

Abstract
We present a novel sampling approach to explore large protein conformational transitions by determining unique substates from instantaneous normal modes calculated from an elastic network model, and applied to a progression of atomistic molecular dynamics snapshots. This unbiased sampling scheme allows us to direct the path sampling between the conformational end states over simulation timescales that are greatly reduced relative to the known experimental timescales. We use adenylate kinase as a test system to show that instantaneous normal modes can be used to identify substates that drive the structural fluctuations of adenylate kinase from its closed to open conformations, in which we observe 16 complete transitions in 4 μs of simulation time, reducing the timescale over conventional simulation timescales by two orders of magnitude. Analysis shows that the unbiased determination of substates is consistent with known pathways determined experimentally.
Introduction
Large conformational transitions play an important role in protein functions, including enzyme catalysis (1), force generation in motor proteins (2), allosteric communication in proteins (3), and changes in the selectivity filter of potassium channels (4). How to understand the molecular origins of these long timescale motions is limited by the considerable timescale heterogeneity of native protein fluctuations that are responsible for functional conformational motions. For example, a recent study on adenylate kinase (AdK) shows that there is a close connection between fast local atomic fluctuations and long timescale conformational transitions. This linkage between local and global fluctuations seems to be a general property of the protein energy landscape (5). A comparison between a thermophilic and mesophilic AdK suggests that fast timescale local fluctuations, although different between the proteins at a given temperature, lead to very similar long timescale collective motions when measured equidistant from their respective melting temperatures (6). Another ensemble-based analysis reveals that local conformational fluctuations facilitate the coupling between proton binding and global structural transitions (7).
Atomistic molecular dynamics (MD) is, in principle, a powerful approach for exploring the molecular mechanisms of functional protein transitions, especially the local conformational fluctuations that occur on short timescales. However, it is still a challenge to obtain a good ensemble estimate of large conformational transitions that often occur on the timescale of milliseconds to seconds. By contrast, global fluctuations can be captured based on collective protein displacements near the native state energy minimum when described by normal modes (8–10). A more general elastic normal mode analysis, which uses a Hookean potential model (11), provides a Gō-like representation of the native reference structure that removes the model dependencies of normal modes derived from empirical protein force fields. This has been successfully used to study protein dynamics by analyzing static experimental x-ray or nuclear magnetic resonance data of various endpoint conformational states (12–14), as well as for predicting large conformational transition pathways (15–19). For example, Miyashita et al. (18) adopted a scheme to iteratively deform the initial native state conformation along one or several lowest frequency modes to explore how cracking or partial unfolding happens during conformational transitions. However, it is still unclear how a protein reaches such large collective motions over long timescales by coupling to higher frequency local fluctuations, especially when the reaction coordinate is unknown a priori.
In this work, we develop a general method that uses atomistic MD simulations to evaluate time progressions of instantaneous low-frequency normal modes generated from an elastic network model, while retaining the underlying anharmonic nature of the atomistic motion. The working hypothesis of our computational approach is the growing appreciation that inherent short timescale fluctuations of the apo form of enzymatic proteins involve a sampling of conformational states that are templated for binding of relevant ligands and other proteins, or at least anticipate some functional states occurring on a longer timescale (20,21). We illustrate the approach using equilibrium MD trajectories of the closed state of AdK (1AKE (22)) to find low-frequency motions that simultaneously probe normal modes that are similar to its open state (4AKE (23)). These instantaneous normal mode trajectories give rise to common intermediate substates, some of which drive the structural fluctuations of AdK from its closed to open state, in which we observe 16 complete conformational transitions in 4 μs of simulation time, compared to the experimental transition timescale for AdK (without ligand) of ∼0.1 ms (20). The key result of our study is that no a priori definition of a reaction coordinate is necessary to characterize transition pathways between two known protein end-states.
Methods
Elastic normal-mode analysis
Normal mode analysis provides an approach for analytically solving the equations of motion under the assumption that the system is well approximated by a harmonic potential energy near the global native minimum (14),
| (1) |
where is the displacement of atomic coordinate i at time t, mi is the corresponding atomic mass, eij represents the ith coordinate of normal mode j, ωj2 corresponds to the frequency of the jth normal mode, and Cj and ϕj are the amplitude and phase of mode j, respectively, which are determined by the initial conditions.
