

Abstract
Intrinsically disordered proteins (IDPs), which lack folded structure and are disordered under nondenaturing conditions, have been shown to perform important functions in a large number of cellular processes. These proteins have interesting structural properties that deviate from the random-coil-like behavior exhibited by chemically denatured proteins. In particular, IDPs are often observed to exhibit significant compaction. In this study, we have analyzed the hydrodynamic radii of a number of IDPs to investigate the sequence determinants of this compaction. Net charge and proline content are observed to be strongly correlated with increased hydrodynamic radii, suggesting that these are the dominant contributors to compaction. Hydrophobicity and secondary structure, on the other hand, appear to have negligible effects on compaction, which implies that the determinants of structure in folded and intrinsically disordered proteins are profoundly different. Finally, we observe that polyhistidine tags seem to increase IDP compaction, which suggests that these tags have significant perturbing effects and thus should be removed before any structural characterizations of IDPs. Using the relationships observed in this analysis, we have developed a sequence-based predictor of hydrodynamic radius for IDPs that shows substantial improvement over a simple model based upon chain length alone.
Introduction
The focus of structural biology has been dramatically expanded in recent years with the widespread interest in intrinsically disordered proteins (IDPs). These proteins have been associated with a large number of important cellular processes and, in particular, are often involved in regulatory macromolecular interactions (1). Whereas chemically denatured proteins typically exhibit behavior consistent with simple random or statistical coil models (2,3), IDPs can possess significant nonrandom structure. In particular, they are often more compact than is expected or observed for chemically denatured proteins of the same length (4). Although numerous recent studies have attempted to characterize the structural properties of disordered states of proteins (5–7), the precise nature of structure in IDPs is still an important question. Although transient secondary structure is common and can be studied in detail with NMR techniques (8,9), the prevalence and importance of tertiary contacts is less certain. In addition, the origin of the variation in compaction seen in IDPs is unclear: does it arise from differences in secondary and/or tertiary structure? If so, what is the nature of this structure?
The sequence differences between folded and intrinsically disordered proteins have been studied extensively (10). IDPs tend to be deficient in the hydrophobic residues necessary for folding and rich in charged residues (11). Various methods have been developed that can quite effectively predict intrinsically disordered regions of proteins from primary amino acid sequences alone (12–15). However, despite the observation that there is significant structural variation between different IDPs (4), there has been little investigation into the sequence determinants of this structure.
A simple way to assess structure in a disordered protein is to measure its hydrodynamic radius (Rh). The Rh is the radius of an idealized sphere that would diffuse at the same rate as the molecule of interest, and is based on the Stokes-Einstein relation in Eq. 1, where kB is the Boltzmann constant, T is the temperature, η is the viscosity, and D is the translational diffusion coefficient. Thus, although the Rh is not a true measure of the radius of a nonglobular protein, as its diffusion is related to its nonspherical shape, it is very useful as a simple measure of compaction in disordered proteins.
| (1) |
Commonly used methods for measuring Rh in IDPs include size exclusion chromatography (SEC) and pulsed-field-gradient (PFG) NMR. In SEC experiments, the size exclusion column is calibrated using folded protein standards of known molecular weight, which allows the apparent molecular weight of the protein of interest to be measured. The Rh is then simply determined as the Rh expected for a folded protein of that molecular weight, for which simple relations exist (16). In PFG NMR experiments, the translational diffusion coefficient of the protein can be directly measured and compared to a standard of known Rh, allowing simple determination of the Rh (17).
In this study, we have compiled Rh measurements and amino acid sequences for a sizeable set of IDPs, which has allowed us to investigate the sequence determinants of compaction in these proteins. We show that the number of proline residues and the net charge seem to be the primary natural determinants of compaction in IDPs. This has important implications for understanding the nature of disordered-state structure and suggests a limited role for hydrophobic contacts. In addition, we also show that polyhistidine tags appear to have a large effect on compaction, suggesting that they should be removed before structural studies of IDPs are performed. Finally, we have used our findings to develop a new method for predicting the Rh of an IDP from the amino acid sequence that provides a substantial improvement over methods based on chain length alone.
