

Abstract
Structure-function relationships in the tetrameric enzyme urate oxidase were investigated using pressure perturbation. As the active sites are located at the interfaces between monomers, enzyme activity is directly related to the integrity of the tetramer. The effect of hydrostatic pressure on the enzyme was investigated by x-ray crystallography, small-angle x-ray scattering, and fluorescence spectroscopy. Enzymatic activity was also measured under pressure and after decompression. A global model, consistent with all measurements, discloses structural and functional details of the pressure-induced dissociation of the tetramer. Before dissociating, the pressurized protein adopts a conformational substate characterized by an expansion of its substrate binding pocket at the expense of a large neighboring hydrophobic cavity. This substate should be adopted by the enzyme during its catalytic mechanism, where the active site has to accommodate larger intermediates and product. The approach, combining several high-pressure techniques, offers a new (to our knowledge) means of exploring structural and functional properties of transient states relevant to protein mechanisms.
Introduction
Hydrostatic pressure perturbation is a powerful tool for exploring the physicochemical characteristics and the functional mechanism of a macromolecule. It allows the Gibbs free energy of the system under study to be modified smoothly and continuously in a controlled manner, and it can be used to enhance the concentration of high-energy conformers and to perturb ligand binding (1–9). Another classical effect of pressure is the destabilization of the quaternary structure of oligomeric proteins (10–12), which is generally observed at low pressure (100–300 MPa).
In this high-pressure (HP) study, these possibilities have been explored for the enzyme urate oxidase (UOX; Enzyme Commission (EC) number 1.7.3.3), also known as uricase, from Aspergillus flavus in complex with 8-azaxanthine (8-aza), which is active as a homotetramer. UOX catalyzes the oxidation of uric acid in the presence of molecular oxygen to a primary intermediate, 5-hydroxyisourate. A wealth of information is available concerning the ligand binding mode and function of this enzyme (13–17), but the structural basis of the catalytic mechanism of this cofactorless oxidase is still not fully understood. Since 8-aza is localized to the same position as the uric acid substrate (16), the structural information drawn from the UOX/8-aza complex can be extended to the enzyme in the presence of its substrate.
Each of the four monomers (A–D) features an antiparallel eight-stranded β-sheet with four helices on the convex side of the β-sheet (18,19). The monomer fold is likely to be unstable and the protein is structurally stabilized through oligomeric association (20). The AB dimer is a 16-stranded β-barrel with helices flanking the exterior of the barrel. Two head-to-tail β-barrels form the tetramer, which is traversed by a central tunnel (Fig. 1 A). The tetramer displays three types of interface between monomers. The AB-type interface, between A and B or C and D monomers, has the largest buried surface area (∼6000 Å2), and is stabilized by main-chain hydrogen bonds between adjacent β-strands within the β-barrel. The active site of each monomer is located at this interface. The AC-type interface (between A and C or B and D monomers), which also has a large buried surface area (∼5200 Å2), is formed between two monomers along the tunnel axis and is stabilized by hydrogen bonds between the extremities of the β-strands forming the β-barrel. The AD-type interface (between A and D or B and C monomers), much more limited in area (buried area ∼800 Å2), delineates the zone where the substrate has access to the active site. Crystal packing induces two very small interfaces between A and a symmetrical C subunit and between A and a symmetrical D subunit (buried area ∼300 Å2).
Figure 1.
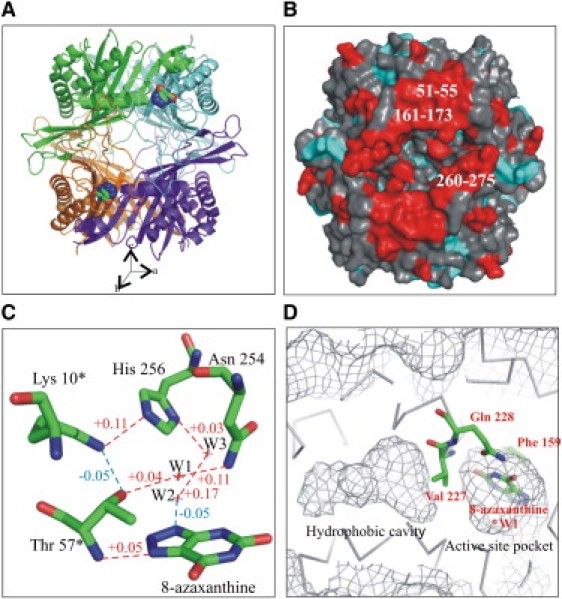
Molecular structure of UOX/8-aza complex under pressure. (A) View along the b unit-cell axis of the four chains of UOX, represented in cartoon format (green, A; blue, B; orange, C; and purple, D). The UOX tetramer has a spherical shape ∼70 Å in diameter, with a central tunnel 15 Å in diameter. The four 8-aza inhibitor molecules (colored atoms) are located at the interfaces between the A and B subunits, and between the C and D subunits (only two inhibitors are visible here; the other two are located on the face behind the tetramer and are thus hidden in this representation). The tunnel axis is along the crystallographic c axis. (B) View of the surface of UOX tetramer along the b unit-cell axis colored red or blue when the relative B-factor difference between AP and HP structures is higher or lower, respectively, than 1 standard deviation with respect to the average. (C) Active site residues and water molecules involved in the catalytic mechanism. A chain of hydrogen bonds involved in catalysis connects Asn154, W1, Thr57∗, Lys10∗, His256, W3, W2, and the N9 atom of the ligand. The hydrogen bonds are shown with dotted lines, colored red or blue when they are elongated or shortened, respectively, by pressure. (D) View of the active-site pocket and the hydrophobic cavity. The inhibitor 8-aza (like the substrate) is stacked above Phe159 and is hydrogen-bonded to the enzyme through molecular tweezers composed of the side chains of Arg176 and Gln228. Val227 is hydrogen-bonded to the ligand by its main-chain nitrogen, and has its hydrophobic side chain lining the hydrophobic cavity.