The solution of Eq. 1 depends on the potential energy model. Tirion (11) introduced a simple pairwise Hookean spring potential based on the known structure of the native protein which naturally corresponds to the global energy minimum. The basic principle of this framework is to treat the interaction potential between two atoms as an elastic spring,
| (2) |
where and denote the position vectors for atom i and j, and . The zero superscript indicates the reference structure, and C is a phenomenological constant. Expanding Eq. 2 to second order about yields
| (3) |
where . The total potential energy within a molecule is then given by
| (4) |
where Rc is an interaction cutoff parameter which effectively defines the number of elastic springs. Within the harmonic approximation, the second derivative of potential energy with respect to atomic coordinate results in the formulation
| (5) |
where is the 3N-dimentional vector representing the Cartesian coordinates of N atoms, and H is the Hessian matrix, that when diagonalized, generates the normal mode eigenvectors, , and corresponding eigenvalues (frequencies), ωj2.
Structural similarity measured by normal-mode overlaps
The overlap of two normal modes is simply defined as the inner product between two normal mode eigenvectors, and ,
| (6) |
In our study, we use the normal mode overlap to define the structural similarity between two different configurations a and b of a given molecule; the normal modes and frequencies of each conformation can be calculated from an elastic network model under the assumption that every configuration is treated as a reference structure independently. We then fix the frequency order of all normal modes, , in set a, and then search for the normal mode in set b, with the highest overlap to the low-frequency mode , i.e.,
| (7) |
The structural similarities between conformation a and b are then defined as weighted normal mode overlaps,
| (8) |
where (ωia)2 is the frequency of target normal mode eia, and tc is number of chosen normal modes. The value of sa,b varies between 0 and 1, where larger values correspond to greater similarity than smaller values.
Fluctuation correlation coefficient
The fluctuation correlation, mij, between the ith and jth α-carbon atom atoms is defined as
| (9) |
where xi and xj are coordinate vectors of a given snapshot, and are the corresponding average coordinate vectors evaluated across the trajectory, and 〈…〉 denotes an ensemble average. Negative values of mij correspond to residues i and j moving in opposite directions, whereas positive values correspond to the same directional movement between residues i and j.
Molecular dynamics and instantaneous normal mode simulations
Molecular dynamics (MD) simulations were run on the closed state of Escherichia coli adenylate kinase using only chain A without the ligands and crystal waters found in PDB entry 1AKE. Simulations were run with the GROMACS 4.0.3 package (24) using the OPLS-AA all-atom force field (25) with 14,714 TIP4P water molecules (26) and four Na+ ions to neutralize overall charge. We employed cubic periodic boundary conditions as well as particle-mesh Ewald (27) using a 1-nm real-space cutoff for the electrostatic interactions, and a 1.4-nm cutoff was used for van der Waals interactions. The simulations were run in the NPT ensemble at constant temperature (300 K) and pressure (1 bar) using the Berendsen coupling scheme (28) for both temperature and pressure, with a step size of 2 fs, and all bonds were constrained with the LINCS algorithm (29). A 500-ps position restrained (heavy atoms) molecular dynamics (MD) simulation was carried out at target temperature to solvate waters and ions after energy minimization procedure. The initial 2 ns of the MD trajectory starting from 1AKE were defined to be part of the equilibration phase, and the subsequent 8 ns were used to sample 15 frames to launch 20 independent simulations for each frame by regenerating velocities. These 300 initial conditions were simulated for a further 5 ns; ∼10% of these trajectories were continued for another 15 ns for a total of 20 ns. From these production simulations, normal modes were generated from the elastic network model every 10 ps over each 5-ns or 20-ns simulation. The cutoff we used for the elastic network model was 15 Å, which allowed for the best differentiation between the lowest frequency normal modes between 1AKE and 4AKE, and we utilized the three lowest frequency normal modes to calculate normal mode similarity (Eq. 8) to the 1AKE and 4AKE reference states.
Results
Fig. 1 a illustrates the time progression of the instantaneous normal mode (INM) similarity of the initial trajectory starting from the 1AKE PDB structure. The INMs decay quickly from the closed (1AKE) start state (i.e., values of ∼1.0), but remain stable around relatively high similarity values of ∼0.7 to the closed state and relatively low similarity of ∼0.55 to the open (4AKE) state, clearly indicating that no transition has occurred. These INM similarity values reflect largely fluctuations in the 1AKE native basin. Fig. 1 b illustrates the root-mean-square deviation (RMSD) progression with respect to the 1AKE and 4AKE reference structures. Clearly the 1AKE ensemble is broad and with large fluctuations around its native basin, but clearly no transition is evident as measured by the complete lack of RMSD similarity to 4AKE. Another independent trajectory (data not shown) shows the same trend.
Figure 1.