Methods
Compilation of hydrodynamic radius measurements and protein sequences
A search of the literature was conducted to identify Rh measurements of IDPs and their associated amino acid sequences. To avoid sequence bias from homologous proteins, the program Needle from the EMBOSS suite (18) was used to identify sequences with >50% similarity to each other; only the longer of two homologous sequences was retained. In addition, only proteins with <300 residues were chosen, because we expected that larger proteins would be more likely to contain a mixture of folded and disordered regions. Table S1 in the Supporting Material presents all of the proteins used in this study, along with their Rh values and amino acid sequences. In total, we used 32 Rh measurements, 12 determined by PFG NMR and 20 by SEC. We did not use any dynamic light-scattering measurements of Rh, because the number of IDP measurements we found was too small to adequately assess their similarity to PFG NMR and SEC measurements. For some proteins, the precise amino acid sequence could not be obtained due to insufficient details regarding the protein expression construct represented by X in Table S1. pH values for each Rh measurement were obtained for the purpose of determining the charge state of histidine residues by assuming a side-chain pKa of 6.8 (19). We also repeated all relevant calculations assuming a histidine side-chain pKa of 6.0; in this case, our results were nearly identical (not shown). Finally, we compiled Rh measurements for a number of folded and chemically denatured proteins; these are listed in Table S2 and Table S3.
Calculation of raa values and associated error bars
To determine error bars for the calculated raa values, which describe the correlation between different amino acid residues and increased compaction or expansion (defined later), we employed a simple bootstrapping analysis whereby random sets of 32 proteins were selected from the full data set (importantly, allowing for multiples of each protein so that each set is different). The raa values were calculated from each random set and the procedure was performed 10,000 times; error bars represent the standard deviations of raa from these replicates. The source code for these calculations is provided in the Supporting Material.
Results
Intrinsically disordered proteins have a greater range of compaction than chemically denatured proteins
Previous studies have shown that there is a strong correspondence between the number of residues in a folded or disordered protein and its molecular size as measured by Rh or radius of gyration (Rg). The simple power-law scaling relationship in Eq. 2 has been used to provide remarkably good predictions; R is the Rg or Rh, N is the number of residues in the protein, and R0 and v are constants (2,20). In an excluded-volume random coil, v is predicted to be 0.588 (21). This is very close to the empirically determined value of 0.598 based upon small-angle x-ray scattering measurements of Rg for a number of chemically denatured proteins, thus providing one of the strongest arguments for the random-coil-like behavior of chemically denatured proteins (2).
| (2) |
In Fig. 1, we plot the Rh values versus the number of residues for a large number of folded, chemically denatured, and intrinsically disordered proteins. Each class of protein has been fit to Eq. 2, providing the following relationships between Rh and number of residues:
| (3) |
| (4) |
and
| (5) |
Figure 1.
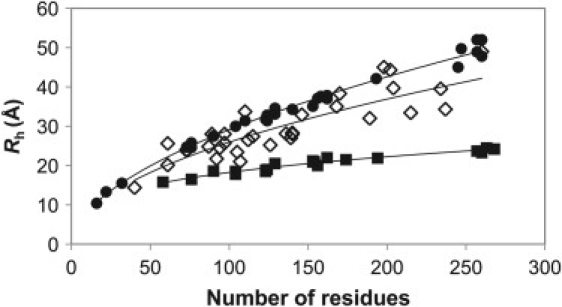
Number of residues versus Rh for 20 folded (solid squares), 27 chemically denatured (solid circles), and 32 intrinsically disordered (open diamonds) proteins.
These relations are in very good agreement with the commonly used values determined by Wilkins et al. (R0 = 4.75 and v = 0.29 for folded; R0 = 2.21 and v = 0.57 for denatured) (20). Rh values predicted from the above relations agree very well with their experimentally determined values for folded and chemically denatured proteins, with root-mean-squared deviations (RMSDs) of 0.56 and 1.20 Å, respectively. However, for IDPs, a much greater range in compaction is observed, and the agreement is much worse (RMSD = 3.85 Å). Overall, the IDPs tend to be more compact than chemically denatured proteins, as has been recognized previously (4), although they can occasionally be even more expanded. Clearly, then, there must be some heterogeneity in the structural properties of IDPs to account for this divergence from the simple power-law relationship that describes folded and chemically denatured proteins so well. The main goal of this study is to relate this variation in compaction to primary amino acid sequences.