Since substrate or inhibitor molecules bind at sites located at the AB-type interfaces, their affinity is intimately related to the interface structure. In this context, applying pressure perturbation to UOX in both the apo and inhibitor-complex forms would be particularly interesting. First, this is an effective and elegant way to study the pressure dissociation of this tetrameric protein in relation to inhibitor binding and catalytic activity. Second, the promotion of higher-energy conformational substates by pressure may reveal conformational changes associated with substrate binding and enzyme function, and a structural check of this hypothesis would be of particular interest.
The 2.3-Å resolution structure of UOX complexed with a uric-acid-like competitive inhibitor, 8-aza, was previously determined at 140 MPa by high-pressure macromolecular crystallography (HPMX) (21). That work provided a clear incentive for a deeper investigation of the effect of pressure perturbation on UOX. After a further HPMX experiment at a slightly higher pressure (150 MPa) and a better resolution (1.8 Å), complementary high-pressure experiments in solution were carried out using small-angle x-ray scattering (SAXS) and fluorescence spectroscopy. Enzymatic activity was also measured both under high pressure and at atmospheric pressure (AP) after pressure incubation of UOX to correlate activity to the observed pressure-induced structural modifications. We propose a model for tetramer dissociation consistent with all measurements and a structural explanation of the pressure-induced loss of affinity for the substrate. We also propose that the pressure-promoted conformer is likely to exist during the enzyme catalytic mechanism, and we highlight the role of a hydrophobic cavity in the enzyme catalytic mechanism.
Materials and Methods
UOX from Aspergillus flavus expressed in S. cerevisiae, and ligands were kindly provided by Sanofi-Aventis (Paris, France).
Fluorescence spectroscopy experiments
UOX contains seven tryptophan residues/subunit: Trp186, Trp188, and Trp208, which are buried within the core of each monomer; Trp160 and Trp174, which are solvent-accessible; and Trp106 and Trp264, located at the AC and AD interfaces, respectively.
The fluorescence experiments and light-scattering measurements were carried out at 298 K using an SLM Series 2 luminescence spectrometer (Aminco Bowman, Rochester, NY) modified to accommodate a high-pressure cell. For equilibrium and kinetics studies, the wavelength excitation of tryptophan was set to 295 nm (8-nm slit). Emission spectra (8-nm slit) were recorded, in triplicate accumulation mode, from 310 to 400 nm after incubation for 5 min after each increment of pressure. Fluorescence spectral changes were quantified by determining the spectral center of mass, <υp> (22,23):
| (1) |
where Fi is the intensity of fluorescence emitted at wavenumber υi. This quantity was then transformed to the average fluorescence wavelength, λp (24):
| (2) |
In turn, a spectral shift of tryptophan fluorescence to higher wavelengths is characterized by an increased average fluorescence wavelength, reflecting an exposure of tryptophan residues to water (Fig. 2, A and C).
Figure 2.

Effect of pressure on the conformational stability and activity of urate oxidase. (A) Time dependence of the average emission wavelength of UOX at 50 (●), 100 (■), 150 (▴), 175 (♦), 185 (●), 195 (+) and 200 MPa (▾). (B) Pressure dependence of the catalytic parameters at 25°C. The reaction was monitored at 292 nm after rapid mixing of a solution of 1–2 μg/ml UOX with a solution of uric acid. Double reciprocal plots of initial rates, kobs, versus uric acid concentrations (36, 54, 72, 110, and 150 μM) were drawn for each experimental pressure. Steady-state kinetic parameters KM (●) and kcat (○) were extracted from these plots by linear regression analysis using a Lineweaver and Burk representation. (C) The stability of UOX with and without inhibitors, probed by changes in the average emission wavelength with pressure: without inhibitor (●), and with 8-aza in equimolar (■) or saturating (▴) concentrations.
The protein concentration was 0.9 mg·ml−1. Experiments were done in 50 mM Tris-HCl buffer, pH 8. Various conditions were tested, with or without inhibitor (8-aza, 9-methyl uric acid, and 8-nitroxanthine) in equimolar or saturating concentrations, with the three inhibitors giving similar results.
Enzyme assays and kinetics
Data were fitted to the Michaelis-Menten equation using least-squares regression analysis to determine the inverse of the apparent affinity constant, KM, for the substrate, expressed in μM, and the catalytic rate constant, kcat, expressed in s−1 (Fig. 2 B). Based on the catalytic mechanism of urate oxidase isolated from soybean root nodules (25,26), we assume that for urate oxidase from Aspergillus flavus, too, the rate-determining step of the reaction is the step associated with the formation of the product, 5-hydroxyisourate (26).
The pressure dependence of the measured rate constant, kobs, at any substrate concentration [S] is described by Eq. 3:
| (3) |
In the case of a simple Michaelis-Menten model, the observed activation volume, ΔV‡obs, can be separated into two parts associated with the catalytic and productive substrate binding steps, respectively. The relevant equation (see, e.g., Morild (27)) is
| (4) |
where
| (5) |
and
| (6) |
SAXS experiments
Data were collected on the ID2 beamline at the European Synchrotron Radiation Facility (Grenoble, France). Scattering intensity was measured as a function of Q, defined as Q = 2πs = (4π/λ)sin θ, where s is the amplitude of the scattering vector s, λ is the x-ray wavelength, and 2 θ the scattering angle.
Pressure experiments were performed at λ = 0.751 Å, to reduce absorption from diamond windows and sample path. A thermostatic high-pressure cell of 3- or 4-mm diameter with 1-mm-thick diamond windows was connected to a 700-MPa pressure control system (Nova Swiss), using distilled water as the pressure medium. A biologically compatible sample holder was used to reduce the sample volume to <100 μl and to avoid direct contact of the sample with the cell and the pressure medium, as described in Skouri-Panet et al. (12).
Simulated scattering curves of the monomer, the AB and AC types of dimer, and the tetramer were computed from the UOX tetramer coordinates of the Protein Data Bank entry 1R56, using CRYSOL (28) with standard values for the hydration layer. The models used to fit the experimental data consisted of linear combinations of these curves. For each experimental curve, the best linear combination was determined by checking that the residual curve randomly crossed the 0 intensity line.
