

Abstract
New particle formation in the atmosphere is an important parameter in governing the radiative forcing of atmospheric aerosols. However, detailed nucleation mechanisms remain ambiguous, as laboratory data have so far not been successful in explaining atmospheric nucleation. We investigated the formation of new particles in a smog chamber simulating the photochemical formation of H2SO4 and organic condensable species. Nucleation occurs at H2SO4 concentrations similar to those found in the ambient atmosphere during nucleation events. The measured particle formation rates are proportional to the product of the concentrations of H2SO4 and an organic molecule. This suggests that only one H2SO4 molecule and one organic molecule are involved in the rate-limiting step of the observed nucleation process. Parameterizing this process in a global aerosol model results in substantially better agreement with ambient observations compared to control runs.
Keywords: aerosol particles, atmospheric nucleation, new particle formation, sulfuric acid
Atmospheric aerosols affect the radiative balance in the Earth’s atmosphere and influence cloud formation, thereby playing a central role in climate forcing. They also have an important impact on visibility and human health. Many of these effects depend on the particle size distribution, which is governed by the emission of primary particles on the one hand and formation of new particles on the other hand. New particle formation events have been observed frequently and worldwide, in boreal forests, coastal, rural, and urban regions, as well as the free troposphere (1). Their contribution to the regional and global budget of atmospheric particles is likely to be significant though it is still poorly constrained (2–5). A detailed understanding of atmospheric nucleation processes is therefore needed.
Observations in the planetary boundary layer revealed a consistent correlation between sulfuric acid (H2SO4) and the concentration of newly formed particles (6–9), where the particle formation rate can be described with a simple power law:
| [1] |
The exponent m was found to consistently vary between 1 and 2. According to the nucleation theorem (10), this suggests that the critical cluster (the smallest stable “particle”) contains only one or two H2SO4 or sulfuric acid–containing molecules. Classical binary (H2SO4-water) and ternary (H2SO4-NH3-water) mechanisms predict much higher values of the exponent and fail to explain the ambient observations (11, 12). Therefore, new approaches such as H2SO4 cluster activation (13) (for m = 1) and kinetic nucleation (14) (for m = 2) have been developed trying to explain the observed new particle formation events. Recently the formation of organosulfate clusters was suggested to explain the chemistry behind the cluster activation or kinetic mechanisms and thus atmospheric nucleation (15). From detailed analyses of nucleation and growth it was inferred that sesquiterpenes might be involved in new particle formation. On the other hand, a number of studies conclude that the role of organics is mainly important for the growth of the nucleated particles rather than for the nucleation itself (e.g., 16, 17).
Furthermore, the role of ion-induced nucleation (IIN) is still unclear. Based on ambient observations and modeling studies, IIN—although energetically favored—seems to play only a minor role in the continental boundary layer and globally (4, 18–22). However, some studies suggest that IIN could be important in the cold upper troposphere (23), over oceans, and even worldwide (24).
The fact that nucleation takes place at sizes below the detection limit of traditional instruments and is therefore not directly accessible to measurements has so far hampered the elucidation of the nucleation process. Despite these experimental challenges and conflicting results, it seems evident that H2SO4 plays a central role in atmospheric nucleation. However, detailed nucleation mechanisms remain still ambiguous because laboratory data have thus far not been successful in explaining atmospheric nucleation.
Results and Discussion
Chamber Experiments—The Role of Sulfuric Acid.