Relating sequence to compaction in intrinsically disordered proteins
To express compaction, we define the term relative Rh as , where comes from Eq. 5. It is important to note that Rrel shows no significant correlation with the number of amino acids for the proteins in the data set (r = 0.03), so it is independent of chain length. To assess the sequence dependence of compaction, we calculate the Pearson correlation coefficient, raa, between the fractional content of each type of amino acid (e.g., if alanines constituted 10% of the residues in a protein, this would be 0.1) and Rrel for each protein. Amino acids that tend to be associated with more expanded proteins will have positive values of raa, whereas those associated with increased compaction will have negative values.
In Fig. 2 A, we plot the raa for each type of amino acid. The first notable feature of this figure is the residue histidine, which has the lowest raa value. Given that 9 of the 32 proteins in the data set contain polyhistidine tags used for the affinity purification, we wondered whether the presence of these tags might be associated with increased compaction. In Fig. 2 B, we have treated histidine residues present in a polyhistidine tag separately from other histidine residues; they are identified by an asterisk. In this plot, we see that polyhistidine residues have an even stronger association with compaction, whereas other histidine residues are not significantly associated with compaction, suggesting that polyhistidine tags can have a major effect on the compaction of IDPs in vitro. This can be seen in more detail in Fig. 3 A, where the Rrel for each protein is plotted versus the fraction of polyhistidine tag residues in each protein (zero for most proteins). There is a remarkable tendency for the nine polyhistidine-tagged proteins to be more compact on average than nontagged proteins.
Figure 2.
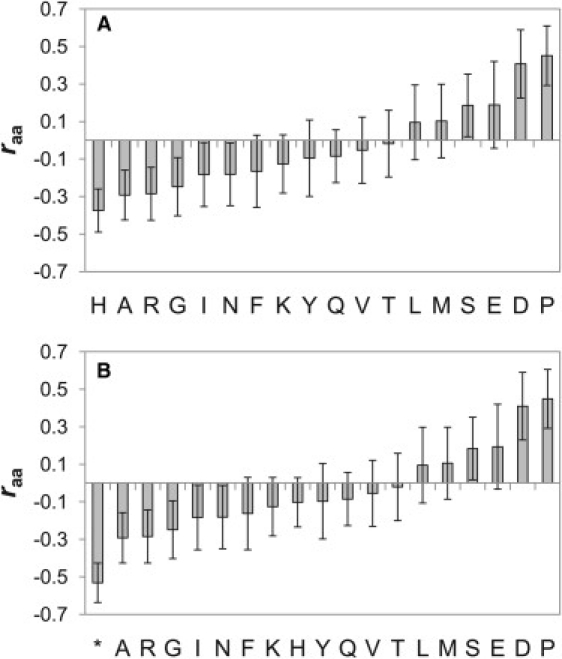
(A) Correlation (raa) between the fractional content of each amino acid from each protein and Rrel. (B) Same as A, except that histidine residues present in a polyhistidine tag are considered separately (asterisk). Error bars were calculated with a bootstrapping procedure (see Methods). W and C are not shown because the number of these residues in the data set was very low.
Figure 3.
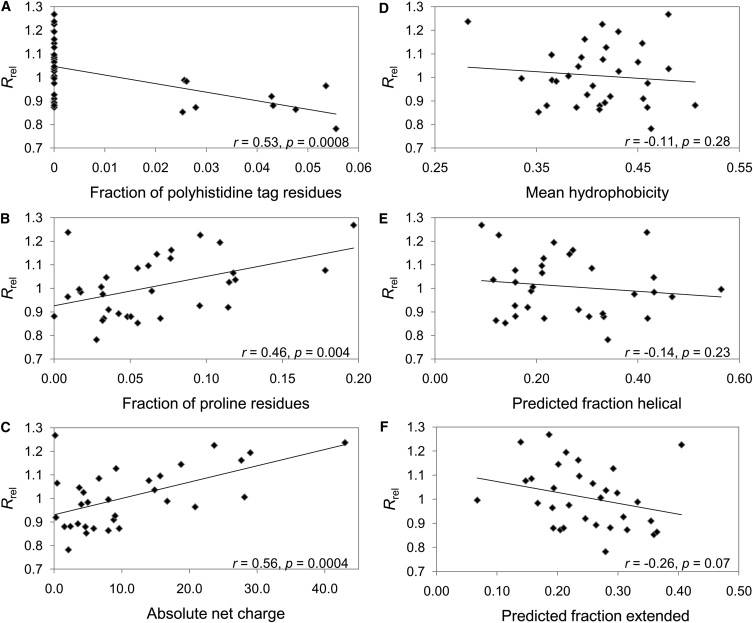
Comparisons of Rrel for all proteins in the IDP data set to the fraction of polyhistidine tag residues (A), fraction of proline residues (B), absolute net charge (C), mean hydrophobicity (D), predicted fraction of helical residues (E), and predicted fraction of extended residues (F).