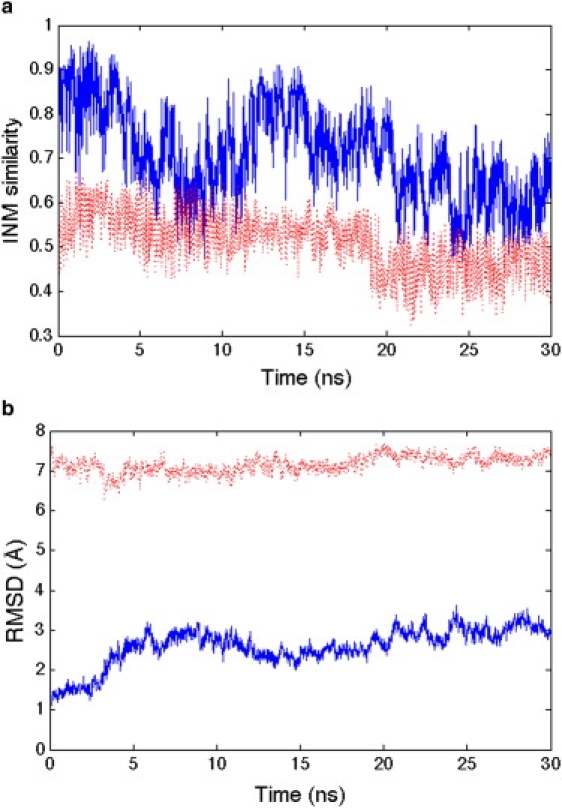
(a) Instantaneous normal mode similarity and (b) RMSD with respect to both reference states 1AKE (blue solid line) and 4AKE (red dashed line) over 30 ns. The INM values are largely measuring equilibrium fluctuations in the 1AKE basin.
We picked 15 snapshots from the 1AKE equilibrium ensemble in the first 10 ns, and launched 20 independent 5-ns trajectories for each snapshot to look for INM similarity change that signals a possible transition away from 1AKE toward the 4AKE end state. A vast majority of these 300 short simulations largely sample the 1AKE basin, with INM similarity values to 1AKE remaining ≥0.7, while the INM similarity to 4AKE is ≤0.6. But for ∼25% of trajectories, the time progressions of the INM similarity when projected against the 1AKE and 4AKE states show high-frequency motions that progress to large conformational changes of three types. Specifically, a majority of trajectories (∼20%) measure a normal mode similarity to 1AKE that is small and remains stable at <0.6, but this does not correspond to any greater similarity to 4AKE (Case 1). A much smaller number of trajectories (∼4%) show large fluctuations of INM similarity between the 1AKE and 4AKE reference states that are nearly anticorrelated (Case 2). Finally, a different case (<1%) corresponds to a small and stable value of the INM similarity to 1AKE, whereas the INM similarity to 4AKE is quite flexible, fluctuating greatly between values of 0.5 and 0.9 (Case 3). These are the primary candidates for productive reaction coordinates, distinguished by either greater INM dissimilarity to 1AKE and/or increased INM similarity to 4AKE. To verify whether the three cases are relatively stable substates, 25 of these trajectories were continued for 20 ns to clarify that the three general cases held for longer simulation times, which they did as shown in Fig. 2, a–c; in fact, one of trajectories restarted from the 1AKE ensemble reached the 4AKE ensemble within 20 ns.
Figure 2.
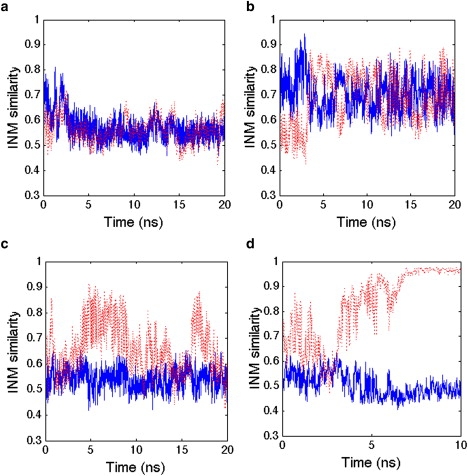
Instantaneous normal mode (INM) similarities to both reference states 1AKE and 4AKE along 20-ns MD trajectories. (Solid blue line and dashed red line) INM similarity to 1AKE and 4AKE, respectively. (a) Case 1 shows no INM similarity to either state; (b) Case 2 shows anticorrelated motions of the INM similarity metric; (c) Case 3 shows low similarity to the 1AKE reference and a developing resemblance to 4AKE; and (d) a trajectory starting from Case 3 transitions to the 4AKE ensemble.
