

Abstract
A number of recent studies have shown that iron dissolution in Fe-containing dust aerosol can be linked to source material (mineral or anthropogenic), mineralogy, and iron speciation. All of these factors need to be incorporated into atmospheric chemistry models if these models are to accurately predict the impact of Fe-containing dusts into open ocean waters. In this report, we combine dissolution measurements along with spectroscopy and microscopy to focus on nanoscale size effects in the dissolution of Fe-containing minerals in low-pH environments and the importance of acid type, including HNO3, H2SO4, and HCl, on dissolution. All of these acids are present in the atmosphere, and dust particles have been shown to be associated with nitrate, sulfate, and/or chloride. These measurements are done under light and dark conditions so as to simulate and distinguish between daytime and nighttime atmospheric chemical processing. Both size (nano- versus micron-sized particles) and anion (nitrate, sulfate, and chloride) are found to play significant roles in the dissolution of α-FeOOH under both light and dark conditions. The current study highlights these important, yet unconsidered, factors in the atmospheric processing of iron-containing mineral dust aerosol.
Keywords: mineral dust aerosol, iron-containing dust, nanoscale iron oxides, acid dissolution, photoinduced dissolution
Iron is an essential element for all biological organisms including those in marine environments. It has been suggested that 30% of the oceans are comprised with high-nutrient low-chlorophyll regions where phytoplankton primary productivity is limited by the amount of bioavailable iron (1–3). Iron is the fourth most abundant element on the Earth’s crust and is mostly found in its thermodynamic stable oxidation state, Fe(III). Fe(III) is relatively insoluble in oxic pH 8 seawater, thus limiting the inorganic concentration to 0.1 nM; any inorganic iron above this concentration will form an iron oxide solid phase (4). Despite this limitation, ocean total iron concentrations range from 0.1–2 nM. Therefore, iron addition to surface waters from dissolved iron in Fe-containing dust deposition are thought to elevate the total amount of dissolved, and bioavailable, iron (5).
Mineral dust aerosol , mainly desert dust and dust from volcanic eruptions, is a source of iron and has been previously thought to account for approximately 95% of the globally averaged atmospheric iron budget (6, 7). Approximately 450 Tg of mineral dust is annually deposited into ocean waters (3, 8). Among many different iron-containing solid phases found in mineral dust, hematite (α-Fe2O3) has been thought to be the most important form for soluble iron and biological utilization (9), and it is currently the only form of iron minerals incorporated into atmospheric chemistry models. However, a number of very recent studies indicate that this approach is far too simplistic and does not accurately reflect both the complexities of atmospheric sources of iron and/or the most important iron-mineral phases involved in dissolution. For example, Journet et al. showed that Fe-containing clays accounted for more than 90% of soluble iron (10). Cwiertny et al. proposed that iron in the form of Fe2+ in clay minerals is a highly labile source of iron (11). Other studies highlight the contribution from anthropogenic sources of soluble iron, mainly from combustion sources (3, 12, 13). These combustion sources are concentrated in both industrialized and biomass burning regions and can represent up to 5–30% of the total iron deposited into ocean regions (3).
Since heterogeneous chemistry and atmospheric processing can impact the amount of soluble iron in Fe-containing dusts, the atmospheric lifetime of iron dust particles is another important factor. Atmospheric lifetimes of dust particles can vary depending on particle size from days for particles greater than 2 μm to weeks for the smaller particles (3), and, as a result, the size distribution of the aerosol changes as it is transported through the atmosphere (14). In addition, it has been recently suggested that cloud processing can cause the formation of nanosize Fe particles (15). Thus, size effects and small particles below 100 nm can play an important role in the atmospheric chemistry of iron-containing dust particles.
In this report, we investigate the dissolution of Fe-containing minerals under simulated atmospheric conditions. Given that iron oxyhydroxides are a major fraction of Fe-containing solid phases in mineral dust, goethite, α-FeOOH, can be used as a model to begin to understand various aspects of size effects in iron dissolution. The current study investigates the size-dependent solubility of α-FeOOH in aqueous suspensions at pH 2 to mimic a deliquescent layer and the low-pH environment of an aerosol due to the uptake of acidic gases in the atmosphere (16). The effects of acid type (HCl, HNO3, and H2SO4) and associated inorganic anion (Cl-,  , and
, and 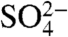 ) on iron dissolution are investigated because dust particles have been found to be associated with sulfate, nitrate, and chloride in the atmosphere. Experiments are done under both light and dark so as to simulate and differentiate between daytime and nighttime atmospheric processing. Dissolution measurements of well-characterized samples are combined with spectroscopy and microscopy to reveal some important, yet unconsidered, processes that can occur in the atmosphere for iron-containing dusts and, as such, some of the complexities of iron-mobilization in the global environment.
) on iron dissolution are investigated because dust particles have been found to be associated with sulfate, nitrate, and chloride in the atmosphere. Experiments are done under both light and dark so as to simulate and differentiate between daytime and nighttime atmospheric processing. Dissolution measurements of well-characterized samples are combined with spectroscopy and microscopy to reveal some important, yet unconsidered, processes that can occur in the atmosphere for iron-containing dusts and, as such, some of the complexities of iron-mobilization in the global environment.