Proline has the highest raa value in Fig. 2, A and B, and thus has the strongest association with more highly expanded proteins. This is further demonstrated in Fig. 3 B, where we plot Rrel for all proteins in the data set versus the fraction of proline residues in each protein. There is a fairly strong, statistically significant correlation (r = 0.46, p = 0.004), suggesting that proline residues play an important role in determining the compaction of IDPs.
Another very interesting aspect of Fig. 2, A and B, is the distribution of charged residues. Aspartate and glutamate residues have the second- and third-highest raa values, suggesting that negatively charged side chains are associated with more expanded states. It appears that positively charged residues may have a slight tendency to associate with more compact states, with arginine having one of the lowest raa values and lysine and histidine also having negative raa values. This apparent discrepancy between positively and negatively charged residues can be explained if one considers that the IDPs in our data set have a greater tendency to be negatively charged than positively charged (19 of 32 IDPs in our data set have a net negative charge). Thus, positively charged residues will tend to reduce the overall net charge of the protein. In Fig. 3 C, we plot Rrel versus the absolute net charge for all proteins in the data set and observe a significant correlation (r = 0.56, p = 0.0004). This correlation is retained even when all histidine residues are ignored (r = 0.58, not shown), demonstrating that this association with net charge is not related to the presence of polyhistidine tags. We also compared Rrel to the average number of charged residues (treating histidine as its fractional charge state). In this case, very little correlation was observed (r = 0.15, p = 0.21) (not shown). These results strongly suggest that the overall net charge, but not simply the number of charged residues alone, is a key determinant of IDP compaction.
Given the importance of hydrophobic interactions in the structure of folded proteins, hydrophobicity might be expected to show a strong association with IDP compaction. However, in Fig. 2, A and B, there is no obvious relation that can be discerned for the hydrophobic residues. In Fig. 3 D, we plot the average Kyte-Doolitle hydrophobicity (22) versus Rrel for the IDPs in the data set. We observe only a very weak correlation between increased hydrophobicity and compaction that is not statistically significant (r = −0.10, p = 0.29). Closely related to hydrophobicity is the average area buried upon folding (AABUF) (23), which was recently used to predict transient collapsed structure in acid-denatured apomyoglobin (24). AABUF shows a stronger correlation with Rrel (r = −0.25, p = 0.08) (not shown). However, the correlations for hydrophobicity and AABUF reduce to −0.02 and −0.01, respectively, if charged residues and prolines are ignored. Although these results do not rule out some role for hydrophobic contacts in the compaction of IDPs, they suggest that any contribution must be quite limited.
We also investigated whether intrinsic propensities for forming secondary structure might be correlated with compaction. In Fig. 3, E and F, we plot the predicted GOR3 (25) helical and extended secondary structure versus Rrel for each protein. There are slight negative correlations for both helical (r = −0.14, p = 0.23) and extended (r = −0.26, p = 0.07) secondary structure, suggesting that there may be a relationship between increased secondary structure and reduced Rh. However, the evidence is not strong enough to say with certainty that there is a statistically significant relationship between secondary structure and compaction. In addition, when charged residues and prolines are ignored in the GOR3 predictions, the correlations reduce to 0.049 and −0.17 for helical and extended structures, respectively (not shown).
Comparison of PFG NMR and SEC measurements
In this study, we have combined measurements of Rh that were made using very different experimental methods: PFG NMR and SEC. Therefore, it is important to address whether these different methods give similar measurements. In Fig. 4, we show the Rh values versus number of residues for PFG NMR and SEC measurements, as well as their best-fit lines (Eq. 1). There are no obvious divergences between the two data sets, and the best-fit lines are very similar, supporting the similarity of the results obtained by these methods. In addition, we have also compared the correlations identified earlier using only proteins from the separate data sets. The PFG NMR and SEC data sets have correlations of 0.56 and 0.35, respectively, between the fraction of proline residues and Rrel, 0.52 and 0.66 between absolute net charge and Rrel, and −0.18 and −0.78 between the fraction of polyhistidine residues and Rrel (there are only three polyhistidine-tagged proteins in the PFG NMR data set, so there is little significance to this weak correlation). The fact that relatively similar correlations exist in these independent data sets (given the limited numbers of measurements available) both confirms the significance of these correlations and supports the validity of combining PFG NMR and SEC measurements in our full data set.