We performed a series of photooxidation experiments in the 27-m3 Paul Scherrer Institute environmental chamber investigating new particle formation in the presence of 1,3,5-trimethylbenzene (TMB), NOx and SO2 at various mixing ratios (SI Text and Table S1). TMB, a known anthropogenic secondary organic aerosol (SOA) precursor, is especially suited for this study because of the low tendency of its oxidation products to nucleate by themselves even at high mixing ratios. As in the atmosphere, after irradiation of this mixture OH radicals oxidize SO2 and TMB producing H2SO4 and a variety of organic products. The production of low-volatility products leads to formation of SOA. Fig. 1 shows the temporal evolution of the aerosol number (A) and mass concentrations (B) of five experiments with similar initial mixing ratios of TMB and nitrogen oxides (Fig. S1), clearly demonstrating the effect of varying SO2 mixing ratio. Without addition of SO2, nucleation occurs after approximately 2 h when the nitrogen oxide concentration approaches zero. With increasing the SO2 mixing ratio, nucleation occurs earlier, and the particle number concentration (diameter Dp > 3 nm) increases from 103 to 105 cm-3. The particle number concentration decreases due to wall loss (WL) and coagulation. The final aerosol mass produced, which is derived from the measured number size distribution, is independent of the SO2 mixing ratio, except for the experiment in which no SO2 is present, where aerosol formation is delayed.
Fig. 1.

(A) Aerosol particle number (Dp ≳ 3 nm) and (B) mass concentration (not corrected for WLs) of five individual experiments at 250 ppb TMB and varying SO2 mixing ratios of 0 to 20 ppb. (C) Calculated concentrations of H2SO4 (solid lines) and NucOrg (dashed lines). Variation of NucOrg is due to slightly different reactivity in the initial phase of the experiment and different condensation sink. (D) Appearance rate of 3-nm particles.
For further analysis we derived the particle formation rates as well as the concentrations of H2SO4 and low-volatility organic compounds assuming that the fraction of the organic oxidation products with the lowest volatility (called NucOrg) can participate in the particle formation process.
Fig. 1C shows the calculated H2SO4 and NucOrg concentrations for the five individual experiments with different initial SO2 mixing ratios. H2SO4 rises sharply after the start of an experiment. In the initial phase its concentration is limited by the loss to the wall. As soon as particles are formed, condensation onto particles overcomes the WL and limits the H2SO4 concentration. As shown in Fig. 1D, the 3-nm particle appearance rate (J3, J3 ∼ dN/dt) follows the rise in H2SO4 with a delay of 39, 21, 18, and 9 min for SO2 mixing ratios of 0.4, 2, 5, and 20 ppb, respectively. The time delay is attributed to the time needed for the nucleated clusters to grow to the detectable size (3 nm) (6–8). The threshold concentration of sulfuric acid when 3-nm particles started to appear was approximately 5 × 106 molecules cm-3. This is in line with ambient observations, which consistently show that 106–107 molecules cm-3 of H2SO4 are necessary to observe particle formation events (e.g., 7, 25–27).
Nucleation Rates in Comparison with Ambient Data.
The actual nucleation rate is the rate of critical cluster formation. We assume the mobility diameter of the critical cluster to be 1.5 nm, in line with recent observations of a stable cluster mode around that size (28). Fig. 2 shows the dependence of J1.5 (Eq. S8) from the gaseous H2SO4 concentration and compares this with ambient observations from the boreal forest in Hyytiälä, Finland (7). The scatter of the atmospheric nucleation rates is attributed to varying temperature, humidity, or varying contributions of unknown species in the nucleation process (29). Our laboratory data agree surprisingly well with nucleation rates observed in the atmosphere. Due to the well-defined conditions of the chamber experiments, the lab data show less scatter and a distinct pattern where data from experiments with higher SO2 are shifted to higher H2SO4 and J1.5 values. Data are color coded with the amount of NucOrg showing an increase of NucOrg with increasing H2SO4, which will be discussed further below.
Fig. 2.

Estimated nucleation rates (J1.5) from this study and atmospheric nucleation rates from Hyytiälä, Finland (7) as a function of gaseous H2SO4 concentration. Colored symbols: this study; gray symbols: ambient data. The black lines represent log(J1.5) = m· log[H2SO4] + b with slopes of m = 1 and 2. The colored lines indicate the slope of the nucleation rate as a function of the H2SO4 concentration at roughly constant NucOrg, which is binned to concentration intervals [blue: < 8·106 cm-3 (m = 0.9); light blue: 8·106 - 1.5·107 cm-3 (m = 1); green: 1.5·107–2.2·107 cm-3 (m = 1.2); yellow: 2.2·107–2.9·107 cm-3 (m = 1.3); red: > 2.9·107 cm-3 (m = 1.35)].