HPMX experiments and structure comparisons
For HPMX, crystals were obtained by the hanging drop technique, at room temperature, using an initial crystallization drop consisting of 3.2 mg.ml−1 protein with an equimolar concentration of inhibitor and a small amount of azide, 6% PEG 8000, 50 mM Tris buffer, pH 8, and 100 mM NaCl in equilibrium with a reservoir. Orthorhombic crystals grew in 1 week and were loaded into a diamond anvil cell (DAC), as described in Fourme et al. (29). The solution used as a compression medium consisted of 15% PEG 8000 in 50 mM Tris, pH 8, and 100 mM NaCl.
Data were collected on the ID27 beamline at the European Synchrotron Radiation Facility using unfocused radiation at λ = 0.374 Å and a MAR165 CCD detector at 320 mm from the sample. Pressure within the DAC compression chamber was monitored using the fluorescence from a ruby chip (29). Slits were adjusted to obtain a 50 × 50-μm beam on the sample. Exposure time was 15 s per image and oscillation angle was 0.25°. The resolution of diffraction data declined beyond 175 MPa without a significant increase in mosaicity. Diffraction disappeared beyond 200–220 MPa, before significant modifications of the crystal habit. Two data sets were collected using the same protocol at atmospheric pressure and at 150 MPa. Only one crystal was used for each data set, using a new large-aperture DAC (30). Each crystal was translated in the beam several times to irradiate successively fresh portions of the crystal. A summary of data collection and refinement statistics is reported in Table S1 in the Supporting Material.
The UOX/8-aza complex crystallized in the orthorhombic space group I222 with one monomer in the asymmetric unit, the tetramer complying with the 222 crystal symmetry. The relative B-factor difference for each atom was calculated using the formula ((BHP – 〈BHP〉) – (BAP – 〈BAP〉))/(〈BHP〉 – 〈BAP〉), which leads to a distribution with a zero mean value and limits bias from refinements (Fig. 1 B). The Voronoi cells were calculated with Voro3D (31) and the correlation matrix with AMBER (32) (Fig. S3). Hydrogen bonds within the monomer were analyzed with ACT, and the interactions between monomers with CONTACT, both from the CCP4 package. Volumes of the internal cavity and of the active site pocket were calculated with CASTp (probe radius 1.3 Å) (33). The root-mean-square deviations between atoms of HP and AP structures were calculated with CNS (34).
Fig. 1 was produced with the visualization software PYMOL (DeLano Scientific, San Carlos, CA).
Atomic coordinates and structure factors of urate oxidase under 150 MPa pressure have been deposited in the Protein Data Bank (3F2M).
Complete details of the different methods can be found in the Supporting Material.
Results
Pressure stability of apo-UOX
Tryptophan fluorescence and SAXS were used to probe protein stability under pressure. At atmospheric pressure, the emission spectra of the protein excited at 295 nm exhibited a maximum at 327 nm. After compression to 150 MPa, a slight decrease in fluorescence intensity at 327 nm was observed (Fig. S1). No further conformational changes were detected, even after incubation under pressure for 20 h at 25°C, as shown by the plot of average emission wavelength (see Materials and Methods) as a function of incubation time (Fig. 2 A). At higher pressures, a significant decrease in signal intensity and a spectral red shift from 328 nm to 342 nm were observed (Fig. S1). As shown in Fig. 2 A, these spectral changes took several hours. After depressurization, a fraction of the protein had aggregated (Table 1), suggesting that at pressures >150 MPa, the protein approaches a threshold of irreversible quaternary structural change. The remaining soluble protein was still tetrameric after the pressure treatment (Fig. S2), but displayed a pressure-dependent decrease in specific activity that culminated at 200 MPa with complete loss of activity (Table 1). However, at pressures up to 150 MPa, this loss of activity could be fully restored upon addition of excess substrate. To gain insight into the effect of pressure on enzyme activity, assays were performed under pressure in addition to those after decompression. Indeed, in the case of UOX, enzymatic activity is a suitable indicator for probing the structural integrity of the tetramer, since, as previously mentioned, the active sites are located at subunit interfaces. UOX activity was therefore measured at pressures up to 190 MPa and the pressure dependence of the catalytic parameters (kcat and KM) was determined (Fig. 2 B). At atmospheric pressure, the apparent affinity for uric acid was estimated to be 11 μM, in agreement with steady-state kinetic studies (25,26). In the 10–175 MPa pressure range, plots of lnKM and lnkcat as a function of pressure were linear. From these plots, ΔV‡kcat and ΔVpb (27) (Eqs. 5 and 6) were determined as +0.13 ± 0.05 ml.mol−1 and −11.7 ± 1.5 ml.mol−1, respectively. These results show that substrate binding is attenuated by pressure, as reflected by the negative value of ΔVpb. In contrast, catalytic activity is only marginally affected by pressure, as reflected by the near-zero value of ΔV‡kcat. It is interesting that above 175 MPa, the linearity of the pressure-dependent plots of lnKM and lnkcat is significantly disrupted. Due to the composite character of volume changes, this behavior might reflect different pressure contributions, such as changes in the rate-determining step, pressure-induced unfolding, or structural modifications at the active site.
Table 1.
Enzymatic activity of UOX measured at decompression after compression at different pressure values
| Pressure (MPa) | Specific activity (μmol.min−1.mg−1 protein) |
Soluble protein (%)∗ |
Loss of activity (%)† |
|
|---|---|---|---|---|
| Before compression | After decompression | After decompression | ||
| 50 | 6.47 | 6.47 | 100 | 0 |
| 100 | 6.51 | 4.91 | 100 | 25 |
| 150 | 6.47 | 4.42 | 85 | 32 |
| 175 | 6.41 | 3.85 | 55 | 40 |
| 185 | 6.47 | 2.70 | 47 | 58 |
| 195 | 6.49 | 1.84 | 30 | 72 |
| 200 | 6.51 | 0 | 8 | 100 |
Pressure was applied for 20 h in all cases.
