

Abstract
Many of the molecules that mediate G-protein signaling are thought to constitutively associate with each other in variably stable signaling complexes. Much of the evidence for signaling complexes has come from Förster resonance energy transfer and bioluminescence resonance energy transfer (BRET) studies. However, detection of constitutive protein association with these methods is hampered by nonspecific energy transfer that occurs when donor and acceptor molecules are in close proximity by chance. We show that chemically-induced recruitment of local third-party BRET donors or acceptors reliably separates nonspecific and specific BRET. We use this method to reexamine the constitutive association of class A G-protein-coupled receptors (GPCRs) with other GPCRs and with heterotrimeric G-proteins. We find that β2 adrenoreceptors constitutively associate with each other and with several other class A GPCRs. In contrast, GPCRs and G-proteins are unlikely to exist in stable constitutive preassembled complexes.
Introduction
Many cellular functions, including signal transduction, depend on static or dynamic protein-protein association. Unfortunately, few methods are available to assess protein-protein interactions in living cells. Among the most useful are methods based on resonance energy transfer (RET) between proteins labeled with genetically-encoded fluorescent or luminescent molecules. These methods are especially useful for detecting changes in protein association or conformation due to physiological or pharmacological manipulation. However, it is more difficult to use RET techniques to determine if proteins are constitutively associated because both random collisional encounters and specific protein interactions generate RET signals (1). Nonspecific RET due to random interactions is particularly problematic in live cells when the labeled proteins are overexpressed or are confined to a subcellular compartment such as the plasma membrane. In principle, specific and nonspecific RET can be distinguished by changing either the donor/acceptor ratio (saturation RET) or the total concentration of these proteins, or by introducing an unlabeled competitor (2–4). These approaches require graded control of protein expression across a broad range of concentrations (including physiological concentrations), accurate measurement of expression levels, and knowledge of the subcellular location of the expressed proteins. These requirements apply to control proteins as well as the proteins of interest, thus the appropriate choice of controls is critically important. In practice, these conditions can not always be met, and interpretation of results is not always straightforward. As a consequence, significant disagreement regarding the existence of some constitutive multiprotein complexes persists (3,5,6).
One instance where previous studies have reached conflicting conclusions is the self-association of class A G-protein-coupled receptors (GPCRs). Most RET studies have concluded that these receptors form dimers or higher-order oligomers both in reconstituted systems and living cells (7), in agreement with earlier biochemical studies (8). However, a few RET studies have reached the opposite conclusion (3,9), and have criticized the methods used to show class A GPCR self-association (3). Similarly, conflicting conclusions have been drawn from RET studies of preassembly of GPCRs and heterotrimeric G-proteins. It has long been thought that the interaction between GPCRs and their cognate G-proteins in intact cells occurs transiently, and that productive receptor-G-protein complexes form after receptor activation (10,11). This collision coupling model has been challenged in large part by studies reporting constitutive RET between GPCRs and G-proteins (12–15). The model put forward to account for this observation is that GPCRs and G-proteins are physically associated before, during, and after receptor activation, and signaling is mediated by structural rearrangement of stable receptor-G-protein complexes (12). However, other studies have failed to observe constitutive RET between GPCRs and G-proteins (16), and no physiological role for preassembled GPCR-G-protein complexes has yet been shown.
In this study, we describe a new method for detecting specific constitutive association of proteins that are confined to subcellular compartments. We validate this method using engineered monomeric and dimeric membrane-associated proteins, then use this method to reexamine constitutive association of class A GPCRs with other class A GPCRs and with heterotrimeric G-proteins.
Materials and Methods
Plasmid DNA constructs
A plasmid encoding Rluc8 (17) was provided by Dr. Sanjiv Sam Gambhir (Stanford University, Palo Alto, CA). FRB and FKBP were provided by Dr. Stephen R. Ikeda (NIAAA, Rockville, MD). Kir3.1 was provided by Dr. Eitan Reuveny (Weizmann Institute of Science, Rehovot, Israel), and Kir3.2 was provided by Dr. Lily Jan (UCSF, San Francisco, CA). The α2AR was provided by Dr. Andrew Tinker (UCL, London, UK). D2R-V was provided by Dr. Jonathan Javitch (Columbia University, New York, NY). Various acceptor, donor and recruiter constructs contained the following peptide sequences:
mem (from GAP-43): MLCCLRRTKQVEKNDEDQKI;
link: GGGGSGGGGSGGGSGGELRGGELE;
zip: MNTEAARRSRARKLQRMKQLEDKVEELLSKNYHLENEVARLKKLVGERID; and
kras: RKHKEKMSKDGKKKKKKSKTKCVIM.
FRB-Gγ2 included a GGSGG linker between the C-terminus of FRB and the N-terminus of Gγ2. The α2AR-V was made by subcloning α2AR into mVenus-N1 with KpnI and HindIII. V-GIRK1 was made by subcloning Kir3.1 into mVenus-C1 with BsrGI and SacI; cotransfection of V-GIRK1 and Kir3.2 produced GIRK1/2-V channels. Caveolin1-V was made by subcloning rat caveolin1α into mVenus-N1 with XhoI and SacII. The construction of GαoA-V, Gβ1γ2-V, masGRK3ct-V, and C-TM-V have been described previously (18,19). All constructs were verified by automated sequencing.
Cell culture and transfection
HEK 293 cells (ATCC, Manassas, VA) were propagated in plastic flasks, in 6-well plates and on polylysine-coated glass coverslips according to the supplier's protocol. Cells were transfected in growth medium using linear polyethylenimine (MW 25,000; Polysciences, Warrington, PA) at an N/P ratio of 20; ≤3 μg of plasmid DNA was transfected per well of a 6-well plate.
Guanine nucleotide depletion
For experiments such as that shown in Fig. 3 cells were resuspended in buffer containing 140 mM potassium gluconate, 5 mM KCl, 10 mM HEPES, 1 mM EGTA, 0.3 mM CaCl2, and 1 mM MgCl2 (pH 7.2), permeabilized with either 1000 U mL−1 α-hemolysin (Sigma, St. Louis, MO; H9395) or 10 μM digitonin, and incubated with 5 mM KCN for ∼15 min before making measurements. In some experiments 0.5 mM GTPγS was added at the same time as rapamycin.