Results and Discussion
Role of Particle Size in the Dissolution of α-FeOOH at pH 2 Under Light and Dark Conditions.
In natural systems, nanoscale-sized iron oxide and iron oxyhydroxides particles are of great interest because they are produced in the environment and may exhibit dissolution properties that differ from larger-sized particles (17). Here, the dissolution of α-FeOOH was investigated in terms of total iron dissolution and speciation [i.e., Fe(II) (aq) and Fe(III) (aq)] in low-pH environments at pH 2. Goethite forms rod-shaped particles, and, to investigate size effects, different sized rods were synthesized. Synthesized nanorods were 75( ± 20) nm by 6( ± 2) nm with N2-adsorption measured surface areas of 119( ± 3) m2 g-1, whereas larger microrods were 1006( ± 55) nm by 25( ± 6) nm and with a lower surface area of 39( ± 2) m2 g-1. Dissolution of these different sized rods was investigated in low-pH environments to examine size-dependent behavior in simulated nighttime and daytime conditions. For nighttime conditions, experiments were done in the dark, whereas simulation of daytime mineral dust chemistry was accomplished through the use of a solar simulator to investigate the effect of light on iron dissolution of nanorods as compared to larger microrods.
The data presented in Fig. 1 show a comparison of total iron dissolution of nanorods versus microrods at pH 2 under dark and light conditions. These data have been normalized to both the total mass and surface area of the initial α-FeOOH in solution. The rate of iron dissolution and the production of dissolved ions are greatest initially (t < 10 hr), followed by a slower rate of change over time. In both mass normalized and surface area normalized data, and under both dark and light conditions, it can be clearly seen that nanorods dissolve faster and to a greater extent relative to microrods. Under dark conditions, the initial rate of total iron dissolution for nanorods, determined from linear regression, is 55.4( ± 4) μmol g-1 h-1, whereas for microrods this is 6.1( ± 0.4) μmol g-1 h-1. No Fe(II) is detected in these dark experiments, so the total iron is in the form of Fe(III). After normalizing to surface area differences (Fig. 1B), there is still an enhancement in the dissolution of nanorods compared to microrods by nearly a factor of three. Fig. 1 C and D compare results for total iron dissolution of nanorods versus microrods at pH 2 in irradiated suspensions on a per mass basis and per surface area basis, respectively. The initial rate of total iron dissolution under light conditions for nanorods is 119( ± 4) μmol g-1 h-1, whereas for microrods this is 15.4( ± 2.7) μmol g-1 h-1.
Fig. 1.

Proton-promoted dissolution of rod-shaped α-FeOOH particles is monitored by the formation of total soluble Fe [Fe(III) only for dark conditions and Fe(III) and Fe(II) for light conditions] in solution at pH 2. The solutions were acidified with HNO3. These plots compare the dissolution of nanorods to microrods under dark and light conditions on a per mass (A, C, and E) and per surface area (B, D, and F) basis.
The production of dissolved Fe(II) is shown in Fig. 1 E and F on a per mass and per surface area basis, respectively, under these same conditions. As seen in Fig. 1 E and F, the Fe(II) production is greater in irradiated nanorod suspensions compared to that of microrods. Additionally, there are differences in the ratio of Fe(II) to total Fe dissolution between nanorods and microrods. The initial (t < 10 hr) Fe(II) fraction with respect to total dissolved Fe is approximately 0.50 for nanorods, but for microrods the Fe(II) fraction is only approximately 0.25. Over time, the rate changes and levels off with an Fe(II) fraction of approximately 0.7 for nanorods and approximately 0.5 for microrods.
Mechanisms for Enhanced Dissolution of Nanoscale α-FeOOH at pH 2 Under Dark and Light Conditions.
In earlier studies, it has been shown that surface area normalized rates for low-pH, proton-promoted dissolution of α-FeOOH increased with decreasing particle size (18, 19) and that higher rates of iron dissolution in smaller particles partially come from their largest fraction of reactive surface planes. According to Cornell et al. (18, 19), the nonreductive iron dissolution involves
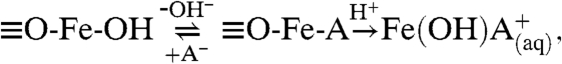 |
[1] |
where A- is the inorganic anion. In this proposed mechanism, acid anion primarily reacts with the surface O-H groups followed by a proton attack yielding surface protonation. This polarizes and weakens the Fe-O bond followed by bond dissociation enabling the detachment of the Fe cation that enters into the solution (10, 20). In goethite rod-shaped particles, the end of the rods that expose (021) surface planes are more reactive as a result of coordination sites with higher concentration of surface O-H groups. Thus, the presence of more reactive sites in crystal faces, i.e., (021), with highly concentrated surface O-H groups results in enhanced reactivity. Nanorods have a greater number of exposed (021) surface planes on a per mass and per surface area basis.