Soluble protein corresponds to the estimation of the UOX remaining in solution after compression and centrifugation (see Materials and Methods).
Loss of activity, expressed as a ratio, reflects the difference in activity of the soluble protein remaining after compression.
At atmospheric pressure, the SAXS form factor exhibited a marked minimum at Q = ∼1.1 nm−1, characteristic of the UOX tetramer. One major change in the scattering intensity as a function of pressure (Fig. 3 A) was the progressive disappearance of this minimum, suggesting the dissociation of the tetramer and the simultaneous presence of lower oligomeric states. The dissociation started between 150 and 175 MPa and was particularly pronounced above 200 MPa (Fig. 3 B).
Figure 3.
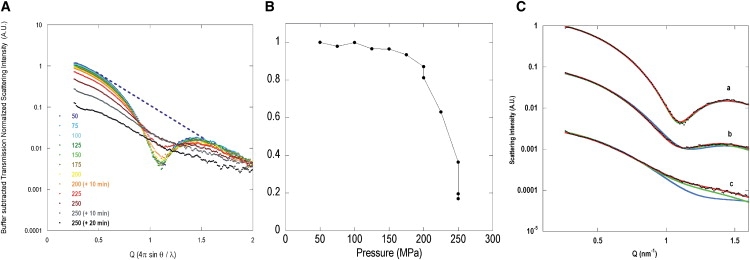
SAXS measurement results. (A) Changes in apo-UOX SAXS curves with increasing pressure. At P = 200 MPa and P = 250 MPa, additional data were collected at 10 min and at 10 and 20 min, respectively, after setting the pressure. (B) Ad hoc indicator of dissociation, evaluated from the area of the intensity minimum at Q ∼ 1.1 nm−1. Each value corresponds to the area delimited from Q1 to Q2, by log(Iexp) and a straight line joining the points (Q1,log(Iexp(Q1)) and (Q2,log(Iexp(Q2)), where Q1 = 0.482 nm−1 and Q2 = 1.467 nm−1. All values are normalized to the value at 50 MPa. The dashed blue line in A shows the upper limit for the integration area at P = 50 MPa. (C) Typical best-possible fits of experimental SAXS curves at three pressures. For the sake of clarity, the Q-range was restricted compared to A. The models consist of a weighted sum of two components: tetramer/AB dimer (blue), tetramer/AC dimer (green), and tetramer/monomer (red) for three pressures, P = 175 MPa (a), P = 225 MPa (b), and P = 250 MPa + 10 min (c). Only the tetramer/monomer mixture provides a satisfactory fit at all pressures.
More information can be gained about the lower oligomeric states observed at high pressure by comparing the experimental SAXS data to model scattering curves derived from the protein atomic coordinates in different potential oligomeric states. Three two-component systems were considered: 1), a mixture of tetramers and AB-type dimers; 2), a mixture of tetramers and AC-type dimers; and 3), a mixture of tetramers and monomers. For each of these mixtures, and at each pressure, a model curve was calculated by optimizing the proportion of tetrameric species to fit the experimental data. Fig. 3 C shows one such fit at three pressures. Since the proportion of tetramer decreased with increasing pressure, the three model curves were clearly distinguishable. Only the curves derived from the tetramer/monomer mixture fitted the experimental data, whatever the pressure, favoring the model whereby the tetramer dissociates into monomers. Based on this hypothesis, Table 2 shows the change in monomer fraction as a function of pressure. The monomer proportion increased with pressure, and with time at a given pressure. The apparent dissociation rate was higher at P = 250 MPa than at 200 MPa. Both the time-dependent changes and increased dissociation rates at high pressure are consistent with the pressure-dependent fluorescence measurements.
Table 2.
Changes in the monomer fraction as a function of pressure, derived from SAXS a few seconds after setting a new pressure value
| Pressure (MPa) | 50 | 75 | 100 | 125 | 150 | 175 | 200 | 200+10 min∗ | 225 | 250 | 250+10 min∗ | 250+20 min∗ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fraction of monomer (%) | 1 | 1 | 1 | 1 | 2.5 | 3.5 | 5 | 10 | 25 | 51 | 76 | 89 |
Measurements were repeated 10 min later at 200 MPa and at 10 and 20 min later at 250 MPa. Precision of the monomer fraction is ∼1%.
The overall decrease in scattering intensity detected in the SAXS patterns, especially at >250 MPa, can be attributed to the progressive precipitation of dissociation products, which results in less coherently scattering material in the beam and explains why the overall process is irreversible. Moreover, some aggregation preceding precipitation was suggested by intensity increases at very small angles, particularly at high pressures (Fig. 3 A).
Increased stability of UOX in the presence of inhibitors
Fluorescence measurements showed that 8-aza, a uric-acid-like inhibitor, increased the stability of the tetramer against pressure. The presence of 8-aza at equimolar concentrations shifted the curve of the average emission wavelength of apo-UOX to higher pressures by ∼35 MPa (Fig. 2 C). With 8-aza at saturating concentrations, this shift was even larger (∼85 MPa). Supporting evidence from SAXS experiments agreed with the fluorescence results and indicated that pressure-induced dissociation of the tetramer was shifted from 175–200 MPa without inhibitor to 250–275 MPa with an excess of inhibitor (data not shown).
Main structural modifications induced by pressure
HPMX experiments were carried out on crystals of the UOX/8-aza complex. Diffraction resolution degraded progressively above 175 MPa and was lost entirely around 200–220 MPa. High-resolution (1.8 Å) data sets were collected at AP and at 150 MPa (HP). The same batch of crystals and protocols for data collection, data analysis, and refinement were used at both pressures to facilitate differential measurements and to minimize systematic errors.