Figure 3.
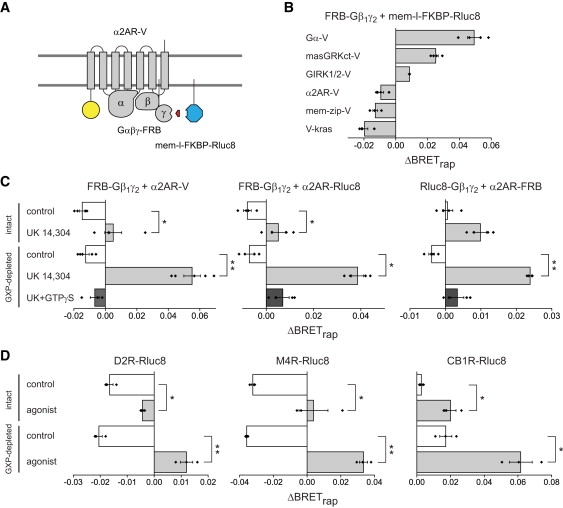
Class A GPCRs do not preassemble with G-protein heterotrimers. (A) Schematic illustrating the use of third-party BRET to detect association of G-proteins with GPCRs. (B) Interactions between FRB-Gβ1γ2 dimers and a Gα subunit, effectors and α2AR-V; in all experiments mem-l-FKBP-Rluc8 was the third-party donor. Rapamycin significantly increases BRET between FRB-Gβ1γ2 (FRB-Gγ2 + Gβ1) and GαoA-V, and the Gβγ-binding proteins masGRKct-V and GIRK1/2-V, but not the control proteins mem-zip-V or V-kras (n = 4). Rapamycin also did not increase BRET between FRB-Gβ1γ2 and the α2AR-V receptor in the presence of Gαi1. (C) The α2AR-V agonist UK 14,304 (10 μM) converted the rapamycin-induced BRET decrease into a small increase in intact cells, and a highly-significant increase in guanine nucleotide (GXP)-depleted (permeabilized) cells (n = 4). This change was abolished by 0.5 mM GTPγS. (D) Experiments analogous to A with D2 dopamine receptors (D2R-Rluc8; left; n = 3), M4 muscarinic acetylcholine receptors (M4R-Rluc8; middle; n = 3) and CB1 cannabinoid receptors (CB1R-Rluc8; right; n=3). Agonists were quinpirole (10 μM), carbachol (100 μM) and WIN 55,212 (1 μM); ∗p < 0.05; ∗∗p < 0.005, unpaired t-test. Bars represent the mean ± SE, and black diamonds indicate data points from individual experiments.
Bioluminescence resonance energy transfer measurements
Cells were detached from plates by rinsing with PBS-EDTA and triturating in PBS 16–24 h after transfection. Suspended cells were transferred to black 96-well microplates. Rapamycin (5 μM; LC Laboratories, Woburn, MA) was present in 50% of the wells, and coelenterazine h (5 μM; Nanolight Technologies, Pinetop, AZ) was added to all wells immediately before making measurements. Luminescence measurements were made using a photon-counting plate reader (Mithras LB940; Berthold Technologies GmbH, Bad Wildbad, Germany).
Confocal imaging
Confocal images (Fig. S1 in the Supporting Material) were acquired using a Leica (Wetzlar, Germany) SP2 scanning confocal microscope and a 63×, 1.4 NA objective. Venus was excited with the 514 nm line of an ArKr laser, and detected at 520–550 nm.
Statistical analysis
Two different methods were used to calculate ΔBRETrap and to test the hypothesis that this value was not equal to zero. In the first method the raw bioluminescence resonance energy transfer (BRET) ratio (Em535/480) was calculated as the number of photons counted at 535 nm divided by the number of photons counted at 480 nm for each replicate; the number of photons counted at each wavelength was generally >105, and was always >104. BRETbasal for each replicate was this ratio minus the Em535/480 measured from cells expressing only the BRET donor, and ΔBRETrap for each replicate was Em535/480 measured in the presence of rapamycin minus Em535/480 in the absence of rapamycin. Values of BRETbasal and ΔBRETrap reported in the text and in Tables S1–S5 in the Supporting Material represent the mean ± SE of replicates for a particular condition. To test whether or not ΔBRETrap was significantly different from zero, a paired t-test was carried out between within-replicate values of Em535/480 measured in the presence of rapamycin and Em535/480 in the absence of rapamycin for each condition. In each case the distributions of Em535/480 passed the Shapiro-Wilk test for normality with p > 0.05. Comparisons between conditions were made using both Student's t-test assuming equal variance, and the variant of this test (a.k.a. Welch's t-test) that is insensitive to unequal variance; p values reported in the text were derived from the latter.
In the second method (Tables S6–S12 in the Supporting Material) a weighted mean of photons collected at each wavelength was calculated for each condition according to:
| (1) |
where μ′ is the weighted mean, xi is the number of photons counted for the ith replicate, and σi is the error for the ith replicate, defined as (20). This definition assumes Poisson error in each photon count. The weighted variance of the photon counts at each wavelength was calculated as:
| (2) |
The weighted mean and variance at each wavelength was then used to calculate a weighted Em535/480 ratio ± propagated error for each condition, and ΔBRETrap for each condition was calculated as the weighted Em535/480 in the presence of rapamycin (Em535/480rap) minus the weighted Em535/480 in the absence of rapamycin (Em535/480basal). The propagated error in ΔBRETrap for each condition multiplied by the critical value of Student's t-distribution for the appropriate degrees of freedom gave the 95% confidence interval; ΔBRETrap was considered to be significantly different from zero if the 95% confidence interval did not encompass zero. Agonist-induced BRET (Fig. 4 was calculated using the same procedure).
Figure 4.