Given these three stable substates on the 20-ns timescale, we took 10 snapshots over one of the longer 20-ns trajectory for each case, and ran 10 independent 5-ns simulations for each snapshot to push further along the three possible INM reaction coordinates. For trajectories launched from Case 3, we recorded two transition events for the 100 independent 5-ns simulations that appear to unambiguously reach the open state based on small and stable INM similarity values of 0.5 to 1AKE and high similarity values to 4AKE that remain stable at >0.9 (Fig. 2 d). Forty-one trajectories starting from Case 3 reverted to the Case 1 substate, while another 28 remained as a Case 3 substate. Another 29 of the 5-ns trajectories for Case 3 showed evidence for time progression toward even larger INM similarity values to 4AKE, and when these trajectories were continued-out for an additional 15 ns, eight of them showed a complete transition to a now stable 4AKE ensemble. By contrast, ∼76% of the trajectories starting from Case 2 continued to show strongly anticorrelated INM similarity trends, ∼18% reverted back to the 1AKE ensemble, and 6% of these trajectories converted to the Case 3 substate. When we continued 20 of the 5-ns trajectories for an additional 15 ns, starting from Case 2 as well as the 6% trajectories that transitioned to the Case 3 substate, we measured no clear transition to the 4AKE ensemble. When they were continued-out to 40 ns, however, two of them transitioned to 4AKE. Finally, whereas most of the Case 1 trajectories continued to maintain the low INM similarity to both 1AKE and 4AKE, 12 trajectories evolved to Case 3, and when these were continued for an additional 15 ns, three trajectories showed a transition to the 4AKE reference state. The details of these INM trajectories are reported in Table 1. Note that we only follow the normal-mode similarity to the 1AKE and 4AKE end-states, and thus do not dictate a reaction coordinate a priori. In what follows, we analyze the INM trajectories more carefully to determine the origin of the substates and pathways that we observe.
Table 1.
Conformational transitions among substates, 1AKE, and 4AKE by following instantaneous normal mode similarity
| INM Similarity type | 1AKE ensemble | Case 1 | Case 2 | Case 3 | 4AKE ensemble |
||
|---|---|---|---|---|---|---|---|
| <20 ns | 30 ns | 40 ns | |||||
| Case 1 | 0 | 88 | 0 | 12 | 3 | ||
| Case 2 | 18 | 0 | 76 | 6 | 1 | 1 | |
| Case 3 | 0 | 41 | 0 | 57 | 10 | ||
Case numbers refer to different INM similarity trends to 1AKE and 4AKE (see text).
INM trajectory analysis
We have shown that instantaneous normal modes from an elastic network model provide an excellent reaction coordinate to monitor progress of large and long timescale conformational transitions between known end-states, and hence will be general to arbitrary protein system. Based on the information in Table 1, the transitions based on INM similarity suggest a mechanism as outlined in Fig. 3 a.
Figure 3.
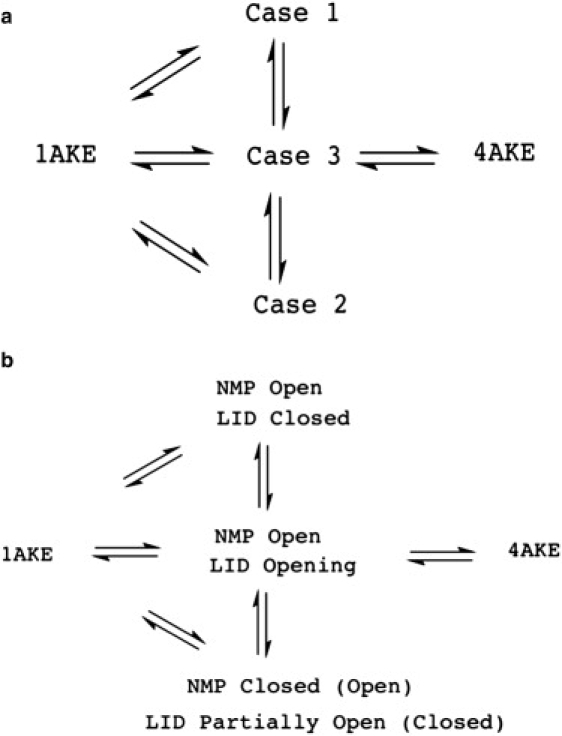
Mechanistic information to complete a transition from the closed (1AKE) to open (4AKE) states. (a) Mechanistic information based on INM similarities to both reference states 1AKE and 4AKE. (b) Mechanistic information based on combining INM similarity trajectories and domain distances to complete a transition from the closed to open states.
We now explore the specific biological implications of INM similarity trajectories for the 1AKE to 4AKE transition for AdK by analyzing MD simulation trajectories for relevant domain motions by measuring average domain distances, defined as the geometric center distance between two domains (based only on α-carbons), to mechanistically understand the corresponding structural changes. AdK is composed of three domains comprising a central and stable CORE domain (residues 1–29, 68–117, and 161–214) and the flexible LID (residues 118–160) and NMP (residues 30–67) domains. In going from the closed to the open state, domain distance from NMP to CORE must increase from 18 Å to 22 Å, and that from LID to CORE must change from 21 Å to 30 Å.