The unit-cell volume of the HP form was reduced by 1.3% and the tetramer volume by 0.3%. The mean root-mean-square deviation between positions of the main-chain atoms within the tetramer was 0.15 Å, with a higher value for residues within α-helices (0.19 Å) and a lower value for residues within β-strands (0.11 Å). The mean volume of the Voronoi cells calculated for the whole tetramer was reduced by 1.7 Å3 in the HP structure, with a larger variation for residues within α-helices (−2.4 Å3) than for residues within β-strands (−1.6 Å3). The sensitivity to pressure is greater for α-helices than for β-strands, as previously observed for other proteins (35–37). Mean hydrogen-bond lengths were slightly elongated at the AC interface, by 0.03 Å (average for 48 H-bonds), but were almost unchanged at the AB interface (0.01 Å) (average for 120 H-bonds). The distance variation between two atoms was smaller than, or comparable to, the precision of the distance obtained from crystallographic results, which was estimated to be ∼0.05 Å. Nevertheless, calculations of mean values provided statistically significant information on overall structural evolutions under HP. The overall B-factor of UOX increased by ∼5 Å2 at HP. Three loops located at interfaces (residues 51–55, 161–173, and 260–275) were found to be particularly pressure-sensitive, as they exhibited the largest increase in B-values between the HP and AP structures. These three loops all reside on the same side of the tetramer (along the b axis), delimiting access to the active site and comprising residues that are involved in the AD interface (Fig. 1 B). On the other two sides of the tetramer (along the a and c axes of the unit cell) there was no particular increase in B-factors.
No significant modification of the crystallographic hydration shell of the enzyme was observed. However, electron density lost with pressure corresponded to 17 water molecules, none of which were in the central tunnel, and electron density gained with pressure corresponded to 14 water molecules, 5 of which were in the central tunnel.
A large hydrophobic cavity of ∼190 Å3 is buried within each monomer and separated from the active-site pocket by a valine residue (Fig. 1 D). No electron density that could be assigned to ordered water molecules and no residual density in the Fo-Fc electron density map were observed in this cavity at either atmospheric pressure or 150 MPa. The volume of this cavity decreased by 16%. It is interesting that the volume of the nearby polar active-site pocket concomitantly increased by 11%.
Comparative analysis of the AP and HP structures revealed correlated displacements of the loop preceding the sixth β-strand (residues 148–182) and the fourth helix (residues 215–240), both containing residues lining the cavity and the active-site pocket (Fig. S3). These pressure-induced displacements are related to the combined contraction of the hydrophobic cavity and swelling of the active-site pocket.
Within the active-site pocket, distances between residues from two different subunits (between His256 and Lys10∗ and between Asn254 and Thr57∗) increased, as did the distance between 8-aza and Thr57∗ from the symmetrical subunit (Fig. 1 C), but remained compatible with H-bond distances. In contrast, the H-bond length between Lys10∗ and Thr57∗ within the same subunit decreased by 0.05 Å and the distances between 8-aza and W1 and 8-aza and the aromatic residue Phe159 (which is stacked below the inhibitor) decreased by 0.05 and 0.06 Å, respectively. In summary, pressure induced a distortion in the active site, with a shortening of distances between atoms within the same subunit and an elongation of distances between atoms belonging to different subunits.
Discussion
A coherent model for pressure-induced destabilization of the functional UOX tetramer can be proposed from the various experimental results with apo- and inhibitor-bound forms (Fig. 4).
Figure 4.
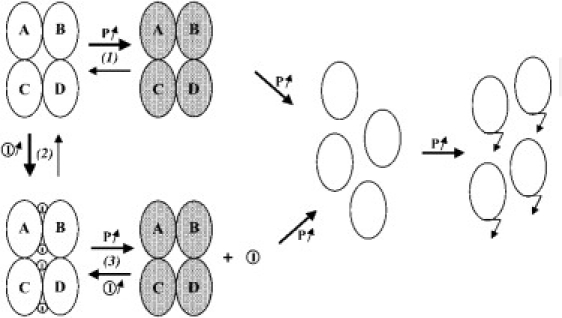
Schematic changes in UOX under high pressure, from the native tetramer with A, B, C, and D subunits to the aggregation of monomers. Monomers are shown as ellipses (white, native substate; gray, pressure-perturbed substate). Ligand molecules (substrate or substrate-like inhibitor) are depicted as circled I.
SAXS, fluorescence spectroscopy experiments, and enzymatic activity measurements reveal differences in the behavior of the enzyme below and above 150–175 MPa (Figs. 2, A and B, and 3 B, and Table 1). For apo-UOX incubated below 150 MPa, enzymatic activity measured at atmospheric pressure after depressurization was gradually lost as the incubation pressure was increased. Activity could be fully restored by increasing the substrate concentration. When assayed under pressure, the enzyme exhibited a reduced substrate affinity, whereas its catalytic efficiency remained intact (Fig. 2 B). All fluorescent spectral changes were reversible up to 150–175 MPa but became irreversible beyond that limit. We thus propose that pressure below 150–175 MPa modifies the protein structure, leading to pressure-perturbed tetrameric conformational states of decreased substrate affinity. The time course of pressure perturbation is driven by the equilibrium between the native apo-state and less active pressure-perturbed apo-states (Fig. 4, (1)). Pressure shifts the equilibrium toward perturbed states and gradually enhances the population of the most perturbed states up to a point where dissociation commences (150–175 MPa). Increasing pressure beyond ∼175 MPa leads to the irreversible dissociation of the tetramer, followed by aggregation. Since the reactions occurring above 150–175 MPa do not correspond to transitions between equilibrium states, their thermodynamic parameters cannot be determined. The dissociation process was also time-dependent, since the dissociation rate increased with time at a given pressure (Fig. 3 B and Table 2). The pressure at which the apo-UOX tetramer dissociated is in agreement with general observations for the pressure-induced dissociation of oligomers (10–12). When simulating SAXS curves with a variety of two-component models, only the monomer/tetramer model gave a good fit to the experimental data over the pressure range. Accordingly, before aggregation, pressure-induced tetramer dissociation generated monomeric intermediates, which were sufficiently stable to be observed during the course of a SAXS experiment.