Involvement of Organic Compounds in Nucleation.
The exponent m of the power law model (Eq. 1) can be obtained as the slope of log(J) versus log(H2SO4) (Fig. 2). Evidently, the ambient as well as the laboratory data are confined within the slopes m = 1 and 2. A linear regression fit to all our laboratory data exhibits a slope of 1.84 ± 0.06 (R = 0.85), whereas the slopes of the individual experiments range from 1.7 to 3.4. These slopes are in close agreement with ambient observations, in contrast to previous studies, where higher slopes were typically found (e.g., 6–9). According to the nucleation theorem, a slope (exponent m) of 2 indicates that the critical cluster contains two sulfuric acid molecules. The nucleation theorem is a very robust theorem that applies irrespective of a specific nucleation mechanism (10, 30). However, it only applies when other variables of influence (temperature and gas phase concentrations of other species participating in the nucleation process) remain constant. This restriction is typically neglected because there are not enough data to obtain meaningful correlations at narrow temperature and gas concentration intervals, and most other compounds that might be involved in nucleation are not known (29). Within an individual experiment, H2SO4 and organic photooxidation products are expected to be highly correlated because their formation and loss processes are highly similar. This can be seen from the color coding in Fig. 2. Even more importantly, besides merely correlating with H2SO4, organics clearly affect the nucleation rate: For a constant H2SO4 concentration, higher NucOrg results in higher nucleation rates. Thus, a given nucleation rate, J1.5, can be sustained at lower H2SO4 levels if there is more organic vapor, and vice versa, indicating that both species contribute to nucleation in a kinetically limited process. We advocate that the same dependence also exists in most nucleation events in the ambient atmosphere. Constraining the analysis to H2SO4 only may therefore be misleading.
Disentangling the Roles of Sulfuric Acid and Organic Compounds in Nucleation.
In the following, we will attempt to disentangle the influence of organic oxidation products from that of sulfuric acid using two different approaches. First, a nonlinear regression analysis was applied to our data as suggested by ref. 29. As the nucleation rate may depend on both H2SO4 and NucOrg, Eq. 1 is rewritten as:
| [2] |
The least-squares fit yielded m = 1.0 ± 0.04, n = 0.8 ± 0.04, and k = 7.2 ± 4.4 × 10-13 cm3 s-1. This indicates an overall dependency of the nucleation rate on H2SO4 and NucOrg each close to the power of one. Constraining both n and m to one leads to a prefactor k of 7.5 ± 0.3 × 10-14 cm3 s-1.
Second, the whole dataset was binned relative to the NucOrg concentration. The power law model (Eq. 1) was then applied to each bin, which represents a dataset at roughly constant NucOrg. Slopes between 1 and 1.3 are obtained as seen in Fig. 2 (colored lines). This shows that eliminating the confounding correlation between the concentrations of H2SO4 and NucOrg results in slopes of log(J1.5) versus log[H2SO4] clearly below 2. This is also shown by the isopleths plot log[H2SO4] versus log[NucOrg] (Fig. S2); it is impossible to explain our data with a dependence of either H2SO4 or NucOrg alone (which would mean horizontal or vertical regression lines in that plot). Based on these results, we propose that the attachment of a H2SO4 molecule with a NucOrg molecule forms the first stage clusters, which then grow by condensation and coagulation.
Discussion
It has previously been shown that in the ambient atmosphere a pool of sub-3-nm clusters is always present with a mobility ranging between 0.7 and 1.7 cm2 V-1 s-1, which corresponds to mobility diameters of 1.6 and 1.0 nm, respectively (28). The primary oxidation step of TMB leads to products with a molecular weight of up to at least 231 g mol-1 (31). The addition of one H2SO4 molecule to such a molecule [similar to a pathway proposed by Bonn et al. (15)] would thus result in a cluster with a molecular weight well above 300 g mol-1. According to Mäkelä et al. (32), this mass corresponds to a mobility of approximately 1.1 cm2 V-1 s-1, well within the boundaries of a typical ambient cluster mode.