In the initial simulation around 1AKE (Fig. 1), we find the INM similarity is measuring fluctuations of the NMP-CORE distance, whereas the LID-CORE distance remains virtually unchanged from the closed state. For Case 1, the NMP domain is open, but the LID domain largely fluctuates around the closed state (Fig. 4 a). For Case 2, the NMP domain fluctuates between partially open and closed state, whereas the LID domain motion is anticorrelated to the given NMP state: when the LID is closed, then NMP is partially open; and when LID is half open, then the NMP state is closed (Fig. 4 b). For Case 3, the NMP domain is stably open, whereas the LID domain progresses to the open state (Fig. 4 c). Finally, when the INM similarity records evidence of a complete and stable transition to 4AKE, both the NMP and LID domains have settled into their open conformations with respect to the CORE domain (Fig. 4 d, which corresponds to Fig. 2 d).
Figure 4.
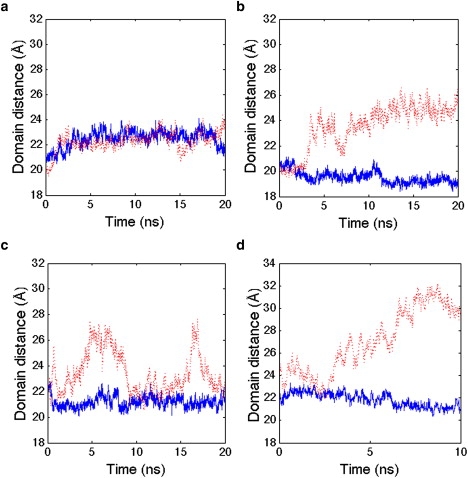
Domain distance changes along trajectories. (Solid blue line) Interdomain distance between the NMP and CORE domains. (Dashed red line) Interdomain distance between the LID and CORE domains. (a) Case 1 substate; (b) Case 2 substate; (c) Case 3 substate; and (d) a trajectory starting from Case 3 transitions to the 4AKE ensemble.
Combining the INM similarity mechanism in Fig. 3 a with the corresponding domain distance trends in Fig. 4, we can now assign a more detailed mechanism for AdK in which the 16 complete transition pathways from the closed to open state always involve the NMP domain opening first, accompanied by significant fluctuation of the LID domain to a partially open state (Case 3). We also observe a substate in which the NMP domain is open and the LID domain is closed (Case 1), and the Case 2 substate showing anticorrelated motions of a closed (partially open) NMP and partially open (closed) LID domain. Fig. 3 b details the mechanism from 1AKE to 4AKE based on domain transition pathways and the contents of Table 1, all derived from INM similarity trajectories based solely on information of the protein end-states.
Fluctuation correlation analysis
We use a fluctuation correlation analysis to explore the coupling among anharmonic motions of residues in the various substates to enable us to understand how they couple to the global scale conformational transition. The resulting correlations are biased by the scheme of how the simulation trajectory is aligned to the reference structure, which we attempted to mitigate by aligning all trajectories to the CORE domain, as nuclear magnetic resonance data showed that there are no significant internal conformational changes in this domain (20), unlike the residues in the hinge regions and NMP and LID domains. Equilibrium trajectories were selected to calculate residue fluctuation correlations using Eq. 9 for each substate type outlined in Fig. 3 a.
We found that residue pairs do not show strong negative Pearson correlation coefficients, and therefore do not show any differences among different substates (data not shown here). However, strong positive correlations are observed that do, in fact, differentiate the substates. Fig. 5 illustrates the statistics of positive correlations only, using a cutoff of 0.4 that is large enough to detect same directional movements without the statistical noise of using a lower value. Generally, fluctuations in different substates share many similarities. Apparently, the NMP and LID domains move concertedly in the same direction, except for small regions—which is highly consistent with the gross transition mechanisms of AdK. However, the correlation metric also defines new regions such as C1 labeled in Fig. 5, which involves the connectivity among four adjacent β-strands (residues 1–7, 28–31, 81–85, and 193–198) that make up a β-sheet in CORE domain and also connect to the NMP and LID domains. It has been experimentally shown that the thermal stability of AdK is not significantly influenced by NMP and LID domains, but is largely defined by the CORE domain (30). The strong positive correlations in Fig. 5, C1, imply that this β-sheet probably plays an important role in the thermal stability of the protein because it is observed in all substates.
Figure 5.