Spectroscopic and SAXS measurements showed that the presence of a ligand at the AB-type interface prevented tetramer dissociation. At atmospheric pressure, the equilibrium between the enzyme in its apo-form and in complex with its ligand was shifted toward the ligand-bound tetramer by increasing the ligand concentration (Fig. 4, (2)). The pressure of dissociation increased by ∼35 MPa (Fig. 2 C) in the presence of 8-aza at an equimolar concentration. The stability is even increased by ∼85 MPa in the presence of a saturated solution of inhibitor. This can be explained by a free-energy linkage effect (38) between ligand binding to the active site located at the AB interface and tetramer stability. It should be noted that the inhibitor concentration dependence suggests that one of the initial effects of pressure is likely to be a ligand release produced by localized destabilization of the active site (Fig. 4, (3)). An early destabilization of the whole interface would indeed be independent of the inhibitor concentration. Finally, the ligand release leads to the perturbed apo-UOX states, which then dissociate into monomers before aggregating.
The origin of the pressure-induced unfolding of proteins is a matter of debate that has been attributed to either the filling of internal cavities by water molecules (39,40) or the collapse of solvent-excluded void volumes (41,42). The case of UOX rather supports the latter mechanism. No water molecules are present in the large hydrophobic cavity, even at high pressure, in contrast to the scenario for T4 lysozyme (39,40), and the reduction in volume of this void hydrophobic cavity is one order of magnitude larger than for the entire tetramer (16% vs. ∼0.3%). The pressure-induced dissociation of the UOX tetramer leads to transient monomers with a high ratio of solvent-exposed hydrophobic residues, which tend to aggregate and precipitate.
The comparison between the HP and AP structures showed an increase of the average B-factor, instead of a decrease, as generally observed (43,44), and a slight increase of average H-bond distances at the main interfaces (AC and AB). The expected variation would be a contraction of ∼0.015 Å, since the value ascribed in proteins for elastic compression is ∼0.1 Å.GPa−1 (43).
At 150 MPa, the enzyme showed a marked decrease of activity. For instance, with a substrate concentration of 150 μM, the value of kobs decreased by ∼25% when compared to its value at atmospheric pressure. As shown in Fig. 2 B, this was coming with an increase of KM from 11 to 25 μM. After pressure release, the enzyme was still soluble (with 85% in the tetrameric form). Thus, we propose that the structure solved at 150 MPa reflects the pressure-perturbed UOX state described previously. As previously mentioned (Table 1 and Fig. 2 B), the pressure-induced loss of activity, which occurred before oligomeric dissociation, is related to less efficient substrate binding and subtle modifications of the active site. As previously described for citrine (45), small structural perturbations in the region of the active site may cause a partial loss of activity. The region delimiting the entrance to the active site displays the largest increases in B-values under pressure, revealing a particular flexibility (35,46). This flexibility could modulate substrate access to the active site, leading to partial inactivation of the enzyme in its pressure-perturbed states. The pressure would simply close the entrance door somehow, leading to an increase of KM. Moreover, the active site swells under pressure, with increased distances between atoms belonging to different subunits and decreased distances between atoms within the same subunit. The hydrophobic cavity contracts concomitantly. Therefore, we suggest that the pressure-induced loss of affinity for the substrate could also be related to the contraction of the hydrophobic cavity, which restrains active-site flexibility and thereby perturbs substrate docking.
It is of interest that the conformation of the enzyme at 150 MPa, with a larger active site than at atmospheric pressure, probably corresponds to an enzyme substate where the active site can accommodate a larger ligand, which is necessary for the enzymatic mechanism. UOX catalyzes the oxidation of uric acid in the presence of molecular oxygen to a primary intermediate, 5-hydroxyisourate, which is nonplanar. During the reaction, swelling is thus necessary to accommodate the product. Pressure changes the Gibbs free energy of the compressed system, and promotes higher-energy substates of decreased partial specific volumes, based on Le Chatelier's rule (1,5,7–9). Accordingly, the UOX 150-MPa structure would represent a conformational substate of the enzyme related to its function, demonstrating that pressure has permitted the trapping of a UOX conformation state that should exist during the catalytic mechanism of the enzyme.
It has been proposed that cavities in the vicinity of active sites play a role in protein flexibility, a mechanism related to functional efficiency (47). At atmospheric pressure, the active-site pocket flexibility would then be balanced by the neighboring hydrophobic cavity during the binding of the substrate. This hypothesis could be verified by activity measurements in two ways, either on a protein under inert gas pressure, since it has been shown that this cavity accommodates xenon or nitrous oxide (48), or on a protein engineered with mutations that modify the cavity volume and shape. The UOX 150 MPa x-ray structure analysis suggests that the necessary enlargement of the active site during the catalytic mechanism is favored by a contraction of the neighboring hydrophobic cavity.
Conclusions
The use of complementary methods gives a detailed picture of the effects of high pressure on the tetrameric enzyme UOX. The 150-MPa crystal structure reveals complex modifications related to substates, which may be close to those actually involved in the mechanism of the enzyme. This high-pressure study highlights the role played by a large neighboring hydrophobic cavity on the flexibility of the active site during catalysis. Accordingly, this work confirms that high-pressure perturbation in molecular biophysics potentially enables the trapping of protein conformation states of biological significance.
Supporting Material
Methods and Materials, three figures, and one table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00223-7.
Supporting Material
Acknowledgments
The authors thank Bertrand Castro and Mohamed El Hajji (Sanofi-Aventis, Montpellier, France) for kindly supplying urate oxidase, Jean-Claude Chervin and co-workers (Institut de Minéralogie et de Physique des Milieux Condensés, Paris, France) for the HPMX high-aperture DAC, Mikaël Naveau (Centre d'Imagerie - Neurosciences et d'Applications aux Pathologies, Caen, France) for programming assistance, and Gavin Fox (Institut de Biologie Structurale, Grenoble, France) for English corrections.
G.M. was supported by a grant from the Ministère de l'Education et de la Recherche, M.G. by a grant from Sanofi-Aventis, and F.D. by a grant from the Conseil Régional de Basse-Normandie.
