

Abstract
In aqueous solution, aldehydes, and to a lesser extent ketones, hydrate to form geminal diols. We investigate the hydration of methylglyoxal (MG) in the gas phase, a process not previously considered to occur in water-restricted environments. In this study, we spectroscopically identified methylglyoxal diol (MGD) and obtained the gas-phase partial pressures of MG and MGD. These results, in conjunction with the relative humidity, were used to obtain the equilibrium constant, KP, for the water-mediated hydration of MG in the gas phase. The Gibbs free energy for this process, ΔG°, obtained as a result, suggests a larger than expected gas-phase diol concentration. This may have significant implications for understanding the role of organics in atmospheric chemistry.
Keywords: hydration, equilibrium constant, water clusters
Atmospheric aerosols are a major topic of current atmospheric studies, given the important, but not fully understood, role that aerosols play in the Earth’s radiative balance (1). Aerosols affect global radiative forcing directly by absorbing or scattering radiation and indirectly by enhancing cloud albedo (1). Because of their changing chemical composition, aerosol optical and physiochemical properties vary, which greatly complicates the quantification of their global radiative forcing. Efforts at modeling aerosol effects on climate, as assessed by the Intergovernmental Panel on Climate Change (1), are primarily based on sulfate aerosol studies, though there is growing evidence that organic aerosols play an important role in climate change (2–4). Organic molecules formed by the oxidation of biogenic and anthropogenic organic emissions have been identified as important components of atmospheric aerosols (2, 5). The formation pathways of these secondary organic aerosols (SOA) remain highly speculative, leading to uncertainties in predictions of atmospheric models (6–8). A large SOA source is missing from models as illustrated by simultaneous field measurements of volatile organic compounds (VOCs) and aerosol particles (6, 7, 9, 10).
Organic acids, especially oxalic and pyruvic acid, are found in SOA, though the origin of these acids is not predicted correctly by the gas-phase chemistry considered in models (11–14). Recent studies suggest that these acids are made in the aqueous phase, particularly in cloud water, by the oxidation of aldehydes (e.g., glyoxal and methylglyoxal) with hydroxyl radicals and other aqueous radical species (14–17). Methylglyoxal (MG) is one of the most abundant α-dicarbonyls present in the atmosphere and is produced from VOCs of both biogenic and anthropogenic origin (14, 18–20).
Water-mediated aldehyde chemistry is expected to have important consequences to the formation of SOA. For example, laboratory studies of MG hydration performed using theoretical (21) and spectroscopic techniques (14, 22–24) suggest that in aqueous environments MG can become hydrated to form methylglyoxal diol (MGD) via reaction 1 and undergo further reactions to form oligomers (14, 21, 24). Like other aldehydes and ketones in aqueous solution, MG hydrates through proton addition to the aldehyde carbonyl and reaction to form MGD (21–23, 25–27). MG has both an aldehydic and ketonic C = O and it was determined computationally that the aldehydic C = O is more favorably hydrated in solution (ΔG = -1.4 kcal mol-1) than the ketonic C = O (ΔG = +2.5 kcal mol-1) (21). In solution, MG is present primarily as MGD (60% diol to 40% tetrol) with the aldehydic group forming a geminal diol (21, 23). MGD has a lower vapor pressure than MG, which allows the molecule to partition more easily into the particle phase, lending to the formation of SOA.
 R1
R1
Although the hydration of small aldehydes in aqueous solutions is known to be extensive, gas-phase hydration of carbonyls has not yet been considered in atmospheric models because it is commonly believed that there is not enough water present to make such reactions favorable. Gas-phase studies of glyoxylic acid performed in our laboratory observed hydration of glyoxylic acid and allowed for the identification of the gem diol through IR spectroscopy, suggesting this chemistry can occur in water restricted environments (28). In a CCl4 matrix with restricted water present, similar results were obtained for the hydration of pyruvic acid.
In this IR spectroscopic study, we identify and assign the vibrational features of gas-phase MGD and characterize the water-mediated gas-phase equilibrium between MG and MGD. The equilibrium constant, KP, is calculated using spectroscopically determined concentrations of MG, MGD, and water. This study shows gas-phase hydration to be significant even under relatively dry environmental conditions. This affects the gas-phase/aqueous particle partitioning of MG and could provide insight into the discrepancy between measured and modeled amounts of SOA (29, 30).
Results and Discussion
Spectral Identification of MG and MGD.
Fundamental gas-phase spectra MG and MGD.
The fundamental gas-phase IR spectrum of MG has been previously observed (31) and provides the basis for determining the presence of MGD in this study (Fig. 1). In the lower energy region of the MG spectrum (Fig. 1A), from 1,050 to 1,500 cm-1, there are three distinguishable bands which are assigned to the CCC asymmetric stretch at 1,240 cm-1 (ν4), the CH bend (ν6) at 1,373 cm-1, and the CH3 bends (ν7) in the 1,415 to 1,440 cm-1 region (Table 1). MG contains a ketonic and aldehydic C = O, which are present in the MG spectrum at 1,723 cm-1 (ν8) and 1,741 cm-1 (ν9), respectively (Fig. 1A). The CH stretching vibration of MG has been previously assigned (31) and occurs at 2,835 cm-1 (ν11). The MG CH stretch is very prominent and was used to compare the MG and MGD spectra and to identify MGD. The peak at 3,450 cm-1, which normally corresponds to the OH stretching region, we assign to the first overtone, 2ν8 and 2ν9, of the ν8 and ν9 C = O stretches as seen in Fig. 1A. This peak has been previously observed, but not assigned (31).