Figure 4.
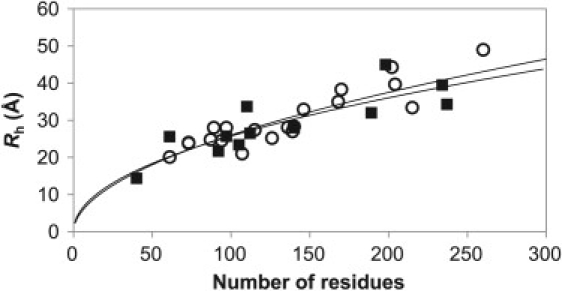
Number of residues versus Rh values measured with SEC (open circles) and PFG NMR (solid squares). The best-fit power-law scaling lines (Eq. 2) are shown for SEC measurements (upper line) and PFG NMR measurements (lower line).
Improved prediction of hydrodynamic radius using sequence information
As discussed above, Rh values for folded and chemically denatured proteins can be predicted extremely well from the number of residues alone using a simple power-law relation (Eq. 1). IDPs, on the on the other hand, show much greater variation in compaction, which we observe to be correlated with their proline content, net charge, and the presence or absence of a polyhistidine tag. Therefore, we decided to investigate whether this sequence information could be used to improve Rh predictions of IDPs.
In Eq. 6, we have extended the simple form of Eq. 2 to account for the fraction of proline residues (Ppro), the absolute net charge (|Q|) and the presence of a polyhistidine tag, where A–D are constants that are fit from the slopes (A and C) and offsets (B and D) of the linear fits between Rrel and Ppro or |Q|, and Shis∗ is a scaling factor applied if the protein has a polyhistidine tag (Shis∗ is 1 if no tag is present). These constants are then optimized to maximize the agreement between the predicted and experimental Rh using a simple Monte Carlo procedure. The best-fit parameters for Eq. 6 from all 32 proteins in the data set are A = 1.24, B = 0.904, C = 0.00759, D = 0.963, Shis∗ = 0.901, R0 = 2.49, and v = 0.509.
| (6) |
To evaluate the ability of this new expression to predict the Rh of IDPs, an all-but-one procedure was performed in which, for each protein, all of the other proteins were used to fit the parameters in both Eqs. 2 and 6. These were then used to predict the Rh of that protein in an unbiased manner. Fig. 5 shows the differences between experimental Rh measurements and values predicted using both methods. We see that the new, sequence-based method (Eq. 6) shows a substantial improvement over the simple power-law relation (Eq. 2), with an RMSD of 2.37 Å between experimental and predicted Rh, compared to 4.13 Å for Eq. 2 (note that this is different from the value of 3.85 Å given earlier, because it comes from the unbiased all-but-one fitting instead of from all data points). In Table 1, we present the comparisons of Rh predictions using varying subsets of the sequence information (i.e., proline content, net charge, and polyhistidine tags). These results demonstrate quite clearly that net charge is the most useful information for predicting Rh, whereas proline content and the presence of a polyhistidine tag are of roughly similar utility. However, the best results are obtained when all sources of information are combined.
Figure 5.
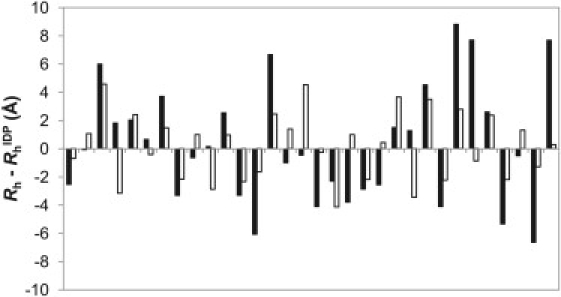
Differences between experimentally determined Rh and predicted with the simple power-law model (solid bars) and the new sequence-based model (open bars) for all 32 IDPs in the data set.
Table 1.
Comparison of RMSDs between experimental Rh values and values predicted using varying subsets of sequence information
| Pro | Q | His∗ | RMSD (Å) |
|---|---|---|---|
| 4.13 | |||
| X | 3.82 | ||
| X | 3.74 | ||
| X | X | 3.29 | |
| X | 3.25 | ||
| X | X | 2.78 | |
| X | X | 2.71 | |
| X | X | X | 2.37 |
Pro, proline content; Q, net charge; His∗, presence of a polyhistidine tag; X indicates that the applicable term from Eq. 6 was utilized for the calculation of Rh values.