Footnotes
Stéphanie Finet's present address is IMPMC, UMR 7590 CNRS UPMC, 4 place Jussieu, 75252 Paris cedex 05, France.
Marion Giffard's present address is Novartis Pharma AG, CH-4056 Basel, Switzerland.
Fabienne Dulin's present address is CERMN, UCBN, bd Becquerel, 14032 Caen, France.
Contributor Information
Eric Girard, Email: eric.girard@ibs.fr.
Nathalie Colloc'h, Email: colloch@cyceron.fr.
References
- 1.Frauenfelder H., Alberding N.A., Uyue K.T. Proteins and pressure. J. Phys. Chem. 1990;94:1024–1037. [Google Scholar]
- 2.Gross M., Jaenicke R. Proteins under pressure. The influence of high hydrostatic pressure on structure, function and assembly of proteins and protein complexes. Eur. J. Biochem. 1994;221:617–630. doi: 10.1111/j.1432-1033.1994.tb18774.x. [DOI] [PubMed] [Google Scholar]
- 3.Mozhaev V.V., Heremans K., Balny C. High pressure effects on protein structure and function. Proteins. 1996;24:81–91. doi: 10.1002/(SICI)1097-0134(199601)24:1<81::AID-PROT6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 4.Boonyaratanakornkit B.B., Park C.B., Clark D.S. Pressure effects on intra- and intermolecular interactions within proteins. Biochim. Biophys. Acta. 2002;1595:235–249. doi: 10.1016/s0167-4838(01)00347-8. [DOI] [PubMed] [Google Scholar]
- 5.Urayama P., Phillips G.N., Gruner S.M. Probing substates in sperm whale myoglobin using high-pressure crystallography. Structure. 2002;10:51–60. doi: 10.1016/s0969-2126(01)00699-2. [DOI] [PubMed] [Google Scholar]
- 6.Kornblatt M.J., Lange R., Balny C. Use of hydrostatic pressure to produce “native” monomers of yeast enolase. Eur. J. Biochem. 2004;271:3897–3904. doi: 10.1111/j.1432-1033.2004.04326.x. [DOI] [PubMed] [Google Scholar]
- 7.Marchal S., Torrent J., Balny C. The powerful high pressure tool for protein conformational studies. Braz. J. Med. Biol. Res. 2005;38:1175–1183. doi: 10.1590/s0100-879x2005000800004. [DOI] [PubMed] [Google Scholar]
- 8.Akasaka K. Probing conformational fluctuation of proteins by pressure perturbation. Chem. Rev. 2006;106:1814–1835. doi: 10.1021/cr040440z. [DOI] [PubMed] [Google Scholar]
- 9.Fourme R., Girard E., Ascone I. High-pressure macromolecular crystallography (HPMX): status and prospects. Biochim. Biophys. Acta. 2006;1764:384–390. doi: 10.1016/j.bbapap.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Weber G. Springer; New York: 1992. Protein Interactions. [Google Scholar]
- 11.Weber G. Thermodynamics of the association and the pressure dissociation of oligomeric proteins. J. Phys. Chem. 1992;97:7108–7115. [Google Scholar]
- 12.Skouri-Panet F., Quevillon-Cheruel S., Finet S. sHSPs under temperature and pressure: the opposite behaviour of lens α-crystallins and yeast HSP26. Biochim. Biophys. Acta. 2006;1764:372–383. doi: 10.1016/j.bbapap.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Bayol A., Capdevielle J., Ferrara P. Modification of a reactive cysteine explains differences between rasburicase and Uricozyme, a natural Aspergillus flavus uricase. Biotechnol. Appl. Biochem. 2002;36:21–31. doi: 10.1042/ba20010083. [DOI] [PubMed] [Google Scholar]
- 14.Imhoff R.D., Power N.P., Tipton P.A. General base catalysis in the urate oxidase reaction: evidence for a novel Thr-Lys catalytic diad. Biochemistry. 2003;42:4094–4100. doi: 10.1021/bi027377x. [DOI] [PubMed] [Google Scholar]
- 15.Colloc'h N., Gabison L., Prangé T. Oxygen pressurized x-ray crystallography: probing the dioxygen binding site in cofactorless urate oxidase and implications for its catalytic mechanism. Biophys. J. 2008;95:2415–2422. doi: 10.1529/biophysj.107.122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabison L., Prangé T., Chiadmi M. Structural analysis of urate oxidase in complex with its natural substrate inhibited by cyanide: mechanistic implications. BMC Struct. Biol. 2008;8:32–39. doi: 10.1186/1472-6807-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juan E.C.M., Hoque M.M., Takenaka A. Structures of Arthrobacter globiformis urate oxidase-ligand complexes. Acta Crystallogr. 2008;D64:815–822. doi: 10.1107/S0907444908013590. [DOI] [PubMed] [Google Scholar]
- 18.Colloc'h N., el Hajji M., Mornon J.P. Crystal structure of the protein drug urate oxidase-inhibitor complex at 2.05 Å resolution. Nat. Struct. Biol. 1997;4:947–952. doi: 10.1038/nsb1197-947. [DOI] [PubMed] [Google Scholar]
- 19.Retailleau P., Colloc'h N., Prangé T. Complexed and ligand-free high-resolution structures of urate oxidase (UOX) from Aspergillus flavus: a reassignment of the active-site binding mode. Acta Crystallogr. 2004;D60:453–462. doi: 10.1107/S0907444903029718. [DOI] [PubMed] [Google Scholar]
- 20.Colloc'h N., Poupon A., Mornon J.P. Sequence and structural features of the T-fold, an original tunnelling building unit. Proteins. 2000;39:142–154. doi: 10.1002/(sici)1097-0134(20000501)39:2<142::aid-prot4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 21.Colloc'h N., Girard E., Fourme R. High pressure macromolecular crystallography: the 140-MPa crystal structure at 2.3 Å resolution of urate oxidase, a 135-kDa tetrameric assembly. Biochim. Biophys. Acta. 2006;1764:391–397. doi: 10.1016/j.bbapap.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Silva J.L., Miles E.W., Weber G. Pressure dissociation and conformational drift of the beta dimer of tryptophan synthase. Biochemistry. 1986;25:5780–5786. doi: 10.1021/bi00367a065. [DOI] [PubMed] [Google Scholar]