Similarly, in light experiments, the observed trends in the production of Fe(II) lend insights into the enhancement in the photochemical dissolution of nanoscale α-FeOOH. For these semiconductor iron oxyhydroxide particles, photoexcitation leads to the formation of electron-hole (e-/h+) pairs. In the absence of surface complexing organic ligands, the photoredox reaction proceeds through surface hydroxyl groups donating an electron to a photoexcited Fe(III) surface atom, resulting in surface-bound Fe(II) and subsequent detachment of Fe(II) into solution (21). Therefore, in irradiated systems, the enhanced Fe(II) production in nanorods can also be a result of the (021) surface planes with higher density of surface O-H groups. Recent field and laboratory studies have shown that, in marine surface waters and in acidic surface waters, light-induced reduction of Fe(III) to Fe(II) is a key process and serves to increase the solubility of marine aerosol Fe (22, 23). As such, any processes that enhance production of soluble Fe(II) are important and increase total iron dissolution. Furthermore, at longer time scales, the Fe(II) production in nanorods is considerably greater than that observed for microrods. For example, after 40 hr of light exposure, the concentration of Fe(II) for nanorods is nearly seven times larger than that of microrods. This difference is again reflected in the total iron dissolution for nanorods compared to microrods. Thus, the results of these studies clearly address size-dependent reactivity under both dark and light conditions, and dissolution of iron-containing minerals on the nanoscale. These results point specifically to the presence of a greater abundance of specific reactive surface planes that can lead to enhancements in reactivity not only on a per mass basis but on a per surface area basis, showing that these nanoscale-sized particles are inherently more reactive under both light and dark conditions.
Interestingly, changes in nanorod morphology were observed upon exposure to low-pH environment. The transmission electron microscope (TEM) images of nanorods before and after the reaction with 0.01 N HNO3 (pH 2.0) in light are shown in Fig. 2 A and B, respectively. Nanorods prior to exposure show sharp, well-defined ends characteristic of their (021) surface planes. In contrast, after completion of 46 hr in the acidic environment, particles show changes in morphology; over time, nanorods became cigar-shaped, developing narrow, rounded ends. Furthermore, analysis of the particle size distribution shows an anisotropic change in the distribution. In particular, a frequency analysis of the length and width of many particles (N = 200) before and at the end of 46 hr of irradiation in acidic solution from additional TEM images besides those presented in Fig. 2 show that the length of nanorods has significantly decreased (approximately 15% on average), whereas the distribution change observed in the width of the nanorods remains the same. The change in the length distribution for nanorods is shown in Fig. 2C. Given, higher fraction of particles with smaller lengths, relative to nanorods before the reaction, clearly indicates that the proton-promoted dissolution takes place on exposed surface planes at the ends of the nanorods. The observed morphological changes support the argument that rates of dissolution are greatest on the (021) faces of α-FeOOH.
Fig. 2.

TEM images of nanorods (A) prior to reaction with HNO3 and (B) after the reaction with 0.01 M HNO3 (pH 2) in the light. The arrows highlight the changes that occur on the ends, (021) faces, of the nanorods as a result of iron dissolution. Analysis of many particles (N = 200) was used to construct distributions for the length and width of nanorods before and after reaction with 0.01 M HNO3 (pH 2) in the light. Only the length distributions showed any change. These are plotted in C for unreacted nanorods (blue) and red reacted nanorods (red).
Role of Acid Type on Iron Dissolution.
Another potentially important factor that has not been previously considered in the dissolution of pure iron oxide phases in the atmosphere is the impact of acid type. Under acidic conditions, the deliquescence layer on particle surfaces often contain high concentration of anions, i.e., sulfate, nitrate, and chloride (24, 25). Therefore, not all dissolved Fe(III) is expected to be present as free ions but in fact complexed and coordinated to anions in the deliquescent layer. Some of these complexes, as well as the inorganic anions themselves, can be photoactive (26, 27), which may play a role in the overall dissolution process for Fe mobilization in sunlight. Furthermore, these anions can coordinate to the surface in different adsorption modes and with different bond strengths to the surface; this too will impact iron dissolution. Here, the role of different atmospherically relevant acids on the generation of soluble iron and iron speciation in nanorod suspensions were investigated with three acids, HNO3, H2SO4, and HCl, at pH 2 under simulated nighttime and daytime atmospheric processing.
Data for total iron dissolution from the three acids studied in dark condition are shown in Fig. 3A. It can be clearly seen that  yields the highest total dissolved Fe, whereas the lowest is in the presence of Cl- and in the case of
yields the highest total dissolved Fe, whereas the lowest is in the presence of Cl- and in the case of 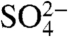 , the total Fe dissolution is just slightly less than that of
, the total Fe dissolution is just slightly less than that of  . In the dark, there is no detectable Fe(II) production. Comparison of the total iron dissolution for the three different acids in the presence of light is shown in Fig. 3B. As shown for HNO3 in Fig. 1, iron dissolution is enhanced in the presence of light for both H2SO4 and HCl. According to Fig. 3B, this behavior is most pronounced for HCl, which had initial rates of total dissolved Fe that are nearly four times greater in the presence of light compared to suspensions acidified with HNO3 and H2SO4 where light induced a twofold enhancement. Fig. 3C compares the photochemical production of dissolved Fe(II) with
. In the dark, there is no detectable Fe(II) production. Comparison of the total iron dissolution for the three different acids in the presence of light is shown in Fig. 3B. As shown for HNO3 in Fig. 1, iron dissolution is enhanced in the presence of light for both H2SO4 and HCl. According to Fig. 3B, this behavior is most pronounced for HCl, which had initial rates of total dissolved Fe that are nearly four times greater in the presence of light compared to suspensions acidified with HNO3 and H2SO4 where light induced a twofold enhancement. Fig. 3C compares the photochemical production of dissolved Fe(II) with  ,
, 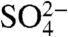 , and Cl-. As seen in Fig. 3C, dissolved Fe(II) concentrations with HNO3 and HCl are found to be similar and significantly higher than that of H2SO4. For suspensions that were acidified with HCl, the Fe(II) to total iron ratio is one, indicating that all of the dissolved iron present in the suspension is present as Fe(II). For the other two acids, Fe(II) fraction [Fe(II)/Total Fe] is much less: For
, and Cl-. As seen in Fig. 3C, dissolved Fe(II) concentrations with HNO3 and HCl are found to be similar and significantly higher than that of H2SO4. For suspensions that were acidified with HCl, the Fe(II) to total iron ratio is one, indicating that all of the dissolved iron present in the suspension is present as Fe(II). For the other two acids, Fe(II) fraction [Fe(II)/Total Fe] is much less: For 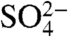 it is approximately 0.3, and for
it is approximately 0.3, and for  this fraction is approximately 0.6. These data show that the production of Fe(II) is strongly dependent on anion type and that sulfate yields the lowest amount of Fe(II).