Clearly, other volatile organic compounds like monoterpenes or sesquiterpenes are more likely to act as precursors for NucOrg under ambient conditions (but the involved functional groups are expected to be similar as for TMB). For example, organosulfate clusters formed via Criegee intermediates from the ozonolysis of sesquiterpenes were proposed to explain new particle formation (15, 30). Others have found only a weak correlation between terpene oxidation products and nucleation (e.g., 29). Some laboratory studies found that the presence of organics enhances the nucleation rate (33–35), whereas others did not show evidence for such an enhancement (36). It may be speculated that in the latter case the residence time was not sufficiently high to show an effect of the organics. From our data we cannot deduce the exact nature of NucOrg. The combined evidence of the contribution of different organics to nucleation from our results and others suggests that the exact nature of the organics (the carbon backbone) may be less important than the specific functional groups required for the cluster formation with H2SO4 (33, 35).
Further evidence for an involvement of organics already in the smallest cluster sizes is gained from the sub-3-nm growth rates (the initial growth between 1.5 and 3 nm, GR1.5–3). These initial growth rates were estimated from the time delays between the rise in H2SO4 and the rise in J3 (e.g., 6, 7) and range from 3–10 nm h-1. Condensation of H2SO4 (37, 38) may only explain up to 20% of GR1.5–3 at high sulfuric acid concentrations and much less at lower concentrations. This indicates that other species than H2SO4, presumably organics, are needed to explain the observed growth. The concentration of nucleating organic vapor (NucOrg) needed in order to explain the observed sub-3-nm growth rates was estimated to be approximately 5 × 107 molecules cm-3, corresponding to a product yield of NucOrg of 0.025%. In the boreal forest, sulfuric acid concentrations are also too low to explain the observed growth, where sulfuric acid could typically explain about 50% of the growth in the diameter range of 1 to 3 nm (e.g., 7). Therefore it was speculated that some other species, presumably organics, were needed to condense on the particles. Furthermore, at Dp = 2–4 nm the particles appeared to be less hygroscopic than pure ammonium sulfate or sulfuric acid (39). This indicates that both organics and H2SO4 (or organosulfates) might be present already in the smallest particles.
The prefactor k in Eq. 2 contains details of the nucleation process. Our analysis yields a k value of 0.7–7 × 10-13 cm3 s-1 from the multilinear regression, which is several orders of magnitude below the hard sphere collision limit (8). This may point to the fact that stable critical cluster formation is governed by the competition between stabilization and decay of an unstable complex formed from the collision of the molecules. Assuming that both sulfuric acid and organics take part in nucleation (Eq. 2) and applying the sub-3-nm growth method to atmospheric data obtained in Hyytiälä, a k value of the order of 1 × 10-13 cm3 s-1 is obtained. This is in fairly good agreement with the above value, considering the uncertainty in the data and the differences in temperature, relative humidity, and chemical composition present.
Global Modeling—Atmospheric Implications
We further tested our proposed mechanism by incorporating it in the three-dimensional chemical transport model GLOMAP, which includes a detailed treatment of aerosol microphysics (40). We modified the model to include the kinetic nucleation involving organics (Eq. 2, using the values of m = 1 and n = 1 found by multilinear regression and k varying from 5 × 10-12 to 5 × 10-14 cm3 s-1), assuming that the organics are emitted by the biosphere, using monoterpene emissions and behavior as a proxy (SI Text). Results were then compared to control runs existing of binary homogeneous nucleation (BHN), kinetic nucleation involving only sulfuric acid (14) (Eq. 1, using m = 2 and k = 4 × 10-13 cm3 s-1), and the cluster activation mechanism (Eq. 1, using m = 1 and k = 5 × 10-7 s-1) (13).