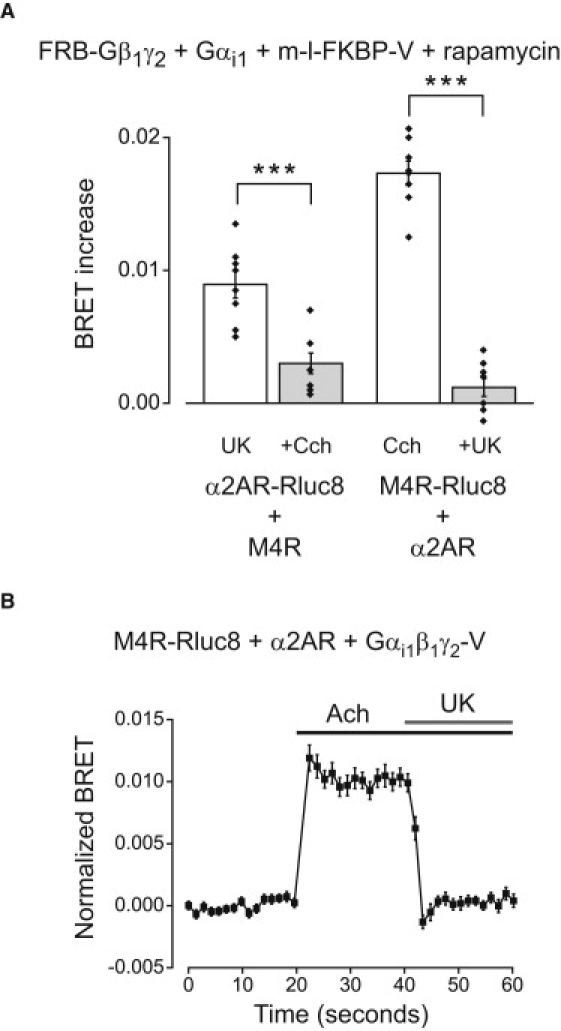
Competition for G-proteins between labeled and unlabeled GPCRs. (A) Agonist induced BRET between α2AR-Rluc8 and Gαi1β1γ2-FRB:mem-l-FKBP-V complexes was significantly reduced by coactivation of unlabeled M4Rs (+Cch, 100 μM carbachol; n = 8). Conversely, agonist induced BRET between M4R-Rluc8 and Gαi1β1γ2-FRB-mem-l-FKBP-V complexes was reduced significantly by coactivation of unlabeled α2ARs (+UK, 10 μM UK 14,304; n = 8). Rapamycin (5 μM) was present throughout. Bars represent the mean ± SE, and black diamonds indicate data points from individual experiments. ∗∗∗p < 0.0005, unpaired t-test. (B) Activation of unlabeled α2ARs with UK 14,304 (10 μM) rapidly decreases acetylcholine (Ach; 100 μM)-induced BRET between M4R-Rluc8 and Gαi1β1γ2-V (n = 24).
These two methods of statistical analysis produced comparable results, although the second method was more conservative with respect to statistical significance. All values were rounded to two decimal places after all calculations were complete. Values reported as 0.00 were >0 and <0.005, whereas values reported as −0.00 were >−0.005 and <0.
Results
Validation of the third-party BRET method
We developed a general method to detect specific constitutive BRET (21) between proteins located in subcellular compartments of living cells. This method (Fig. 1 A) relies on rapamycin-induced dimerization of FK506 binding protein (FKBP) and the FKBP-rapamycin-binding domain of mTOR (FRB). One protein of interest is fused directly to either the RET donor or acceptor, and the other protein of interest (the recruiter) is fused to either FKBP or FRB. The complementary RET acceptor or donor is fused to the cognate FRB/FKBP moiety as well as a peptide sequence that directs the protein to the appropriate compartment. This protein serves as a third-party donor/acceptor. Rapamycin causes FRB and FKBP to dimerize, thus recruiting the third-party RET partner to the recruiter protein of interest. If the two proteins of interest specifically interact, then rapamycin will (under favorable conditions) induce an increase in RET. In contrast, if the two proteins of interest do not specifically interact then rapamycin will not change the proximity of the donor and acceptor, and will not increase RET. Any RET signal that exists before recruitment can be attributed to nonspecific collisional encounters, provided the third-party RET partner does not interact with either protein of interest. A critical requirement is that FKBP-FRB dimerization should not change the concentration of RET donors or acceptors within the compartment that contains the molecules of interest.
Figure 1.
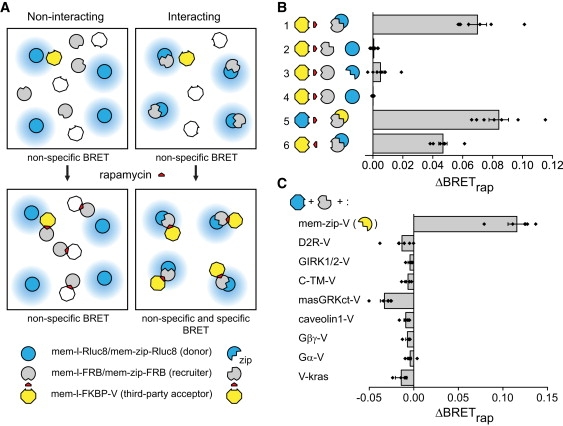
Third-party RET with plasma membrane proteins. (A) Nonspecific BRET is observed between third-party acceptors and both monomeric (left) and dimeric (right) donors (top). Rapamycin-induced dimerization of FRB and FKBP recruits acceptors to the vicinity of dimeric donors only (bottom). (B) Rapamycin increases BRET only when both proteins of interest are dimeric (n = 4–8 experiments, each carried out in quadruplicate). Symbols correspond to the proteins indicated at the bottom and center of A: 1, mem-l-FKBP-V + mem-zip-FRB + mem-zip-Rluc8; 2, mem-l-FKBP-V + mem-zip-FRB + mem-l-Rluc8; 3, mem-l-FKBP-V + mem-l-FRB + mem-zip-Rluc8; 4, mem-l-FKBP-V + mem-l-FRB + mem-l-Rluc8; 5, mem-l-FKBP-Rluc8 + mem-zip-FRB + mem-zip-V; 6, mem-l-FRB-V + mem-zip-FKBP + mem-zip-Rluc8. (C) Rapamycin does not increase BRET when mem-l-FKBP-Rluc8 and mem-zip-FRB are expressed with any of several noninteracting integral or peripheral membrane protein acceptors (n = 5 for each). Bars represent the mean ± SE, and black diamonds indicate data points from individual experiments.