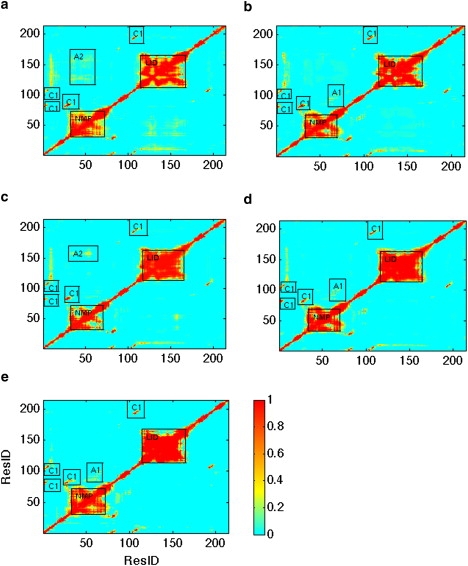
Statistics on residue-to-residue fluctuation correlations. The x axis and y axis are residue numbers. The color bar represents frequency, i.e., the number of trajectories having strong positive correlation (>0.4) divided by the total number of trajectories in such substate. (a–e) 1AKE, Case 1, Case 2, Case 3, and 4AKE ensemble, respectively.
However, differences among different substates also exist as shown in Fig. 5. For the Case 1, Case 3, and 4AKE ensembles, region A1 shows some degree of strong correlations that does not exist in the 1AKE and Case 2 ensembles. By contrast, the 1AKE and Case 2 ensembles show higher frequency of strong positive correlation in region A2 than the other ensembles. These differences can be explained by the fact that residues 81–100 in the CORE domain of region A1 come into close proximity and thereby stabilize the NMP domain in its open state. By contrast, the hinge residues 156–167 between the LID and CORE domains, which define the A2 region, stabilize the NMP domain in its closed state. Overall the LID and NMP domains are highly flexible, although the different substates show regions corresponding to reduced fluctuations (i.e., correlation holes) due to hinge residues. For the LID domain, apparently the positive correlations of these hinge residues must be activated, as they are in Case 3, to reach the 4AKE open state, because they are inactive in Case 1 and hence resemble the 1AKE closed state. The least productive route for conformational transition from 1AKE to 4AKE, through the Case 2 substate, shows that the NMP domain hinge residues show reduced motion consistent with its closed or partially open form.
Discussion
Throughout hundreds of short independent simulations, we find that the dominant transition pathway is
although occasionally 1AKE can directly evolve to Case 3 before transitioning to the 4AKE ensemble. By contrast, we find that the alternative path
is far less populated. Although the LID domain is flexible, our simulated pathways clearly show that the LID domain prefers the closed state, since the reversible rate from
is higher than the corresponding forward rates (Table 1). These results are consistent with experiments on AdK. Hanson et al. (31) used high-resolution single-molecule FRET to quantitatively measure the distance changes between the LID and CORE domains (fluorescent-labeled residues 127 and 194, respectively) in AdK, concluding that equilibrium favors the closed state even in the absence of substrates. Henzler-Wildman et al. (20) also provided experimental domain distance changes between NMP and LID domains (labeled residues 52 and 145), via their use of fluorescence resonance-energy transfer, in which they found that a state similar to the closed state is sampled to a significant fraction even without ligand. As both experiments did not measure the distance changes between NMP and CORE domains, the flexibility of the NMP domain cannot be directly compared to experiment.
Our unforced reaction coordinate method has found mechanisms of conformational transition similar to previous computational studies that have explored conformational transition pathways along predefined reaction coordinates for AdK, either from the open to closed form, or vice versa. Maragakis and Karplus (32) used a coarse-grained mixed potential to distort structure along an energy-minimum path between the open and closed end-state to predict that the LID domain closing precedes the closing of the NMP domain. Instead of using a single multidimensional potential, Chu and Voth (33) utilized a set of interconnected one-dimensional coarse-grained potentials, known as the double-well network model, to introduce roughness into the free energy model. By using different initial structures, they found similar pathways to ours, although their model cannot determine the preference for a particular pathway as we have found here. Arora and Brooks (34) used atomistic MD simulations and umbrella sampling along a predefined reaction coordinate to explore the energy landscape in the presence and absence of ligand, in which they found support for a population-shift mechanism for ligand binding to AdK. By contrast, using a coarse-grained model and mapping of the lowest normal modes of a nonlinear elastic network model to predefined reaction coordinates (domain distance), Whitford et al. (35) showed a mixed mechanism in going from the open to closed state for AdK. In particular, the LID domain motion follows a population-shift mechanism whereas the NMP domain follows a ligand-induced mechanism, with a pathway of binding events in which LID domain motion always precedes NMP motion in either the opening or closing transitions. Lu and Wang (36) also used a coarse-grained potential to study the AdK conformational transition, involving a superposition of microscopic two-well potentials between α-carbons, whose wells are determined by the known open (4AKE) and closed (1ANK) structures. Using trajectories that measure RMSD changes from the 4AKE and 1ANK structures, they determined an intermediate where the LID domain is closed and the NMP domain is open, and a second transient intermediate in which the NMP domain is closed but the LID domain is open. They found that the latter pathway dominated the conformational transition for AdK opening, with the LID domain opening first followed by opening of the NMP domain, contradicting our atomistic MD results presented here. Finally, Kubitzki and de Groot (37) provided an interesting study that used atomistic force fields and replica exchange combined with essential dynamics to explore the transition pathways for AdK, which is the most relevant to the work presented here. They were able to observe two transitions to the open state within 50 ns by enhanced temperature sampling of the first five eigenvectors of the principle components around the 1AKE closed state; they found a dominant pathway in which the half-opening of the NMP domain precedes a partial correlated opening of the LID and NMP domains.