Fig. 3 shows vertical profiles of total particle number concentrations from aircraft observations over North America and US outflow compared to our simulations. The “traditional” kinetic mechanism and the cluster activation mechanism, both including only sulfuric acid, overpredict the aerosol number in the free troposphere (altitude of 2–8 km), unless the mechanisms are restricted to the boundary layer. The kinetic nucleation mechanism involving organics gives very good agreement for the whole vertical profile, producing the typical Z-shaped profile often observed over continental regions.
Fig. 3.

Vertical profiles of total particle number concentrations (Dp > 3 nm) over the continental United States (A: 28°N–53°N, 240°E–269°E) and U.S. outflow (B: 32°N–52°N, 290°E–323°E) observed in summer 2004 during the Intercontinental Chemical Transport Experiment–North America (INTEX-NA) campaign (black symbols: horizontal lines indicating the 5th and 95th percentiles). Also shown are simulated concentrations with different particle formation mechanisms: BHN (black line), activation mechanism (J1.5 = 5 × 10-7 s-1 [H2SO4]) throughout the atmosphere (blue line) and artificially restricted to the boundary layer with BHN above (dotted blue line), traditional kinetic nucleation (J1.5 = 4 × 10-13 cm3 s-1 [H2SO4]) throughout the atmosphere (purple line) and artificially restricted to the boundary layer with BHN above (dotted purple line), and kinetic nucleation mechanism involving organics (J1.5 = k[H2SO4][organic]) and BHN occurring throughout the atmosphere (red line: k = 5 × 10-13 cm3 s-1; red shading shows range where k varied by a factor of 10). All simulations include primary particle emissions and SOA formation.
Fig. 4 shows simulated surface particle number concentrations with these different nucleation mechanisms. Both the traditional activation and kinetic mechanisms result in substantial nucleation over oceanic regions where there are ship emissions of SO2 (e.g., North Atlantic) or oceanic DMS emissions (e.g., Southern Ocean), which is generally not confirmed by field observations (41). The general lack of nucleation in oceanic regions is, however, captured by the new organic activation mechanism, due to a low concentration of organics and subsequently slow nucleation rates.
Fig. 4.

Simulated April surface mean total particle number concentrations (Dp > 3 nm) with different particle formation mechanisms: (A) BHN, (B) activation mechanism restricted to the boundary layer (J1.5 = 5 × 10-7 s-1 [H2SO4]) with BHN above, (C) kinetic mechanism restricted to the boundary layer (J1.5 = 4 × 10-13 cm3 s-1 [H2SO4]2) with BHN above, and (D) kinetic nucleation involving organics (J1.5 = 5 × 10-13 cm3 s-1 [H2SO4][organic]) and BHN occurring throughout the atmosphere.
Conclusions
Our results bridge the long-standing discrepancy between atmospheric observations and laboratory studies. Our results suggest that organic compounds together with sulfuric acid are likely to initiate the nucleation process. Furthermore, both organics and sulfuric acid contribute to the subsequent growth of these clusters. As seen from the results, the nucleation rate is practically linearly dependent on sulfuric acid and organic concentrations with rate coefficients 0.7–7 × 10-13 cm3 s-1. These results allow for a more accurate description of nucleation, leading to the formation of aerosol and—under favorable circumstances—to their growth into the cloud condensation nuclei (CCN) size range.
The mechanism described here may have implications for the role of the biosphere in climate regulation through aerosol radiative forcing. It has previously been shown that a significant fraction of CCN are derived from nucleation (3). The dependence of the nucleation rate and particle growth rate on biogenic emissions may lead to a coupling of CCN to biospheric emissions, which are predicted to increase with changing climate over the coming century (42).