To validate this method we measured basal (BRETbasal) and rapamycin-induced (ΔBRETrap) BRET between engineered monomeric and dimeric proteins and third-party acceptors or donors. Proteins of interest contained a dual palmitoylation sequence to direct expression at the plasma membrane (mem), and either an inert linker peptide (l) or a leucine-zipper peptide from GCN4 (zip), which forms stable constitutive homodimers. These were fused to the Renilla luciferase variant Rluc8 (17) to serve as monomeric (mem-l-Rluc8) or dimeric (mem-zip-Rluc8) BRET donors, or to FRB to serve as monomeric (mem-l-FRB) or dimeric (mem-zip-FRB) recruiters. The fluorescent protein venus (V) fused to FKBP and the mem-linker peptide (mem-l-FKBP-V) served as the third-party acceptor. When dimeric donor and recruiter proteins were coexpressed in HEK 293 cells rapamycin induced a large increase in BRET (Fig. 1 B), consistent with the formation of mem-zip-Rluc8/mem-zip-FRB/mem-l-FKBP-V ternary complexes. In contrast, no BRET increase was observed if either the donor, the recruiter or both lacked the zip peptide. Rapamycin also increased BRET for dimeric proteins when the positions of FKBP and FRB were exchanged, or when the positions of Rluc8 and V were exchanged (Fig. 1 B). Importantly, all donor-acceptor combinations produced substantial BRETbasal (ranging from 0.05 to 0.14) before the addition of rapamycin (Table S1 and Table S6), thus the presence of a basal BRET signal is not a valid indicator of a constitutive protein interaction.
The absence of a change in BRET with noninteracting control proteins (Fig. 1 B) implies that the formation of mem-zip-FRB/mem-l-FKBP-V complexes did not significantly change the abundance or orientation of acceptors at the plasma membrane, as such changes would be indicated by many different membrane-associated acceptors. We verified this implication directly by imaging cells during application of rapamycin. The subcellular distribution of mem-l-FKBP-V was not detectably changed by rapamycin in cells expressing this acceptor together with mem-zip-Rluc8 and mem-zip-FRB (Fig. S1). The normalized ratio of mem-l-FKBP-V intensity at the plasma membrane and the cell interior in the presence of rapamycin was 98 ± 1% of the control value (n = 19, p = 0.16). This suggests that rapamycin does not recruit mem-l-FKBP-V from intracellular compartments to the plasma membrane, consistent with the strong attachment of this third-party acceptor to the plasma membrane via dual palmitoylation.
In cases where the donor and recruiter proteins specifically associate the magnitude of rapamycin-induced BRET should depend on the relative abundance of these molecules. Increasing the relative expression of donors should increase the fraction of donors found in donor/donor complexes as compared to donor/recruiter complexes. Because only the latter can contribute to rapamycin-induced BRET, increasing the relative expression of donors should decrease ΔBRETrap. Indeed, increasing the expression of mem-zip-Rluc8 (while keeping expression of mem-zip-FRB and mem-link-FKBP-V constant) decreased rapamycin-induced BRET (Fig. S2). In contrast, changing the expression of donors and recruiters in parallel (leaving the relative abundance of donor/donor and donor/recruiter complexes unchanged) did not change rapamycin-induced BRET (Fig. S2). Thus the third-party BRET method is predictably sensitive to the donor/recruiter ratio, but is relatively insensitive to the total expression level of the proteins of interest, at least for high-affinity complexes such as those formed by zip peptides.
To assess the ability of the third-party BRET method to reject nonspecific interactions we expressed several integral or peripheral plasma membrane proteins that were not expected to specifically interact with zip peptides. Each was fused to venus, and was coexpressed with the recruiter mem-zip-FRB and the third-party donor mem-l-FKBP-Rluc8. Rapamycin significantly increased BRET with the positive control mem-zip-V, but not with any of the other acceptor proteins (Fig. 1 C). This confirms that the BRET increase observed with mem-zip-V was not simply due to a change in the number or orientation of donors at the plasma membrane. All of these acceptors generated significant BRETbasal (Table S2 and Table S7), underscoring the propensity of membrane-associated pairs to produce nonspecific RET (1). Interestingly, recruitment of a third-party donor or acceptor often decreased BRET slightly when proteins of interest were not expected to specifically interact (Fig. 1 C). The mechanism of this decrease is not clear, although one plausible explanation is that the rapamycin-induced interaction between the recruiter and the third-party partner sterically occludes a fraction of the random encounters that produce nonspecific RET.
Self-association of class A GPCRs
Previous Förster resonance energy transfer and BRET studies have made extensive use of saturation methods to determine the specificity of class A GPCR self-association. As the third-party BRET method is conceptually distinct from saturation methods, we used this method to reexamine the self-association of β2 adrenoreceptors (β2ARs; Fig. 2 A). This receptor was the subject of one of the first reports of class A GPCR dimerization (8), but has also been the subject of conflicting saturation BRET studies (3,6,22). When β2AR-Rluc8 and β2AR-FRB were expressed together with mem-l-FKBP-V, rapamycin reliably induced a significant increase in BRET (Fig. 2 B), consistent with the self-association of β2ARs. No such increase was observed with β2AR-FRB and several other membrane-associated donors, even though substantial BRETbasal was observed with all of these donors (Table S3 and Table S8). A smaller but still highly significant BRET increase was observed when β2AR-V and β2AR-FRB were expressed together with mem-l-FKBP-Rluc8 (ΔBRETrap = 0.01 ± 0.00; p < 0.0001; n = 10). Coexpression of increasing amounts of unlabeled β2AR progressively decreased the magnitude of rapamycin-induced BRET between β2AR-Rluc8 and mem-l-FKBP-V (with β2AR-FRB; Fig. S3), consistent with genuine self-association of β2ARs. Finally, acute activation of β2AR-Rluc8 and β2AR-FRB with 10 μM isoproterenol had no effect on ΔBRETrap (0.04 ± 0.00 and 0.04 ± 0.00, control and isoproterenol, respectively; p = 0.94; n = 5).