Discussion
Our analysis of Rh values for a number of IDPs demonstrates that net charge and proline content are the primary natural determinants of Rh. In addition, the presence of polyhistidine tags in the recombinant protein samples also leads to significantly increased compaction. Hydrophobicity and secondary structure, on the other hand, contribute very little.
The finding that net charge is the most dominant factor contributing to the variation in IDP compaction is interesting but not surprising, as increasing repulsive electrostatic forces would be expected to cause conformational expansion. It is also very interesting when we consider the high fraction of charged residues (10,11) and the high frequency of residues that undergo phosphorylation in IDPs (26). Therefore, phosphorylation or other posttranslational modifications that affect net charge (e.g., ubiquitination or methylation) could represent a simple method for modulating compaction in IDPs. This could be utilized in various regulatory functions. Increased expansion might make binding sites more accessible or modulate the spatial separation between two domains separated by an intrinsically disordered linker. Conversely, increased compaction might bring separate binding elements closer together, thus facilitating an interaction, or sterically block access to another part of the protein.
Despite the strong relationship between compaction and net charge, there is very little correlation with the number of charged residues alone, suggesting that only the net effect of all charged residues is important. Our results are therefore consistent with the recently proposed polyelectrostatic model, in which independent point charges in a disordered protein are averaged in a mean-field interpretation and the overall net charge of the protein is found to be most significant for its interaction properties (27). Although this is obviously a highly simplified model, it has been very effective for describing the phosphorylation dependence of binding a polyvalent IDP to a single ligand (27,28). Of course, at a detailed molecular level, there are likely to be local structural effects due to sequence-dependent charge variations. However, from the perspective of overall molecular compaction, with the resolution available in our data set, only the net charge appears to be significant.
Other examples from the literature provide further support for the dependence of IDP compaction on net charge. For example, intrinsically disordered α-synuclein, which is best known for its aggregating role in Parkinson's disease, has been shown to become more compact at low pH, presumably due to neutralization of charged carboxylate side chains in the acidic C-terminal region (29–31). Another recent study of 14-residue peptides of varying sequence showed that increasing negative charge led to significant conformational expansion (32).
In addition to net charge, the proline content of IDPs also seems to be important for determining their compaction. This is easy to rationalize: proline residues have a strong tendency to adopt backbone dihedral angles in the extended regions of Ramachandran space and, in particular, the highly extended righthand side of the broad β-region, commonly referred to as the polyproline II region (33). This tendency toward extended backbone conformations thus leads to an increased Rh for proline-rich sequences. In addition, one could hypothesize that the tendency of proline residues to undergo cis-trans isomerization might inhibit the formation of more compact elements of structure. It seems very likely that the high proline content associated with IDPs is related to their effect on compaction, i.e., in addition to its role in promoting disorder (10), proline content may be evolutionarily related to the functional requirements for varying IDP compaction.
The lack of a contribution from hydrophobic residues in IDP compaction is a very important result of our analysis. Given that burial of hydrophobic residues is the dominant force driving protein folding, much of the speculation regarding transient structure in IDPs has focused upon the importance of hydrophobic side-chain interactions. However, given the deficiency of hydrophobic residues and abundance of charged residues in IDPs (11), it is perhaps not surprising that the effect of hydrophobic clustering is minimal. Of course, there could still be some contribution to compaction from hydrophobicity that is undetectable, given the limited size and resolution of our data set. In a recent study, Krishnan et al. made 10 phenylalanine-to-alanine mutations in the intrinsically disordered Nup116 FG domain (34). This resulted in a slight increase in Rh, from 25.2 to 27.1 Å, suggesting that hydrophobic interactions between phenylalanine side chains likely play some role in the compaction of this protein. In addition, there is significant evidence for hydrophobic clustering in the unfolded states of some folded proteins, including the drkN SH3 domain (35–37), reduced unfolded lysozyme (38), and acid-denatured apomyoglobin (24). The fact that collapsed structure in acid-denatured apomyoglobin could be predicted quite well using the AABUF parameter (24) although AABUF shows little correlation with our IDP data set suggests that there may be fundamental structural differences between IDPs and the unfolded states of normally folded proteins that are related to their very different sequence characteristics.