this fraction is approximately 0.6. These data show that the production of Fe(II) is strongly dependent on anion type and that sulfate yields the lowest amount of Fe(II).
Fig. 3.

Dissolution of α-FeOOH nanorod particles is monitored by the formation of total soluble Fe and Fe(II) in solution at pH 2.0. These plots compare the dissolution of nanorods in three different acids, HNO3, H2SO4, and HCl under dark and light conditions. (A) Total iron [Fe(III) only] in the dark. (B). Total iron dissolution (Fe(III) and Fe(II)) in light. (C) Fe(II) production in the presence of light.
Mechanisms for Dissolution of α-FeOOH Under Dark and Light Conditions with Different Acids.
In the absence of light, total iron dissolution for the different anions can be related to the strength and mode of anion adsorption, and the ability of the complex to be further protonated, as shown in Eq. 1. Sulfate and nitrate ions are polyatomic oxyanions that can bond to the surface in different adsorption modes, including a bidentate and bridging coordination mode which chloride is unable to form. Bridging and bidentate coordination can potentially enhance iron dissolution. As such, it is the ability of anions to form certain surface complexes that can explain the differences in total iron dissolution in the dark.
The effect of inorganic anion on dissolved Fe(II) concentrations in the presence of light can be explained in part by considering the photochemistry of aqueous iron complexes at low pH. In the absence of other anions, [Fe(OH2)6]3+ is the dominant species at pH less than 2.5, whereas the concentration of [Fe(OH2)5(OH)]2+ is the most important chromophore at visible wavelength with a maximum concentration at approximately pH 3.5 (9). The photoreduction of [Fe(OH2)5(OH)]2+ is given by
| [2] |
In Cl--containing suspensions, [Fe(OH2)5Cl]2+ and [Fe(OH2)4Cl2]+ coexist at low pHs where the former is the dominant species at approximately pH 1, whereas the [Fe(OH2)5(OH)]2+ is only present at a very low level (27–29). Both chloride-containing iron species are photoactive in UV and visible regions of the solar spectrum. As reported by Hsu et al., [Fe(OH2)5Cl]2+ is photoreduced to [Fe(OH2)6]2+ according to
| [3] |
The quantum yield of the photodissociation of [Fe(OH2)5Cl]2+ is approximately twice the value of [Fe(OH2)5(OH)]2+ (29), and photoreduction of [Fe(OH2)6]3+ is not a principal source of OH• because its absorption spectrum does not significantly overlap the solar spectrum (28). Based on above facts and the data presented, it can be suggested that the formation of Fe(III)-Cl complexes would result in higher rate of Fe(II) production in suspensions with the chloride ion. Furthermore, formation of Fe-Cl complexes can result a decrease in the redox potential of Fe(III)/Fe(II) couple and thus enhance the production of Fe(II) in these systems.
In the case of HNO3,  is a weak ligand and does not readily form solution-phase iron complexes leaving [Fe(OH2)6]3+ and [Fe(OH2)5(OH)]2+ as dominant species. Photolysis of [Fe(OH2)5(OH)]2+ efficiently yields OH• radicals, a strong oxidizer, that reoxidizes Fe(II) back to Fe(III). In addition,
is a weak ligand and does not readily form solution-phase iron complexes leaving [Fe(OH2)6]3+ and [Fe(OH2)5(OH)]2+ as dominant species. Photolysis of [Fe(OH2)5(OH)]2+ efficiently yields OH• radicals, a strong oxidizer, that reoxidizes Fe(II) back to Fe(III). In addition,  in acidic waters is a well-known chromophore which absorbs light approximately 305 nm to yield NO2 and
in acidic waters is a well-known chromophore which absorbs light approximately 305 nm to yield NO2 and  and OH• (30) via
and OH• (30) via
| [4] |
| [5] |
On the basis of the above information, it can be suggested that the higher OH• concentration originated from the photolysis of both  and [Fe(OH2)5(OH)]2+ results in a faster transformation of [Fe(OH2)6]2+ back to [Fe(OH2)6]3+; thus, a lower Fe(II) fraction compared to Cl- suspensions.
and [Fe(OH2)5(OH)]2+ results in a faster transformation of [Fe(OH2)6]2+ back to [Fe(OH2)6]3+; thus, a lower Fe(II) fraction compared to Cl- suspensions.