Figure 2.
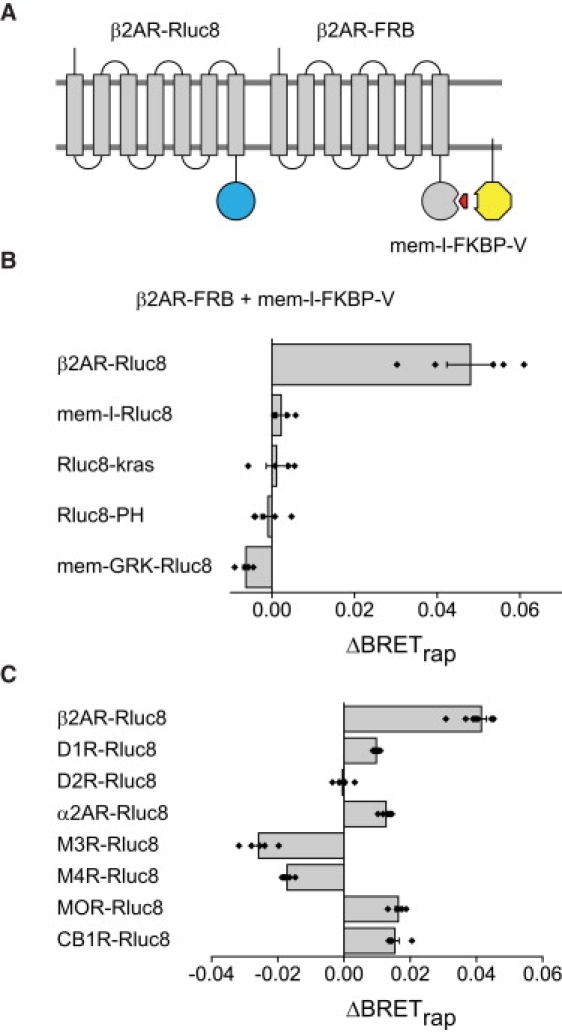
β2 adrenoreceptors self-associate and associate with other class A GPCRs. (A) Schematic illustrating the use of third-party BRET to detect β2AR self-association. (B) Rapamycin significantly increases BRET with β2AR-Rluc8 as the donor and β2AR-FRB as the recruiter (n = 5), but not several other membrane-associated donors (n = 4–5); in all cases mem-l-FKBP-V was the third-party acceptor. (C) Heteromeric association of the recruiter β2AR-FRB with various class A GPCRs as donors and mem-l-FKBP-V as the third-party acceptor. In addition to β2AR-Rluc8 (n = 10), donor GPCRs (n = 5 for each) included α2 adrenoreceptors (α2AR-Rluc8), D2 and D1 dopamine receptors (D2R-Rluc8 and D1R-Rluc8), M3 and M4 muscarinic acetylcholine receptors (M3R-Rluc8 and M4R-Rluc8), μ opioid receptors (MOR-Rluc8), and CB1 cannabinoid receptors (CB1R-Rluc8). Bars represent the mean ± SE, and black diamonds indicate data points from individual experiments.
Several class A GPCRs are thought to associate with β2ARs, including other adrenoreceptors (23,24). Therefore, we examined the association of β2AR-FRB with several other class A GPCR donors. These receptors supported varying levels of BRETbasal before addition of rapamycin (Table S4 and Table S9). Several of these receptors also supported rapamycin-induced BRET, suggesting that they associated with β2AR-FRB (Fig. 2 C). None of the other GPCR donors studied produced ΔBRETrap that approached the magnitude of that produced by β2AR-Rluc8, suggesting that β2ARs may prefer homomeric interactions over heteromeric interactions. Interestingly, ΔBRETrap was strongly negative when either M3 or M4 muscarinic acetylcholine receptors was used as the BRET donor (p = 0.047 and 0.014, respectively). This result suggests that these receptors may be less likely than other class A GPCRs to associate with β2ARs.
Preassembly of GPCRs and G-proteins
We then used this method to reexamine constitutive interactions between GPCRs and heterotrimeric G-proteins (12,13) (Fig. 3 A). FRB was fused to the amino terminus of the Gγ2 subunit to serve as a recruiter, and this was paired with the third-party donor mem-l-FKBP-Rluc8. Gγ and Gβ subunits form obligate dimers, therefore Gβ1 was expressed with FRB-Gγ2 to allow formation of FRB-Gβ1γ2 dimers. Rapamycin increased BRET when these proteins were expressed with GαoA-V (ΔBRETrap = 0.05 ± 0.00; n = 4; p < 0.005; Fig. 3 B), demonstrating that FRB-Gβ1γ2 could form heterotrimers and recruit mem-l-FKBP-Rluc8. Rapamycin also significantly increased BRET when FRB-Gβ1γ2 was expressed with a membrane-associated fragment of G-protein receptor kinase 3 (masGRKct-V) or G-protein-regulated inwardly-rectifying potassium channels (GIRK1/2-V; Fig. 3 B), both of which are known to form transient (lifetime < 1 s) complexes with Gβγ dimers. Thus FRB-Gβ1γ2 dimers were capable of reporting association with several Gβγ-interacting proteins. In contrast, rapamycin decreased BRET when FRB-Gβ1γ2 was expressed with unlabeled Gαi1 and a venus-labeled α2A-adrenoreceptor (α2AR-V). Similarly, rapamycin decreased BRET when either of two negative control acceptor proteins was expressed (Fig. 3 B). All of these acceptor proteins produced significant BRETbasal with mem-l-FKBP-Rluc8 (Table S5 and Table S10).