Fractionally populated secondary structure is known to be common in IDPs (8,9). Therefore, that we do not observe a statistically significant correlation between predicted secondary structure and compaction does not mean it is not an important aspect of IDP structure. Residues that preferentially sample different regions of Ramachandran space could be expected to have some effect on compaction (as is likely the case for proline), and sequences favoring β-turns that reverse the direction of the polypeptide chain should also lead to more compact states. However, these effects cannot be conclusively identified in our limited data set, as their contributions are probably quite small compared to electrostatic interactions.
The observation that polyhistidine tags used for affinity purification of recombinant proteins appear to cause a significant increase in IDP compaction is very interesting but somewhat disturbing, given the frequency with which these tags are used in in vitro studies. The effect is likely due to interactions between the polyhistidine tag and other residues in the protein, possibly to the partially charged (depending on the pH) nature of the histidine side chains. Due to the intrinsic conformational entropy of a disordered polypeptide chain, interactions involving the N- or C-termini are more favorable than internal regions, as was previously shown by Chan and Dill (39) and noted for the unfolded state of the drkN SH3 domain (35). Thus, the N- or C-terminus is the worst possible position for a tag that has a tendency to interact with other regions. Although highly useful for the protein purification procedure, clearly it is important to remove polyhistidine tags before performing any structural or functional studies on IDPs.
A significant limitation of the data set used in this study results from the different conditions under which each measurement was made. For example, although most measurements were made at room temperature, some were made around 4°C which may have a significant effect on any hydrophobic interactions. In addition, widely varying buffer conditions were used. The strong association we identified between electrostatics and compaction suggests that salt concentration should have a substantial effect on IDP structure. Thus, it is likely that the role of charge in IDP compaction is even greater than suggested by the correlations we observe, given the large variations in salt concentration between different experimental measurements. Additional studies of different proteins under uniform experimental conditions or of individual proteins under varying conditions will be extremely useful for more precise assessment of the contributions of different factors to IDP compaction.
The method for prediction of IDP Rh presented in this study should be valuable for future experimental studies of IDPs, as it provides an improved reference to which IDP compaction can be compared. Nevertheless, we still observe significant deviations between predicted and experimentally determined values. Assuming we had a perfect method for Rh prediction, the expected variation arising from experimental error alone can be estimated by looking at the deviation from simple power-law scaling in chemically denatured proteins, where an RMSD of 1.20 Å was observed. Although the chemically denatured proteins have more uniform solvent conditions, the RMSD of 2.37 Å for our predictions of IDP Rh suggests that significant improvement could be made to our prediction method. Increasing the size of the data set and decreasing the variation in sample conditions would likely allow more statistically significant correlations to be observed that could lead to improved predictions. In addition, it is highly likely that IDP structure is not encoded by the fractional residue content alone, but instead is significantly dependent on the order of residues in the primary sequence. For example, local clusters of charged or hydrophobic residues might be expected to have a cooperative effect on compaction compared to a uniform sequence distribution (as is likely the case for the observed effect of polyhistidine tags). Comparing predicted to experimental Rh will be useful for identifying IDPs with such nonrandom or cooperative structural properties.
Our results emphasize the growing recognition that a dynamic continuum from ordered to disordered states exists within proteins, with varying amounts of flexibility and structure being possible within folded and disordered proteins. IDPs do not have homogenous structural properties. Rather, simple sequence properties like charge and proline content can have a large effect on their compaction. The sequence determinants of structure in IDPs are potentially much less complex than in folded proteins, where precise three-dimensional structure must be encoded in the primary amino acid sequence, but nevertheless, they are still likely very important for determining the numerous important biological functions identified for IDPs. As our structural understanding of IDPs improves, so should our ability to relate their structural properties to biological functions.
Supporting Material
Three tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00234-1.
Supporting Material
Acknowledgments
This work was supported by grants from the Canadian Institutes for Health Research.