Methods
The H2SO4 concentration in the chamber was calculated using a simple kinetic model considering reactions 3 and 4:
| [3] |
and
| [4] |
The OH radical concentration was determined from the measured decrease of the TMB concentration (reaction 5):
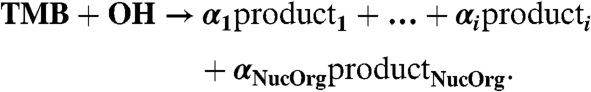 |
[5] |
We assume that the fraction of the organic oxidation products with the lowest volatility can participate in the particle formation process (NucOrg). Its concentration was estimated from the amount of TMB reacted assuming a product yield (αNucOrg) of 0.025%, which was determined from the particle growth rate (see Discussion). WLs and condensation onto particles are the dominant sinks for both H2SO4 and NucOrg.
The largest sources of uncertainty regarding the determination of the gas phase concentrations of H2SO4 and NucOrg are the ill-defined WL rates. We calculated the upper limit of the WL by assuming that the molecules will always stick to the wall surface upon impact and that release from the surface to the gas phase after uptake does not occur. A decrease of the assumed WL rates would therefore shift the concentrations to higher values (by about 1 order of magnitude if WL is neglected; see Fig. S4). Because WL does not vary in time, it merely serves as scaling factor and does not influence the dependency of the nucleation rate from H2SO4 and NucOrg.
The 3-nm particle appearance rate (J3) was obtained from the measured particle number concentration after correction for coagulation, dilution, and WLs. The nucleation rate of 1.5-nm clusters (J1.5) was estimated from the J3 data by accounting for the loss processes that have occurred during the time that the particles spend growing from their nucleating cluster to the measured size range (37, 43).
SI Text contains a full discussion of the methods, data analysis, and uncertainties attributed to the data.
Supplementary Material
Acknowledgments.
This work is supported by the Swiss National Science Foundation, the Natural Environment Research Council, as well as the European Commission projects EUROCHAMP, POLYSOA, EUCAARI, and CLOUD-ITN. We thank the NASA Global Tropospheric Chemistry Program and investigators (Antony Clarke, University of Hawaii, and Bruce Anderson, NASA) for making available the INTEX-NA observations.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911330107/DCSupplemental.
References
- 1.Kulmala M, et al. Formation and growth rates of ultrafine atmospheric particles: A review of observations. J Aerosol Sci. 2004;35(2):143–176. [Google Scholar]
- 2.Spracklen DV, et al. The contribution of boundary layer nucleation events to total particle concentrations on regional and global scales. Atmos Chem Phys. 2006;6:5631–5648. [Google Scholar]
- 3.Spracklen DV, et al. Contribution of particle formation to global cloud condensation nuclei concentrations. Geophys Res Lett. 2008;35:L06808. doi: 10.1029/2007GL033038. [Google Scholar]
- 4.Pierce JR, Adams PJ. Can cosmic rays affect cloud condensation nuclei by altering new particle formation rates? Geophys Res Lett. 2009;36:L09820. doi: 10.1029/2009GL037946. [Google Scholar]
- 5.Makkonen R, et al. Sensitivity of aerosol concentrations and cloud properties to nucleation and secondary organic distribution in ECHAM5-HAM global circulation model. Atmos Chem Phys. 2009;9(5):1747–1766. [Google Scholar]
- 6.Weber RJ, et al. Measurements of new particle formation and ultrafine particle growth rates at a clean continental site. J Geophys Res-Atmos. 1997;102(D4):4375–4385. [Google Scholar]
- 7.Sihto SL, et al. Atmospheric sulphuric acid and aerosol formation: Implications from atmospheric measurements for nucleation and early growth mechanisms. Atmos Chem Phys. 2006;6:4079–4091. [Google Scholar]