Previous studies concluded that inactive α2ARs and G-proteins form preassembled complexes, in part because basal RET was observed between α2ARs and heterotrimers (12,13). Because we did not detect a constitutive interaction between FRB-Gβ1γ2 and α2AR-V, we wanted to confirm that FRB-Gβ1γ2 formed heterotrimers with Gαi1 that could interact with α2AR-V. Activation of α2AR-V receptors with the agonist UK 14,304 converted the rapamycin-induced BRET decrease into a small increase (Fig. 3 C). This finding is consistent with previous reports of agonist-induced RET increases between α2ARs and Gβγ dimers (12,16). The ternary complex model of GPCR signaling predicts that active-state agonist-receptor-G-protein complexes will be particularly stable when G-proteins lack access to guanine nucleotides (11,25). Accordingly, when permeabilized cells were depleted of nucleotides the rapamycin-induced BRET decrease was converted into a large increase, but only when agonist was present (Fig. 3 C). Nucleotide depletion alone (in the absence of agonist) did not affect the rapamycin-induced BRET decrease. The combined effect of agonist and nucleotide depletion was prevented by the poorly-hydrolyzable analog GTPγS. These results are thus consistent with the ternary complex model, and demonstrate that FRB-Gβ1γ2 and Gαi1 formed heterotrimers that interacted with α2AR-V in an agonist-dependent manner. These results were not dependent on a particular configuration of donor, recruiter and acceptor, as similar results were obtained with α2AR-Rluc8 as the donor or with α2AR-FRB as the recruiter (Fig. 3 C).
We also asked if this pattern of BRET changes was unique to α2 adrenoreceptors. Experiments using the same G-protein recruiter (Gαi1 + FRB-Gβ1γ2) and third-party acceptor (mem-l-FKBP-V) were carried out with three additional Gαi/o-coupled GPCRs as donors: dopamine D2 receptors (D2R-Rluc8), M4 muscarinic acetylcholine receptors (M4R-Rluc8), and CB1 cannabinoid receptors (CB1R-Rluc8). Results obtained with D2R-Rluc8 and M4R-Rluc8 were similar to those obtained with α2AR-Rluc8 (Fig. 3 D and Table S11), in that rapamycin decreased BRET in unstimulated cells, stimulation with agonist lessened this decrease or induced a slight increase, and the effect of agonist was enhanced in nucleotide-depleted cells. In contrast, rapamycin produced a small but significant increase in BRET in unstimulated cells expressing CB1R-Rluc8 (ΔBRETrap = 0.01 ± 0.00; p < 0.0005; n = 4), suggesting that some of these receptors were associated with G-proteins containing FRB-Gβ1γ2 in the absence of an agonist. The specific CB1R agonist WIN 55,212 significantly enhanced the rapamycin-induced BRET increase (Fig. 3 D). These findings are consistent with the known properties of CB1 receptors, which exhibit a high degree of constitutive activity that can be enhanced further by a full agonist (26). If constitutive activity was responsible for association of unliganded CB1R-Rluc8 and G-proteins, then nucleotide depletion alone would be expected to enhance this association. Indeed, in contrast to the other GPCRs we studied, nucleotide depletion alone enhanced the rapamycin-induced BRET signal generated by CB1R-Rluc8 (Fig. 3 D). Furthermore, rapamycin failed to significantly increase BRET in cells expressing CB1R-Rluc8 in the presence of the inverse agonist SR 141716A (ΔBRETrap = 0.00 ± 0.00; p = 0.37; n = 4). Taken together, these results show that the third-party BRET method detects active-state complexes but not preassembled inactive-state complexes between several GPCRs and G-proteins.
Finally, the possibility remained that inactive GPCRs and G-proteins formed preassembled inactive-state complexes that did not permit rapamycin-induced BRET, and that agonist activation produced a conformational change that permitted rapamycin-induced BRET. We tested this possibility by introducing a second GPCR to compete for G-proteins. If the agonist-induced increase in BRET in the presence of rapamycin reflected a conformational change of a stable GPCR-Rluc8/G-protein complex, then activation of a second (unlabeled) GPCR should be unable to influence this change. Alternatively, if the agonist-induced increase in BRET in the presence of rapamycin reflected association of GPCR-Rluc8 and G-protein, then activation of a second GPCR should be able to compete for G-proteins and thus inhibit the increase. To test this idea we expressed α2AR-Rluc8, FRB-Gβ1γ2, and Gαi1 together with unlabeled M4R and the third-party acceptor mem-l-FKBP-V. In the continuous presence of rapamycin, the α2AR agonist UK 14,304 induced an increase in BRET, which was reduced significantly when M4Rs were activated with carbachol (100 μM; p < 0.0005; Fig. 4 A and Table S12). We also carried out the converse experiment with M4R-Rluc8 and unlabeled α2ARs with similar results (p < 0.0005; Fig. 4 A). Competition between GPCRs for freely-exchanging G-proteins should occur with a rapid time course. To test this idea, we carried out a similar experiment to that shown in Fig. 4 A, and monitored BRET between M4R-Rluc8 and Gαi1β1γ2-V during application of acetylcholine and the subsequent addition of UK 14,304. As shown in Fig. 4 B, activation of unlabeled α2ARs decreased agonist-induced BRET between M4R-Rluc8 and Gαi1β1γ2-V within a few seconds. These results are difficult to reconcile with the idea that either receptor is permanently associated with G-protein heterotrimers, and imply that the agonist-induced signal reflects an association event rather than a conformational change within a preassembled inactive-state complex.