- 23.Ruan K., Weber G. Hysteresis and conformational drift of pressure-dissociated glyceraldehydephosphate dehydrogenase. Biochemistry. 1989;28:2144–2153. doi: 10.1021/bi00431a028. [DOI] [PubMed] [Google Scholar]
- 24.Vidugiris G.J., Royer C.A. Determination of the volume changes for pressure-induced transitions of apomyoglobin between the native, molten globule, and unfolded states. Biophys. J. 1998;75:463–470. doi: 10.1016/S0006-3495(98)77534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahn K., Tipton P.A. Kinetic mechanism and cofactor content of soybean root nodule urate oxidase. Biochemistry. 1997;36:4731–4738. doi: 10.1021/bi963184w. [DOI] [PubMed] [Google Scholar]
- 26.Kahn K., Tipton P.A. Spectroscopic characterization of intermediates in the urate oxidase reaction. Biochemistry. 1998;37:11651–11659. doi: 10.1021/bi980446g. [DOI] [PubMed] [Google Scholar]
- 27.Morild E. The theory of pressure effects on enzymes. Adv. Protein Chem. 1981;34:93–166. doi: 10.1016/s0065-3233(08)60519-7. [DOI] [PubMed] [Google Scholar]
- 28.Svergun D.I., Barberato C., Koch M.H.J. Crysol: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 29.Fourme R., Kahn R., Ascone I. High-pressure protein crystallography (HPPX): instrumentation, methodology and results on lysozyme crystals. J. Synchrotron Radiat. 2001;8:1149–1156. doi: 10.1107/s0909049501011037. [DOI] [PubMed] [Google Scholar]
- 30.Girard E., Dhaussy A.C., Fourme R. Toward fully fledged high-pressure macromolecular crystallography. J. Appl. Cryst. 2007;40:912–918. [Google Scholar]
- 31.Dupuis F., Sadoc J.F., Mornon J.P. Voro3D: 3D Voronoi tessellations applied to protein structures. Bioinformatics. 2005;21:1715–1716. doi: 10.1093/bioinformatics/bth365. [DOI] [PubMed] [Google Scholar]
- 32.Case D.A., Cheatham T.E., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binkowski T.A., Naghibzadeh S., Liang J. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunger A.T., Adams P.D., Warren G.L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Kundrot C.E., Richards F.M. Crystal structure of hen egg-white lysozyme at a hydrostatic pressure of 1000 atmospheres. J. Mol. Biol. 1987;193:157–170. doi: 10.1016/0022-2836(87)90634-6. [DOI] [PubMed] [Google Scholar]
- 36.Akasaka K., Li H., Woodward C.K. Pressure response of protein backbone structure. Pressure-induced amide 15N chemical shifts in BPTI. Protein Sci. 1999;8:1946–1953. doi: 10.1110/ps.8.10.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canalia M., Malliavin T.E., Kalbitzer H.R. Molecular dynamics simulations of HPr under hydrostatic pressure. Biopolymers. 2004;74:377–388. doi: 10.1002/bip.20089. [DOI] [PubMed] [Google Scholar]
- 38.Suarez M.C., Rocha C.B., Foguel D. Free-energy linkage between folding and calcium binding in EF-hand proteins. Biophys. J. 2008;95:4820–4828. doi: 10.1529/biophysj.108.135715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins M.D., Hummer G., Gruner S.M. Cooperative water filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation. Proc. Natl. Acad. Sci. USA. 2005;102:16668–16671. doi: 10.1073/pnas.0508224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ando N., Barstow B., Gruner S.M. Structural and thermodynamic characterization of T4 lysozyme mutants and the contribution of internal cavities to pressure denaturation. Biochemistry. 2008;47:11097–11109. doi: 10.1021/bi801287m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Royer C.A. Revisiting volume changes in pressure-induced protein unfolding. Biochim. Biophys. Acta. 2002;1595:201–209. doi: 10.1016/s0167-4838(01)00344-2. [DOI] [PubMed] [Google Scholar]
- 42.Royer C.A. Insights into the role of hydration in protein structure and stability obtained through hydrostatic pressure studies. Braz. J. Med. Biol. Res. 2005;38:1167–1173. doi: 10.1590/s0100-879x2005000800003. [DOI] [PubMed] [Google Scholar]
- 43.Girard E., Kahn R., Fourme R. The first crystal structure of a macromolecular assembly under high pressure: CpMV at 330 MPa. Biophys. J. 2005;88:3562–3571. doi: 10.1529/biophysj.104.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girard E., Prangé T., Fourme R. Adaptation of the base-paired double-helix molecular architecture to extreme pressure. Nucleic Acids Res. 2007;35:4800–4808. doi: 10.1093/nar/gkm511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barstow B., Ando N., Gruner S.M. Alteration of citrine structure by hydrostatic pressure explains the accompanying spectral shift. Proc. Natl. Acad. Sci. USA. 2008;105:13362–13366. doi: 10.1073/pnas.0802252105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins M.D., Quillin M.L., Gruner S.M. Structural rigidity of a large cavity-containing protein revealed by high-pressure crystallography. J. Mol. Biol. 2007;367:752–763. doi: 10.1016/j.jmb.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hubbard S.J., Argos P. A functional role for protein cavities in domain: domain motions. J. Mol. Biol. 1996;261:289–300. doi: 10.1006/jmbi.1996.0460. [DOI] [PubMed] [Google Scholar]
- 48.Colloc'h N., Sopkova-de Oliveira Santos J., Abraini J.H. Protein crystallography under xenon and nitrous oxide pressure: comparison with in vivo pharmacology studies and implications for the mechanism of inhaled anesthetic action. Biophys. J. 2007;92:217–224. doi: 10.1529/biophysj.106.093807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.