cholesterol. Biochim. Biophys. Acta. 2008;1778:2710–2716. doi: 10.1016/j.bbamem.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Yip C.M., Darabie A.A., McLaurin J. Abeta42-peptide assembly on lipid bilayers. J. Mol. Biol. 2002;318:97–107. doi: 10.1016/S0022-2836(02)00028-1. [DOI] [PubMed] [Google Scholar]
- 36.Dante S., Hauss T., Dencher N.A. Cholesterol inhibits the insertion of the Alzheimer's peptide A beta(25–35) in lipid bilayers. Eur. Biophys. J. 2006;35:523–531. doi: 10.1007/s00249-006-0062-x. [DOI] [PubMed] [Google Scholar]
- 37.Kremer J.J., Sklansky D.J., Murphy R.M. Profile of changes in lipid bilayer structure caused by beta-amyloid peptide. Biochemistry. 2001;40:8563–8571. doi: 10.1021/bi010417x. [DOI] [PubMed] [Google Scholar]
- 38.Maltseva E., Kerth A., Brezesinski G. Adsorption of amyloid beta (1-40) peptide at phospholipid monolayers. Chembiochem. 2005;6:1817–1824. doi: 10.1002/cbic.200500116. [DOI] [PubMed] [Google Scholar]
- 39.Freites J.A., Ali S., Dennin M.B. Annexin A1 interaction with a zwitterionic phospholipid monolayer: a fluorescence microscopy study. Langmuir. 2004;20:11674–11683. doi: 10.1021/la049713b. [DOI] [PubMed] [Google Scholar]
- 40.Haas H., Mohwald H. Specific and unspecific binding of Concanavalin A at monolayer surfaces. Thin Solid Films. 1989;180:101–110. [Google Scholar]
- 41.Blaurock A.E., Gamble R.C. Small phosphatidylcholine vesicles appear to be faceted below the thermal phase-transition. J. Membr. Biol. 1979;50:187–204. [Google Scholar]
- 42.Dierksen K., Typke D., Baumeister W. Three-dimensional structure of lipid vesicles embedded in vitreous ice and investigated by automated electron tomography. Biophys. J. 1995;68:1416–1422. doi: 10.1016/S0006-3495(95)80314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klement K., Wieligmann K., Fändrich M. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer's abeta(1-40) amyloid fibrils. J. Mol. Biol. 2007;373:1321–1333. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 44.Betts V., Leissring M.A., Walsh D.M. Aggregation and catabolism of disease-associated intra-Abeta mutations: reduced proteolysis of AbetaA21G by neprilysin. Neurobiol. Dis. 2008;31:442–450. doi: 10.1016/j.nbd.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]