Discussion
The widespread use of RET techniques to study protein-protein interactions has led to the refinement of several experimental methods to distinguish signals that arise from specific interactions from those that arise from random interactions (4). The techniques used most commonly measure changes in energy transfer that occur as the ratio of acceptors to donors or the total concentration of acceptors and donors increases, and compare these changes to predictions made by simplified models (2,27,28). Although these methods have proven to be extremely useful, they are not without limitations. For example, specific interactions produce RET that saturates as the abundance of acceptors increases. However, nonspecific interactions also produce RET that is saturable or quasi-linear as acceptor density increases (3,4). No widely accepted criteria exist for objectively distinguishing the two situations. A potential remedy for this problem is to compare proteins of interest to several control proteins that have similar characteristics, but here too criteria defining what constitutes an appropriate control are not applied uniformly.
We developed a new method for distinguishing nonspecific and specific constitutive interactions using RET. The third-party approach is unrelated to existing methods, thus it avoids many of the problems associated with these methods. Third-party RET is conceptually straightforward, and does not require graded expression or quantitation of acceptors or donors. This method does require construction of third-party donors or acceptors, as well as control experiments to ensure that donor or acceptor concentration or orientation do not change. Although in this study we restricted our attention to signaling proteins located at the plasma membrane, by directing third-party RET partners to other subcellular compartments it should be possible to use this method to gain information regarding interactions in these compartments. Finally, although our studies used BRET, this approach is equally applicable to other RET techniques.
The third-party method also has important limitations. The failure to observe a signal with a given pair of proteins does not by itself rule out the existence of a specific interaction. This limitation is shared by all RET methods, but energy transfer involving a third-party is necessarily more complex than direct transfer between proteins of interest, therefore this problem is likely to be worse with third-party RET than with other methods. Similarly, like other RET methods our approach can not distinguish direct protein-protein interactions from indirect interactions that bring proteins of interest into close proximity. Finally, net energy transfer involving a third-party will depend on many factors, including the stoichiometry of the proteins involved and the conformation of the rapamycin-induced ternary complex. Therefore, the absolute magnitude of ΔBRETrap can not be used to draw quantitative inferences regarding the stability or structure of protein complexes. Because of these limitations, third-party RET will, like other methods, be most useful when used in conjunction with other methods to discriminate and characterize specific and nonspecific interactions (e.g., Fig. S3).
In this study, we used third-party BRET to reexamine two questions related to interactions between GPCR signaling molecules about which previous studies have disagreed. Most previous RET studies have concluded that class A GPCRs self-associate as dimers or higher-order oligomers. Recently, however, James et al. (3) reexamined this question with a variant of the saturation methods used by others. They concluded that class A GPCRs do not self-associate, and that prior studies either misinterpreted or incorrectly applied the theory underlying this type of experiment (3,6). The third-party BRET method described here does not rely on the theoretical changes in RET efficiency that underlie saturation methods. Our results support the conclusion that β2 adrenoreceptors self-associate in living cells, in agreement with previous saturation RET studies in cells (22,29) and phospholipid vesicles (30), as well as previous biochemical studies (8). Therefore, the experimental framework proposed by James et al. (3) does not seem to be a significant improvement over other methods based on systematic changes in acceptor and/or donor density. Our results also suggest that β2 adrenoreceptors associate with several other class A GPCRs, including some that have not been reported previously to interact with these receptors. The third-party RET method may thus prove useful for studies designed to detect and monitor GPCR association in intact cells.
We also examined the association of GPCRs and heterotrimeric G-proteins before and during receptor activation. The ternary complex model described by De Lean et al. (11) includes a substantial fraction of precoupled receptor-G-protein complexes that form spontaneously (without agonist occupancy of the receptor). This precoupled population of receptors corresponds to the fraction of high affinity agonist binding sites in membrane preparations. Such precoupled complexes do not accumulate when guanine nucleotides are present, as indicated by the lack of high affinity agonist binding in intact cells (31,32). However, this observation does not rule out the possibility that receptors and G-proteins may be preassembled in structurally distinct inactive-state complexes or colocalized in domains. Several recent studies using conventional RET methods have provided evidence for such preassembled complexes in intact cells (12–15) but see Hein (16) and Azpiazu and Gautam (33). The purported advantages of such an arrangement include facilitation of rapid and specific signaling.
In this study, third-party BRET failed to detect inactive-state complexes containing GPCRs and G-proteins. As pointed out above, by itself this failure is not strong evidence that such complexes do not exist. However, we could detect active-state complexes with third-party BRET using three different recruiter-third-party combinations and with several different GPCRs, suggesting the assay is robust with respect to known ternary complexes. Moreover, we show that the BRET signal that appears with receptor activation results from an association event, rather than a conformational change. Therefore, we conclude that it is unlikely that the receptors we studied form stable preassembled complexes with heterotrimeric G-proteins. Specifically, our results suggest that the receptors we studied do not directly associate with their cognate G-proteins in stable inactive-state complexes. Our results do not speak to the possibility that natively-expressed GPCRs and G-proteins may bind to a common scaffold or be concentrated in a common domain in other cell types. Nevertheless, several studies have shown rapid and specific signaling using the same cells and receptors used in this study (12,16,34). Therefore, a collision coupling mechanism seems to be sufficient to support rapid and specific signaling by GPCRs.
Supporting Material
Twelve tables and three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00232-8.
Supporting Material
Acknowledgments
We thank all the individuals who generously supplied plasmid DNA used in this project.
This work was supported by the National Institutes of Health (GM078319) and the National Science Foundation (MCB0620024).