The effect of sulfate on proton-promoted iron dissolution of α-FeOOH is different from the other anions. According to Fig. 3 B and C, the total iron dissolution in H2SO4 suspensions is enhanced by a factor of approximately 2, equivalent to HNO3 suspensions, whereas the fraction of Fe(II) is only approximately 0.3, half of that observed with HNO3. Therefore, approximately 70% of the dissolved iron acidified with H2SO4 exists in the form of soluble Fe(III). As Fe(III) is less soluble compared to Fe(II), it can be proposed that the dissolve Fe(III) fraction is coordinate with sulfate forming solution-phase Fe(III)-sulfato complexes with different photochemical behavior leading to less Fe(II) formation.
Anion Adsorption on α-FeOOH Nanorod and Microrod Surfaces.
To gain additional molecular level insights into the behavior of α-FeOOH nanorods and microrods in pH 2 solutions, attenuated total reflection–Fourier transform infrared (ATR-FTIR) spectroscopy was used to investigate surface adsorption of nanorods and microrods in the presence of the different acids used in this study. Adsorption of both HCl and HNO3 from solution phase on to the nanorods gave little spectral information in the 1,000 to 4,000 cm-1 spectral range, as expected based on the frequencies of the surface complexes, for chloride frequencies below 1,000 cm-1, and the strength of the interaction with the surface; nitrate forms surface complexes with weak absorption band intensities and frequencies similar to that of nitrates in solution. However, for sulfate, ATR-FTIR spectroscopy revealed distinct absorption bands with clear differences observed between sulfate adsorbed on nanorod compared to microrod surfaces.
Fig. 4 shows several ATR-FTIR spectra for sulfate adsorption on both nanorods and microrods at pH 2. For comparison, a solution-phase spectrum of sulfate anion in the absence of goethite is also shown. As a free anion, sulfate has tetrahedral symmetry and belongs to the Td point group. For this symmetry, only one peak at approximately 1,100 cm-1 is expected for the triply degenerate v3 asymmetric stretch. However, in aqueous solutions several species can exist and the symmetry is reduced, leading to additional absorption bands in this spectral region. For sulfate adsorbed on the α-FeOOH surface, it can be seen that there is an even greater number of absorption bands present in the spectra due to a further reduction in symmetry upon surface adsorption and the presence of different surface complexes. Further inspection of these spectra shown in Fig. 4 shows distinct differences between the ATR-FTIR spectra for sulfate adsorbed on nanorods compared to microrods. For example, the absorption band at 1,240 cm-1 clearly observed for nanorods is barely present in microrod spectra.
Fig. 4.

ATR-FTIR spectra of sulfate adsorption at pH 2. Spectra are shown for six successive additions of 0.01 N H2SO4 in 15 min intervals. The solution-phase spectra, collected in the absence of α-FeOOH, are shown in A. The spectra for adsorbed sulfate on nanorods and microrods are shown in B and C, respectively. Solution-phase absorptions have been subtracted from the spectra shown in B and C.
When sulfate adsorbs on the surface as an inner-sphere surface complex, the symmetry is lowered. In the case of a monodentate inner-sphere complex with C3v symmetry, the ν3 band of free sulfate anion splits into two, and the symmetric stretch, ν1, becomes infrared active with a band at approximately 975 cm-1 (31). Therefore, the observed spectral bands at approximately 1,140; 1,042; and 975 cm-1, shown in Fig. 4B, suggest formation of inner-sphere monodentate complexes of sulfate, as the major adsorbed species, with surfaces of goethite microrods. As seen in Fig. 4C, four distinct peaks can be found in the spectra collected for nanorods. Based on previous studies, sulfate can also form a bidentate binuclear (bridging) surface complex; the symmetry is further lowered to C2v, splitting the ν3 band into three bands between 1,050 and 1,250 cm-1 in addition to the ν1 band at lower wavenumbers. Therefore, the splitting indicates the presence of an inner-sphere bidentate binuclear sulfate complex on the surface of goethite nanorods. Several additional experiments (analysis of supernatant and measurements of iron sulfate standards) were carried out to investigate if there was any solution-phase contribution of dissolved [Fe(III) - SO4]+; these experiments showed no contribution of [Fe(III) - SO4]+, indicating the assignment to adsorbed species only.
Thus it is concluded that higher absorption intensities and formation of binuclear complexes point out the presence of additional, and potentially more reactive, sites for nanorod surface, which can lead to higher dissolution rates, as observed from nanorod suspensions at pH 2. Thus, these spectroscopic results provide evidence that differentiate between the surface chemistry of nanorods compared to microrods. In addition, these results further our understanding of the role of acid anion in forming surface complexes that are unique for α-FeOOH nanorods.