- 8.Riipinen I, et al. Connections between atmospheric sulphuric acid and new particle formation during QUEST III–IV campaigns in Heidelberg and Hyytiälä. Atmos Chem Phys. 2007;7(8):1899–1914. [Google Scholar]
- 9.Kuang C, McMurry PH, McCormick AV, Eisele FL. Dependence of nucleation rates on sulfuric acid vapor concentration in diverse atmospheric locations. J Geophys Res-Atmos. 2008;113:D10209. doi: 10.1029/2007JD009253. [Google Scholar]
- 10.Oxtoby DW, Kashchiev D. A general relation between the nucleation work and the size of the nucleus in multicomponent nucleation. J Chem Phys. 1994;100(10):7665–7671. [Google Scholar]
- 11.Benson DR, Young LH, Kameel FR, Lee SH. Laboratory-measured nucleation rates of sulfuric acid and water binary homogeneous nucleation from the SO2 + OH reaction. Geophys Res Lett. 2008;35:L11801. doi: 10.1029/2008GL033387. [Google Scholar]
- 12.Curtius J. Nucleation of atmospheric aerosol particles. CR Phys. 2006;7(9-10):1027–1045. [Google Scholar]
- 13.Kulmala M, Lehtinen KEJ, Laaksonen A. Cluster activation theory as an explanation of the linear dependence between formation rate of 3 nm particles and sulphuric acid concentration. Atmos Chem Phys. 2006;6:787–793. [Google Scholar]
- 14.McMurry PH, Friedlander SK. New particle formation in the presence of an aerosol. Atmos Environ. 1979;13(12):1635–1651. [Google Scholar]
- 15.Bonn B, Kulmala M, Riipinen I, Sihto SL, Ruuskanen TM. How biogenic terpenes govern the correlation between sulfuric acid concentrations and new particle formation. J Geophys Res-Atmos. 2008;113:D12209. doi: 10.1029/2007JD009327. [Google Scholar]
- 16.Sellegri K, Hanke M, Umann B, Arnold F, Kulmala M. Measurements of organic gases during aerosol formation events in the boreal forest atmosphere during QUEST. Atmos Chem Phys. 2005;5:373–384. [Google Scholar]
- 17.Laaksonen A, et al. The role of VOC oxidation products in continental new particle formation. Atmos Chem Phys. 2008;8(10):2657–2665. [Google Scholar]
- 18.Laakso L, et al. Hot-air balloon as a platform for boundary layer profile measurements during particle formation. Boreal Environ Res. 2007;12:279–294. [Google Scholar]
- 19.Eisele FL, et al. Negative atmospheric ions and their potential role in ion-induced nucleation. J Geophys Res-Atmos. 2006;111:D04305. doi: 10.1029/2005JD006568. [Google Scholar]
- 20.Iida K, et al. Contribution of ion-induced nucleation to new particle formation: Methodology and its application to atmospheric observations in Boulder, Colorado. J Geophys Res-Atmos. 2006;111:D23201. doi: 10.1029/2006JD007167. [Google Scholar]
- 21.Boy M, Kazil J, Lovejoy ER, Guenther A, Kulmala M. Relevance of ion-induced nucleation of sulfuric acid and water in the lower troposphere over the boreal forest at northern latitudes. Atmos Res. 2008;90(2-4):151–158. [Google Scholar]
- 22.Kazil J, Lovejoy ER, Barth MC, O’Brien K. Aerosol nucleation over oceans and the role of galactic cosmic rays. Atmos Chem Phys. 2006;6:4905–4924. [Google Scholar]
- 23.Arnold F. Atmospheric aerosol and cloud condensation nuclei formation: A possible influence of cosmic rays? Space Sci Rev. 2006;125(1-4):169–186. [Google Scholar]
- 24.Yu F, Turco R. Case studies of particle formation events observed in boreal forests: Implications for nucleation mechanisms. Atmos Chem Phys. 2008;8(20):6085–6102. [Google Scholar]
- 25.Weber RJ, et al. New particle formation in the remote troposphere: A comparison of observations at various sites. Geophys Res Lett. 1999;26(3):307–310. [Google Scholar]