References
- 1.Vogel S.S., Thaler C., Koushik S.V. Fanciful FRET. Sci. STKE. 2006;2006:re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- 2.Kenworthy A.K., Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 A using imaging fluorescence resonance energy transfer. J. Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James J.R., Oliveira M.I., Davis S.J. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 4.Marullo S., Bouvier M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol. Sci. 2007;28:362–365. doi: 10.1016/j.tips.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Bouvier M., Heveker N., Milligan G. BRET analysis of GPCR oligomerization: newer does not mean better. Nat. Methods. 2007;4 doi: 10.1038/nmeth0107-3. 3–4, author reply 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salahpour A., Masri B. Experimental challenge to a ‘rigorous’ BRET analysis of GPCR oligomerization. Nat. Methods. 2007;4 doi: 10.1038/nmeth0807-599. 599–600, author reply 601. [DOI] [PubMed] [Google Scholar]
- 7.Milligan G., Bouvier M. Methods to monitor the quaternary structure of G-protein-coupled receptors. FEBS J. 2005;272:2914–2925. doi: 10.1111/j.1742-4658.2005.04731.x. [DOI] [PubMed] [Google Scholar]
- 8.Hebert T.E., Moffett S., Bouvier M. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 1996;271:16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- 9.Meyer B.H., Segura J.M., Vogel H. FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. USA. 2006;103:2138–2143. doi: 10.1073/pnas.0507686103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tolkovsky A.M., Levitzki A. Mode of coupling between the beta-adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry. 1978;17:3795. doi: 10.1021/bi00611a020. [DOI] [PubMed] [Google Scholar]
- 11.De Lean A., Stadel J.M., Lefkowitz R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 12.Gales C., Van Durm J.J., Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G-protein complexes. Nat. Struct. Mol. Biol. 2006;13:776–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- 13.Nobles M., Benians A., Tinker A. Heterotrimeric G-proteins precouple with G-protein-coupled receptors in living cells. Proc. Natl. Acad. Sci. USA. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayoub M.A., Maurel D., Pin J.P. Real-time analysis of agonist-induced activation of protease-activated receptor 1/Galphai1 protein complex measured by bioluminescence resonance energy transfer in living cells. Mol. Pharmacol. 2007;71:1329–1340. doi: 10.1124/mol.106.030304. [DOI] [PubMed] [Google Scholar]
- 15.Audet N., Galés C., Pineyro G. Bioluminescence resonance energy transfer assays reveal ligand-specific conformational changes within preformed signaling complexes containing delta-opioid receptors and heterotrimeric G-proteins. J. Biol. Chem. 2008;283:15078–15088. doi: 10.1074/jbc.M707941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hein P., Frank M., Bünemann M. Dynamics of receptor/G protein coupling in living cells. EMBO J. 2005;24:4106–4114. doi: 10.1038/sj.emboj.7600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loening A.M., Fenn T.D., Gambhir S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. 2006;19:391–400. doi: 10.1093/protein/gzl023. [DOI] [PubMed] [Google Scholar]
- 18.Hollins B., Kuravi S., Lambert N.A. The c-terminus of GRK3 indicates rapid dissociation of G-protein heterotrimers. Cell. Signal. 2009;21:1015–1021. doi: 10.1016/j.cellsig.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin K., Sethi P.R., Lambert N.A. Abundance and stability of complexes containing inactive G-protein-coupled receptors and G proteins. FASEB J. 2008;22:2920–2927. doi: 10.1096/fj.08-105775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bevington P.R., Robinson D.K. McGraw-Hill; New York, NY: 2003. Data Reduction and Error Analysis for the Physical Sciences. [Google Scholar]
- 21.Pfleger K.D., Eidne K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET) Nat. Methods. 2006;3:165–174. doi: 10.1038/nmeth841. [DOI] [PubMed] [Google Scholar]
- 22.Mercier J.F., Salahpour A., Bouvier M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- 23.Xu J., He J., Hall R.A. Heterodimerization of alpha 2A- and beta 1-adrenergic receptors. J. Biol. Chem. 2003;278:10770–10777. doi: 10.1074/jbc.M207968200. [DOI] [PubMed] [Google Scholar]
- 24.Prinster S.C., Hague C., Hall R.A. Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol. Rev. 2005;57:289–298. doi: 10.1124/pr.57.3.1. [DOI] [PubMed] [Google Scholar]
- 25.Limbird L.E., Gill D.M., Lefkowitz R.J. Agonist-promoted coupling of the beta-adrenergic receptor with the guanine nucleotide regulatory protein of the adenylate cyclase system. Proc. Natl. Acad. Sci. USA. 1980;77:775–779. doi: 10.1073/pnas.77.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouaboula M., Perrachon S., Casellas P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J. Biol. Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 27.Veatch W., Stryer L. The dimeric nature of the gramicidin A transmembrane channel: conductance and fluorescence energy transfer studies of hybrid channels. J. Mol. Biol. 1977;113:89–102. doi: 10.1016/0022-2836(77)90042-0. [DOI] [PubMed] [Google Scholar]
- 28.Fung B.K., Stryer L. Surface density determination in membranes by fluorescence energy transfer. Biochemistry. 1978;17:5241–5248. doi: 10.1021/bi00617a025. [DOI] [PubMed] [Google Scholar]
- 29.Angers S., Salahpour A., Bouvier M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET) Proc. Natl. Acad. Sci. USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fung J.J., Deupi X., Kobilka B.K. Ligand-regulated oligomerization of beta(2)-adrenoceptors in a model lipid bilayer. EMBO J. 2009;28:3315–3328. doi: 10.1038/emboj.2009.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kent R.S., De Lean A., Lefkowitz R.J. A quantitative analysis of beta-adrenergic receptor interactions: resolution of high and low affinity states of the receptor by computer modeling of ligand binding data. Mol. Pharmacol. 1980;17:14–23. [PubMed] [Google Scholar]
- 32.Insel P.A., Stoolman L.M. Radioligand binding to beta adrenergic receptors of intact cultured S49 cells. Mol. Pharmacol. 1978;14:549–561. [PubMed] [Google Scholar]
- 33.Azpiazu I., Gautam N. A fluorescence resonance energy transfer-based sensor indicates that receptor access to a G-protein is unrestricted in a living mammalian cell. J. Biol. Chem. 2004;279:27709–27718. doi: 10.1074/jbc.M403712200. [DOI] [PubMed] [Google Scholar]
- 34.Bünemann M., Bücheler M.M., Hein L. Activation and deactivation kinetics of alpha 2A- and alpha 2C-adrenergic receptor-activated G-protein-activated inwardly rectifying K+ channel currents. J. Biol. Chem. 2001;276:47512–47517. doi: 10.1074/jbc.M108652200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.