Conclusions and Environment Implications
Using α-FeOOH as a proxy for a source of iron from mineral dust aerosol, several factors that effect iron dissolution in low-pH environments have been investigated. Some of the key points addressed here include the role of particle size and the mechanisms involved in the enhanced chemical activity of nanoscale iron oxides, in particular, the importance of reactive surface planes that yield enhanced concentrations of dissolved iron during simulated daytime and nighttime atmospheric processing that go beyond surface area considerations. For α-FeOOH, an important aspect is the higher density of surface O-H groups on the (021) plane that leads to both enhanced proton-promoted and photoreductive dissolution and thus greater amounts of soluble iron. To best understand the implications of size-dependent dissolution and increase the amount of soluble and bioavailable forms of iron, the dissolution at longer time scales is most important. Since atmospheric transport of mineral dust is associated with these longer time scales, dissolved iron from nanorods may be a more important long-term source of bioavailable Fe compared to larger microrods. The dominant contribution to total dissolved iron in the presence of light is dissolved Fe(II), which is produced in greater quantities for nanorods relative to microrods. Given the potential importance of heterogeneous photochemical processes on particle surfaces in the atmosphere, redox reactions that enhance the solubility of iron oxides are particularly indispensable in controlling the extent of iron dissolution and speciation in atmospheric aerosols (32, 33).
Furthermore, these results clearly indicate that iron dissolution for nanorods in low-pH environments is greatly influenced by the nature of the acid. Once mineral dust aerosol undergoes atmospheric transport and aging in polluted environments, they contain relatively high concentration of inorganic species. In general, nitrate and sulfate tend to enhance iron dissolution in the dark relative to chloride. These results reveal that iron-containing mineral particles that become mixed with sea salt or become associated with chloride ion in some other way (e.g., heterogeneous uptake of HCl) will yield less total soluble iron in daytime and nighttime atmospheric processing, yet there will be a higher fraction of dissolved Fe(II) during daytime processing relative to the other inorganic anions. Sulfate, in comparison, shows a larger fraction of Fe(III) in irradiated systems. It is also worth noting that changing global emissions of sulfur and nitrogen oxides will influence HNO3 and H2SO4 concentrations. According to recent field studies and model predictions, it is anticipated that global SO2 emissions are to be reduced over the next century, whereas the global NOx emission is expected to increase (34). This will result in more nitrate adsorbed on to mineral dust particle surfaces that will influence iron mobilization from dust. Finally, in order to accurately predict bioavailable iron in acidified mineral dust aerosol plumes and marine environments, these factors that influence iron solubility will need to be included in future atmospheric chemistry models. The current study presents important information that can be further used in atmospheric chemistry models.
Materials and Methods
Source Materials.
Ferric nitrate nonahydrate [Fe(NO3)3·9H2O; Sigma Aldrich; 98%], sodium bicarbonate (NaHCO3, Sigma Aldrich, 99.5%), and potassium hydroxide (KOH, Sigma Aldrich) were used for α-FeOOH synthesis. In iron dissolution experiments, solutions of 0.1 N (pH 1) and 0.01 N (pH 2) were prepared from concentrated HNO3 (70.6% HNO3, Mallinckrodt), H2SO4 (95.9% H2SO4, Mallinckrodt), and HCl (37.6% HCl, Fisher). Measurements of dissolved Fe(II) and total dissolved iron were performed with 1,10-phenanthroline (≥99%, Sigma Aldrich), hydroxylamine hydrochloride (98%, Sigma Aldrich), a buffer from ammonium acetate (98.5%, Fisher), and glacial acetic acid (99.7%, EMD).
Synthesis of Goethite.
α-FeOOH nanoparticles were synthesized according to the method of Anschutz and Penn (35) as previously described (11). Dropwise addition of NaHCO3 to a solution of Fe(NO3)·9H2O results in formation of ferrihydrite nanoparticles; subsequently, these are allowed to age at pH 12 for 24 h at 90 °C to yield the nanoscale goethite. Microrods were synthesized using the method of Cornell and Schwertmann reported in the literature (36).
Goethite Characterization.
Nanorods and microrods were characterized using powder x-ray diffraction Bruker D-5000 diffractometer. Surface areas were determined from a seven-point N2-adsorption isotherm using a Quantachrome Nova 1200 surface area analyzer. Samples for surface area analysis were evacuated overnight (approximately 12 h) at a temperature of 80 °C. Nanorod and microrod dimensions were obtained from single particle analysis with TEM. Suspensions (approximately 0.2 g/L) of nanorods and microrods were prepared in deionized water, and a single drop was applied to a TEM grid. Particle size was determined from the analysis of approximately 500 nanorods and 300 microrods. In addition, to examine how dissolution reactions change the morphology of goethite particles, samples were taken for further TEM analysis.
Dissolution Experiments.
The experiments were carried out in a custom-made glass reactor using synthesized nanorods of α- FeOOH and microrods. Reactor design has been described previously (17). These experiments were conducted in the absence and presence of a solar simulator (150 W xenon lamp, Oriel Corp). The temperature was kept constant through use of a water jacket. The experiments were performed under deoxygenated conditions, suspensions were purged with N2 for approximately 15 min prior to irradiation, and during irradiation the headspace of the reaction vessel was purged to maintain positive N2 pressure. The sample loading was maintained 0.2 g/L of α-FeOOH in solutions of HNO3, H2SO4, and HCl at pH 2. Under these conditions, nanorod and microrod suspensions were stable, and the particles were fairly well dispersed. All dark and light experiments were conducted in triplicate with average measurements reported. Reported errors represent one standard deviation.