References
- 1.Dyson H.J., Wright P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 2.Kohn J.E., Millett I.S., Plaxco K.W. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanford C., Kawahara K., Lapanje S. Proteins in 6-M guanidine hydrochloride. Demonstration of random coil behavior. J. Biol. Chem. 1966;241:1921–1923. [PubMed] [Google Scholar]
- 4.Uversky V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mittag T., Forman-Kay J.D. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Vendruscolo M. Determination of conformationally heterogeneous states of proteins. Curr. Opin. Struct. Biol. 2007;17:15–20. doi: 10.1016/j.sbi.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Meier S., Blackledge M., Grzesiek S. Conformational distributions of unfolded polypeptides from novel NMR techniques. J. Chem. Phys. 2008;128:052204. doi: 10.1063/1.2838167. [DOI] [PubMed] [Google Scholar]
- 8.Jensen M.R., Houben K., Blackledge M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc. 2008;130:8055–8061. doi: 10.1021/ja801332d. [DOI] [PubMed] [Google Scholar]
- 9.Marsh J.A., Singh V.K., Forman-Kay J.D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 11.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Dosztányi Z., Csizmok V., Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 13.Linding R., Jensen L.J., Russell R.B. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Romero P., Obradovic Z., Dunker A.K. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 15.Ward J.J., Sodhi J.S., Jones D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Uversky V.N. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 1993;32:13288–13298. doi: 10.1021/bi00211a042. [DOI] [PubMed] [Google Scholar]
- 17.Jones J.A., Wilkins D.K., Dobson C.M. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR. 1997;10:199–203. [Google Scholar]
- 18.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 19.Cantor C.R., Schimmel P.R. W. H. Freeman; San Francisco: 1980. Biophysical Chemistry, Part I. [Google Scholar]
- 20.Wilkins D.K., Grimshaw S.B., Smith L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 21.LeGuillou J.C., Zinn-Justin J. Critical exponents for the n-vector model in three dimensions from field theory. Phys. Rev. Lett. 1977;39:95–98. [Google Scholar]
- 22.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 23.Rose G.D., Geselowitz A.R., Zehfus M.H. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 24.Felitsky D.J., Lietzow M.A., Wright P.E. Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc. Natl. Acad. Sci. USA. 2008;105:6278–6283. doi: 10.1073/pnas.0710641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garnier J., Gibrat J.F., Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 26.Iakoucheva L.M., Radivojac P., Dunker A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borg M., Mittag T., Chan H.S. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl. Acad. Sci. USA. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittag T., Orlicky S., Forman-Kay J.D. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl. Acad. Sci. USA. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uversky V.N., Li J., Fink A.L. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 30.Wu K.P., Weinstock D.S., Baum J. Structural reorganization of α-synuclein at low pH observed by NMR and REMD simulations. J. Mol. Biol. 2009;391:784–796. doi: 10.1016/j.jmb.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClendon S., Rospigliosi C.C., Eliezer D. Charge neutralization and collapse of the C-terminal tail of α-synuclein at low pH. Protein Sci. 2009;18:1531–1540. doi: 10.1002/pro.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soranno A., Longhi R., Buscaglia M. Kinetics of contact formation and end-to-end distance distributions of swollen disordered peptides. Biophys. J. 2009;96:1515–1528. doi: 10.1016/j.bpj.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adzhubei A.A., Sternberg M.J. Left-handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 1993;229:472–493. doi: 10.1006/jmbi.1993.1047. [DOI] [PubMed] [Google Scholar]
- 34.Krishnan V.V., Lau E.Y., Rexach M.F. Intramolecular cohesion of coils mediated by phenylalanine—glycine motifs in the natively unfolded domain of a nucleoporin. PLOS Comput. Biol. 2008;4:e1000145. doi: 10.1371/journal.pcbi.1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsh J.A., Forman-Kay J.D. Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints. J. Mol. Biol. 2009;391:359–374. doi: 10.1016/j.jmb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Crowhurst K.A., Forman-Kay J.D. Aromatic and methyl NOEs highlight hydrophobic clustering in the unfolded state of an SH3 domain. Biochemistry. 2003;42:8687–8695. doi: 10.1021/bi034601p. [DOI] [PubMed] [Google Scholar]
- 37.Crowhurst K.A., Tollinger M., Forman-Kay J.D. Cooperative interactions and a non-native buried Trp in the unfolded state of an SH3 domain. J. Mol. Biol. 2002;322:163–178. doi: 10.1016/s0022-2836(02)00741-6. [DOI] [PubMed] [Google Scholar]
- 38.Klein-Seetharaman J., Oikawa M., Schwalbe H. Long-range interactions within a nonnative protein. Science. 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]
- 39.Chan H.S., Dill K.A. Interchain loops in polymers: effects of excluded volume. J. Chem. Phys. 1989;90:492–509. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.