Conclusions
In this article, we have introduced a novel method (to our knowledge) for describing the time progression of inherently anharmonic MD local motions. This is done by generating a sequence of instantaneous normal modes from an elastic network model projected against two protein end-states in order to connect them to more global transitions to either intermediate substates or the final end-state. Note that the INM similarity always fluctuates during the trajectory, even in the local equilibrium states, which is a feature of the underlying anharmonic local motions. We would argue that evidence of local cracking, i.e., partial unfolding to aid large conformational change (18,35), is evident in our INM projections onto the two end-states. For example, Fig. 2 d shows that just before the transition to 4AKE, the INMs are highly dissimilar to both end-states, and thus one could analyze the underlying anharmonic MD trajectory to determine the underlying structural origin of possible cracking.
Our INM sampling method shares similarities to transition path sampling (TPS) (38) in which “shooting moves” are repeatedly performed to generate better transition pathway ensembles to move a system from state to state. Clearly our method is less general than the original TPS, as we are focusing on proteins and their hierarchical substates (5,39), in which pathways can be captured in the fluctuation correlations local to a given substate. In TPS the initial pathway connecting the two end-states must be given, and the refinement accuracy and convergence speed will heavily depend on this proposed initial pathway. We suggest that the INM projected against the two end-states, and harvesting from many independent short-time MD simulations, may provide a useful scheme for generating this initial protein transition pathway efficiently without prior knowledge.
Our approach also has great potential for the building of a Markovian state model (40,41) to accurately predict kinetics based on the principle of substate interconversion. Our main goal in this work was to show the ability of INM similarity to identify unique substates and drive local fluctuations to gradually move from start- to end-states, and to provide mechanistic information, such as substate population and pathway distribution, as opposed to that provided by a detailed kinetics model. However, it is similar to a Markovian state model, in that it clusters trajectories based on similar INM trajectory profiles that are legitimate substates that interconvert rapidly, whereas dissimilar INM trajectory profiles defined substates that did not interconvert rapidly.
In summary, we have presented an approach to explore large protein conformational transitions by using INM similarity to identify key substates along transition pathways, and use them to monitor global interconversions among substates to reach the end-state based on short timescale simulations. The fluctuation correlation analysis shows that these identified substates and corresponding short timescale equilibrium trajectories can convey most aspects of the transition mechanism in residue detail, although it may be potentially difficult to use this unforced treatment to observe complete transition pathways if the energy minimum of a given substate is too deep—meaning that local fluctuations may have little similarity to the other end-state. Nevertheless, we believe that the INM framework provides a unique method for the exploration of protein conformational transitions when the concept of hierarchical substates is valid, in which long-time collective motions between substates are manifest in fast local coupled structural fluctuations.
Acknowledgments
We thank the Shanghai Center for Systems Biomedicine at Shanghai Jiao Tong University for providing computing resources.
The work was supported in part by the National Basic Research Program of China (grant No. 2005CB724301) and the Science and Technology Commission of Shanghai Municipality, China (grant No. 08511501701). T.H.-G. thanks the National Institutes of Health for support.
Contributor Information
Cheng Peng, Email: chengpeng@berkeley.edu.
Teresa Head-Gordon, Email: tlhead-gordon@lbl.gov.