Analytical Methods.
Ferrous iron was measured colorimetrically with 1,10-phenanthroline, which forms a complex with Fe(II) that absorbs light at 510 nm (37). For Fe(II) analysis, 200 μL of a 5 mM 1,10-phenanthroline solution and 200 μL of an ammonium acetate buffer were added to 1 mL of sample. Total dissolved iron was determined via a similar protocol, except that 40 μL of 1.5 M hydroxylamine hydrochloride, which reduces Fe(III) to Fe(II), was added to the sample. Fe(III) concentrations were then determined by difference [Total Fe—and Fe(II)].
ATR-FTIR Spectroscopy.
ATR-FTIR spectroscopy was done using a modified horizontal ATR cell (Pike Technologies, Inc.) and a Thermo Nicolet FTIR spectrometer equipped with a mercury cadmium telluride detector. The ATR crystal was coated with a hydrosol of goethite rods (approximately 1.5 mL of a 2 g/L goethite suspension) and allowed to air-dry for 24 h. This process left behind a thin layer of goethite that uniformly coats the crystal. Solution-phase experiments were also conducted in the absence of the thin goethite layer.
Acknowledgements.
The authors would like to thank Dr. Jonas Baltrusaitis for his help with TEM images. This material is based upon work supported by the National Science Foundation (EAR-0506679).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Martin JH, Gordon RM, Fitzwater SE. Iron in Antarctic waters. Nature. 1990;345:156–158. [Google Scholar]
- 2.de Baar HJW, Boyd PW. The Role of Iron in Plankton Ecology and Carbon Dioxide Transfer of the Global Oceans. New York: Cambridge Univ Press; 2000. [Google Scholar]
- 3.Mahowald NM, et al. Atmospheric iron deposition: Global distribution, variability, and human perturbations. Annu Rev Mar Sci. 2009;1:245–278. doi: 10.1146/annurev.marine.010908.163727. [DOI] [PubMed] [Google Scholar]
- 4.Rose AL, Waite TD. Kinetic model for Fe(II) oxidation in seawater in the absence and presence of natural organic matter. Environ Sci Technol. 2002;36:433–444. doi: 10.1021/es0109242. [DOI] [PubMed] [Google Scholar]
- 5.Erel Y, Pehkonen SO, Hoffmann MR. Redox chemistry of iron in fog and stratus clouds. J Geophys Res-Atmos. 1993;98:18423–18434. [Google Scholar]
- 6.Guerzoni S, Molinaroli E, Chester R. Saharan dust inputs to the western Mediterranean Sea: Depositional patterns, geochemistry and sedimentological implications. Deep-Sea Res Pt II. 1997;44:631–654. [Google Scholar]
- 7.Duggen S, Croot P, Schacht U, Hoffmann L. Subduction zone volcanic ash can fertilize the surface ocean and stimulate phytoplankton growth: Evidence from biogeochemical experiments and satellite data. Geophys Res Lett. 2007;34:L01612. doi: 10.1029/2006GL027522. [Google Scholar]
- 8.Jickells TD, et al. Global iron connections between desert dust, ocean biogeochemistry, and climate. Science. 2005;308:67–71. doi: 10.1126/science.1105959. [DOI] [PubMed] [Google Scholar]
- 9.Stefansson A. Iron(III) Hydrolysis and solubility at 25 °C. Environ Sci Technol. 2007;41:6117–6123. doi: 10.1021/es070174h. [DOI] [PubMed] [Google Scholar]
- 10.Journet E, Desboeufs KV, Caquineau S, Colin JL. Mineralogy as a critical factor of dust iron solubility. Geophys Res Lett. 2008;35:L07805. doi: 10.1029/2007GL031589. [Google Scholar]
- 11.Cwiertny DM, et al. Characterization and acid-mobilization study of iron-containing mineral dust source materials. J Geophys Res-Atmos. 2008;113:D05202. doi: 10.1029/2007JD009332. [Google Scholar]
- 12.Guieu C, Bonnet S, Wagener T, Loye-Pilot MD. Biomass burning as a source of dissolved iron to the open ocean? Geophys Res Lett. 2005;32:L19608. doi: 101029/2005GL022962. [Google Scholar]
- 13.Sedwick PN, Sholkovitz ER, Church TM. Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: Evidence from the Sargasso Sea. Geochem Geophy Geosy. 2007;8:Q10Q06. doi: 10.1029/2007GC001586. [Google Scholar]
- 14.Adamopoulos AD, Kambezidis HD, Kaskaoutis DG, Giavis G. A study of aerosol particle sizes in the atmosphere of Athens, Greece, retrieved from solar spectral measurements. Atmos Res. 2007;86:194–206. [Google Scholar]
- 15.Shi Z, et al. Formation of iron nanoparticles and increase in iron reactivity in mineral dust during simulated cloud processing. Environ Sci Technol. 2009;43:6592–6596. doi: 10.1021/es901294g. [DOI] [PubMed] [Google Scholar]
- 16.Meskhidze N, Chameides WL, Nenes A. Dust and pollution: A recipe for enhanced ocean fertilization? J Geophys Res. 2005;110:D03301. doi: 101029/2004JD005082. [Google Scholar]