- 26.Birmili W, Wiedensohler A, Plass-Dulmer C, Berresheim H. Evolution of newly formed aerosol particles in the continental boundary layer: A case study including OH and H2SO4 measurements. Geophys Res Lett. 2000;27(15):2205–2208. [Google Scholar]
- 27.Boy M, et al. New particle formation in the front range of the Colorado Rocky Mountains. Atmos Chem Phys. 2008;8(6):1577–1590. [Google Scholar]
- 28.Kulmala M, et al. Toward direct measurement of atmospheric nucleation. Science. 2007;318:89–92. doi: 10.1126/science.1144124. [DOI] [PubMed] [Google Scholar]
- 29.Laaksonen A, et al. SO2 oxidation products other than H2SO4 as a trigger of new particle formation. Part 2: Comparison of ambient and laboratory measurements, and atmospheric implications. Atmos Chem Phys. 2008;8(23):7255–7264. [Google Scholar]
- 30.MacDowell LG. Formal study of nucleation as described by fluctuation theory. J Chem Phys. 2003;119(1):453–463. [Google Scholar]
- 31.Wyche KP, et al. Gas phase precursors to anthropogenic secondary organic aerosol: Detailed observations of 1,3,5-trimethylbenzene photooxidation. Atmos Chem Phys. 2009;9(2):635–665. [Google Scholar]
- 32.Mäkelä JM, Riihelä M, Ukkonen A, Jokinen V, Keskinen J. Comparison of mobility equivalent diameter with Kelvin-Thomson diameter using ion mobility data. J Chem Phys. 1996;105(4):1562–1571. [Google Scholar]
- 33.Zhang RY, et al. Atmospheric new particle formation enhanced by organic acids. Science. 2004;304(5676):1487–1490. doi: 10.1126/science.1095139. [DOI] [PubMed] [Google Scholar]
- 34.Verheggen B, et al. alpha-Pinene oxidation in the presence of seed aerosol: Estimates of nucleation rates, growth rates, and yield. Environ Sci Technol. 2007;41(17):6046–6051. doi: 10.1021/es070245c. [DOI] [PubMed] [Google Scholar]
- 35.Zhang R, et al. Formation of nanoparticles of blue haze enhanced by anthropogenic pollution. Proc Natl Acad Sci USA. 2009;106(42):17650–17654. doi: 10.1073/pnas.0910125106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berndt T, Boge O, Stratmann F, Heintzenberg J, Kulmala M. Rapid formation of sulfuric acid particles at near-atmospheric conditions. Science. 2005;307(5710):698–700. doi: 10.1126/science.1104054. [DOI] [PubMed] [Google Scholar]
- 37.Verheggen B, Mozurkewich M. An inverse modeling procedure to determine particle growth and nucleation rates from measured aerosol size distributions. Atmos Chem Phys. 2006;6:2927–2942. [Google Scholar]
- 38.Kulmala M, et al. On the formation, growth and composition of nucleation mode particles. Tellus B. 2001;53(4):479–490. [Google Scholar]
- 39.Riipinen I, et al. Applying the Condensation Particle Counter Battery (CPCB) to study the water-affinity of freshly-formed 2–9 nm particles in boreal forest. Atmos Chem Phys. 2009;9(10):3317–3330. [Google Scholar]
- 40.Spracklen DV, Pringle KJ, Carslaw KS, Chipperfield MP, Mann GW. A global off-line model of size-resolved aerosol microphysics: I Model development and prediction of aerosol properties. Atmos Chem Phys. 2005;5:2227–2252. [Google Scholar]
- 41.Heintzenberg J, Birmili W, Wiedensohler A, Nowak A, Tuch T. Structure, variability and persistence of the submicrometre marine aerosol. Tellus B. 2004;56(4):357–367. [Google Scholar]
- 42.Lathiere J, Hauglustaine DA, De Noblet-Ducoudre N, Krinner G, Folberth GA. Past and future changes in biogenic volatile organic compound emissions simulated with a global dynamic vegetation model. Geophys Res Lett. 2005;32:L20818. doi: 10.1029/2005GL024164. [Google Scholar]
- 43.Kerminen VM, Kulmala M. Analytical formulae connecting the “real” and the “apparent” nucleation rate and the nuclei number concentration for atmospheric nucleation events. J Aerosol Sci. 2002;33(4):609–622. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.