References
- 1.Wolf-Watz M., Thai V., Kern D. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 2.Geeves M.A., Holmes K.C. Structural mechanism of muscle contraction. Annu. Rev. Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- 3.Goodey N.M., Benkovic S.J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 4.Lange A., Giller K., Baldus M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 5.Henzler-Wildman K., Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 6.Henzler-Wildman K.A., Lei M., Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 7.Whitten S.T., García-Moreno E B., Hilser V.J. Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins. Proc. Natl. Acad. Sci. USA. 2005;102:4282–4287. doi: 10.1073/pnas.0407499102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levitt M., Sander C., Stern P.S. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- 9.Go N., Noguti T., Nishikawa T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. USA. 1983;80:3696–3700. doi: 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks B., Karplus M. Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tirion M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 12.Bahar I., Rader A.J. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005;15:586–592. doi: 10.1016/j.sbi.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 2005;13:373–380. doi: 10.1016/j.str.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Tama F., Sanejouand Y.H. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Sweet C.R., Petrone P., Izaguirre J.A. Normal mode partitioning of Langevin dynamics for biomolecules. J. Chem. Phys. 2008;128:145101–145113. doi: 10.1063/1.2883966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kantarci-Carsibasi N., Haliloglu T., Doruker P. Conformational transition pathways explored by Monte Carlo simulation integrated with collective modes. Biophys. J. 2008;95:5862–5873. doi: 10.1529/biophysj.107.128447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirillova S., Cortés J., Siméon T. An NMA-guided path planning approach for computing large-amplitude conformational changes in proteins. Proteins. 2008;70:131–143. doi: 10.1002/prot.21570. [DOI] [PubMed] [Google Scholar]
- 18.Miyashita O., Onuchic J.N., Wolynes P.G. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc. Natl. Acad. Sci. USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Z.Y., Shi Y.Y., Liu H.Y. Molecular dynamics simulations of peptides and proteins with amplified collective motions. Biophys. J. 2003;84:3583–3593. doi: 10.1016/S0006-3495(03)75090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henzler-Wildman K.A., Thai V., Kern D. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 21.Vendruscolo M., Dobson C.M. Structural biology. Dynamic visions of enzymatic reactions. Science. 2006;313:1586–1587. doi: 10.1126/science.1132851. [DOI] [PubMed] [Google Scholar]
- 22.Müller C.W., Schulz G.E. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 Å resolution. A model for a catalytic transition state. J. Mol. Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- 23.Müller C.W., Schlauderer G.J., Schulz G.E. Adenylate kinase motions during catalysis: an energetic counterweight balancing substrate binding. Structure. 1996;4:147–156. doi: 10.1016/s0969-2126(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 24.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 25.Kaminski G.A., Friesner R.A., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 26.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 27.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 28.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 29.Hess B., Bekker H., Fraaije J. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 30.Bae E., Phillips G.N. Roles of static and dynamic domains in stability and catalysis of adenylate kinase. Proc. Natl. Acad. Sci. USA. 2006;103:2132–2137. doi: 10.1073/pnas.0507527103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanson J.A., Duderstadt K., Yang H. Illuminating the mechanistic roles of enzyme conformational dynamics. Proc. Natl. Acad. Sci. USA. 2007;104:18055–18060. doi: 10.1073/pnas.0708600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maragakis P., Karplus M. Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J. Mol. Biol. 2005;352:807–822. doi: 10.1016/j.jmb.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 33.Chu J.W., Voth G.A. Coarse-grained free energy functions for studying protein conformational changes: a double-well network model. Biophys. J. 2007;93:3860–3871. doi: 10.1529/biophysj.107.112060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arora K., Brooks C.L. Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitford P.C., Gosavi S., Onuchic J.N. Conformational transitions in adenylate kinase. Allosteric communication reduces misligation. J. Biol. Chem. 2008;283:2042–2048. doi: 10.1074/jbc.M707632200. [DOI] [PubMed] [Google Scholar]
- 36.Lu Q., Wang J. Single molecule conformational dynamics of adenylate kinase: energy landscape, structural correlations, and transition state ensembles. J. Am. Chem. Soc. 2008;130:4772–4783. doi: 10.1021/ja0780481. [DOI] [PubMed] [Google Scholar]
- 37.Kubitzki M.B., de Groot B.L. The atomistic mechanism of conformational transition in adenylate kinase: a TEE-REX molecular dynamics study. Structure. 2008;16:1175–1182. doi: 10.1016/j.str.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 38.Bolhuis P.G., Chandler D., Geissler P.L. Transition path sampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 39.Li C.B., Yang H., Komatsuzaki T. Multiscale complex network of protein conformational fluctuations in single-molecule time series. Proc. Natl. Acad. Sci. USA. 2008;105:536–541. doi: 10.1073/pnas.0707378105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swope W.C., Pitera J.W., Suits F. Describing protein folding kinetics by molecular dynamics simulations. 1. Theory. J. Phys. Chem. B. 2004;108:6571–6581. [Google Scholar]
- 41.Singhal N., Snow C.D., Pande V.S. Using path sampling to build better Markovian state models: predicting the folding rate and mechanism of a tryptophan zipper β hairpin. J. Chem. Phys. 2004;121:415–425. doi: 10.1063/1.1738647. [DOI] [PubMed] [Google Scholar]