- 17.Cwiertny DM, Hunter GJ, Pettibone JM, Scherer MM, Grassian VH. Surface chemistry and dissolution of alpha-FeOOH nanorods and microrods: Environmental implications of size-dependent interactions with oxalate. J Phys Chem C . 2009;113:2175–2186. [Google Scholar]
- 18.Cornell RM, Posner AM, Quirk JP. Crystal morphology and dissolution of goethite. J Inorg Nucl Chem. 1974;36:1937–1946. [Google Scholar]
- 19.Cornell RM, Posner AM, Quirk JP. Kinetics and mechanisms of acid dissolution of goethite alpha-FeOOH. J Inorg Nucl Chem. 1976;38:563–567. [Google Scholar]
- 20.Wiederhold JG, et al. Iron isotope fractionation during proton-promoted, ligand-controlled, and reductive dissolution of goethite. Environ Sci Technol. 2006;40:3787–3793. doi: 10.1021/es052228y. [DOI] [PubMed] [Google Scholar]
- 21.Miller WL, King DW, Lin J, Kester DR. Photochemical redox cycling of iron in coastal seawater. Mar Chem. 1995;50:63–77. [Google Scholar]
- 22.Voelker BM, Morel FMM, Sulzberger B. Iron redox cycling in surface waters: Effects of humic substances and light. Environ Sci Technol. 1997;31:1004–1011. [Google Scholar]
- 23.Borer P, Sulzberger B, Hug SJ, Kraemer SM, Kretzschmar R. Photoreductive dissolution of iron(III) (hydr)oxides in the absence and presence of organic ligands: Experimental studies and kinetic modeling. Environ Sci Technol. 2009;43:1864–1870. doi: 10.1021/es801352k. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan RC, Guazzotti SA, Sodeman DA, Prather KA. Direct observations of the atmospheric processing of Asian mineral dust. Atmos Chem Phys. 2007;7:1213–1236. [Google Scholar]
- 25.Sullivan RC, et al. Mineral dust is a sink for chlorine in the marine boundary layer. Atmos Environ. 2007;41:7166–7179. [Google Scholar]
- 26.Rubasinghege G, Grassian VH. Photochemistry of adsorbed nitrate on aluminum oxide particle surfaces. J Phys Chem A. 2009;113:7818–7825. doi: 10.1021/jp902252s. [DOI] [PubMed] [Google Scholar]
-
27.Hsu C-L, Wang S-L, Tzou Y-M. Photocatalytic reduction of Cr(VI) in the presence of
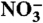 and Cl- electrolytes as influenced by Fe(III) Environ Sci Technol. 2007;41:7907–7914. doi: 10.1021/es0718164. [DOI] [PubMed] [Google Scholar]
and Cl- electrolytes as influenced by Fe(III) Environ Sci Technol. 2007;41:7907–7914. doi: 10.1021/es0718164. [DOI] [PubMed] [Google Scholar] - 28.Nadtochenko VA, Kiwi J. Photolysis of FeOH2+ and FeCl2+ in aqueous solutionPhotodissociation kinetics and quantum yields. Inorg Chem. 1998;37:5233–5238. [Google Scholar]
- 29.Lim M, Chiang K, Amal R. Photochemical synthesis of chlorine gas from iron(III) and chloride solution. J Photoch Photobio A. 2006;183:126–132. [Google Scholar]
- 30.Goldstein S, Rabani J. Mechanism of nitrite formation by nitrate photolysis in aqueous solutions: The role of peroxynitrite, nitrogen dioxide, and hydroxyl radical. J Am Chem Soc. 2007;129:10597–10601. doi: 10.1021/ja073609+. [DOI] [PubMed] [Google Scholar]
- 31.Hug SJ. In situ Fourier transform infrared measurements of sulfate adsorption on hematite in aqueous solutions. J Colloid Interf Sci. 1997;188:415–422. [Google Scholar]
- 32.Fujii M, Rose AL, Waite TD, Omura T. Superoxide-mediated dissolution of amorphous ferric oxyhydroxide in seawater. Environ Sci Technol. 2006;40:880–887. doi: 10.1021/es051622t. [DOI] [PubMed] [Google Scholar]
- 33.Fu H, Xu T, Yang S, Zhang S, Chen J. Photoinduced formation of Fe(III)-sulfato complexes on the surface of α-Fe2O3 and their photochemical performance. J Phys Chem C. 2009;113:11316–11322. [Google Scholar]
- 34.Ooki A, Uematsu M. Chemical interactions between mineral dust particles and acid gases during Asian dust events. J Geophys Res-Atmos. 2005;110:D03201. doi: 10.1029/2004JD004737. [Google Scholar]
- 35.Anschutz AJ, Penn RL. Reduction of crystalline iron(III) oxyhydroxides using hydroquinone: Influence of phase and particle size. Geochem Trans. 2005;6:60–66. doi: 10.1186/1467-4866-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornell RM, Schwertmann U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses. 2nd Ed. Weinheim, Germany: Wiley-VCH; 2003. [Google Scholar]
- 37.Stucki JW, Anderson WL. The quantitative assay of minerals for Fe2+ and Fe3+ using 1,10-Phenanthroline1. Sources of variability. Soil Sci Soc Am J. 1981;45:633–637. [Google Scholar]