Fig. 1.

Fundamental gas-phase spectra from 1,050 cm-1 to 3,600 cm-1 of (A) MG and (B) MG and MGD. MGD frequencies are labeled with δ.
Table 1.
Theoretical and observed experimental frequencies for MG and MGD with vibrational modes assignments
| Theoretical frequency, cm-1 | Theoretical intensity , km mol-1 | Experimental frequency, cm-1 | Mode assignments | |
| 1,099 | 182 | 1,088 | 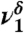 |
Hb C-O stretch |
| 1,185 | 73 | 1,173 | 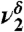 |
COH wag |
| 1,180 | 18 | 1,199 | 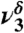 |
CCC stretch |
| 1,207 | 22 | 1,240 | ν4 | CCC stretch |
| 1,275 | 48 | 1,292 | 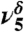 |
Hb COH bend |
| 1,335 | 2 | 1,373 | ν6 | CH bend |
| 1,366–1,415 | — | 1,415–1,440 | ν7 | 3 × CH3 bend |
| 1,754 | 132 | 1,723 | ν8 | C = O stretch |
| 1,774 | 114 | 1,741 | ν9 | HC = O stretch |
| — | — | 1,780 | 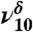 |
Diol |
| 2,811 | 72 | 2,835 | ν11 | CH stretch |
| 2,838 | 43 | 2,895 | 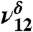 |
CH stretch |
| 3,497 | — | 3,443 | 2ν8 | C = O overtone |
| 3,530 | — | 3,458 | 2ν9 | C = O overtone |
| 3,478 | 86 | 3,505 | 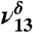 |
Hb OH stretch |
| 3,623 | 55 | 3,585 | 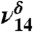 |
Fr OH stretch |
B3LYP/6-31 + G(d,p) theoretical frequencies were scaled using wavenumber linear scaling method. δ, MGD vibration; Hb, hydrogen bonding; Fr, free OH.
The addition of water to the MG sample hydrates MG to form MGD (Fig. 1B). The spectrum in Fig. 1B shows both MG and MGD features under very low relative humidities. In the absence of literature assignments, theoretical frequency and intensity calculations aided in assigning MGD vibrational modes (Table 1). The formation of MGD can be observed by the coincidence of the distinct MGD vibrational modes that appear in the lower energy region from 1,050 to 1,500 cm-1 (Fig. 1B, Inset), the decrease in the ν9 intensity relative to ν8 intensity, and the appearance of OH stretching vibrations at 3,505 cm-1 (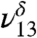 ) and 3,585 cm-1 (
) and 3,585 cm-1 (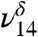 ) (Fig. 1B) with the addition of water vapor. With water present, the CH stretching mode of MGD is observed at 2,895 cm-1 (
) (Fig. 1B) with the addition of water vapor. With water present, the CH stretching mode of MGD is observed at 2,895 cm-1 (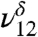 ). Because the lower energy region of the spectra from 1,050 to 1,500 cm-1 becomes increasingly complex due to overlapping bands from the addition of water, the intensities of the ν8, ν9,
). Because the lower energy region of the spectra from 1,050 to 1,500 cm-1 becomes increasingly complex due to overlapping bands from the addition of water, the intensities of the ν8, ν9, 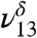 , and
, and 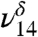 stretches were used to quantify and follow the formation of MGD.
stretches were used to quantify and follow the formation of MGD.
C = O fundamental and overtone stretching region.
With the addition of trace amounts of water, the C = O stretching region is altered by the decrease in intensity of ν9 relative to the ν8, presumably due to the formation of MGD. To better understand these changes in the fundamental C = O stretching region, the intensity and peak width (FWHM) of the fundamental and overtone C = O stretches for MG and MGD were modeled by two Lorenzian curves. The fundamental MG C = O stretching region shows ν8 and ν9 with roughly the same intensity (Fig. 2A). As MG is hydrated to form MGD, the aldehydic C = O is hydrated to form the gem diol, which can be observed spectroscopically by the decrease in the relative intensity of ν9 in Fig. 2B, intensity ν8∶ν9 = 1.0∶0.75. In addition to the MG and MGD fundamental C = O stretches near 1,730 cm-1, there is a third peak around 1,780 cm-1 (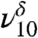 ), which is attributed to the diol because of its intensity increase mirroring the increase in partial pressure of water. The MG and MGD C = O first overtone region near 3,450 cm-1 has the ketonic C = O overtone at 3,443 cm-1 (2ν8) and the aldehydic C = O overtone at 3,458 cm-1 (2ν9). In both MG and MGD spectra, the intensity of the first overtone C = O stretches drops by at least a factor of 10 from the intensity of the fundamental C = O stretches. In the MG C = O first overtone spectrum in Figure 2C, the relative intensity of 2ν8∶2ν9 = 1.00∶0.23, whereas in Figure 2D it is 1.00∶0.17. This relative intensity decrease seen for ν9 in Figure 2D is consistent with the decrease in the fundamental C = O, reinforcing the suggestion that the fundamental aldehydic C = O intensity is decreasing because of hydration of the carbonyl to generate diol.
), which is attributed to the diol because of its intensity increase mirroring the increase in partial pressure of water. The MG and MGD C = O first overtone region near 3,450 cm-1 has the ketonic C = O overtone at 3,443 cm-1 (2ν8) and the aldehydic C = O overtone at 3,458 cm-1 (2ν9). In both MG and MGD spectra, the intensity of the first overtone C = O stretches drops by at least a factor of 10 from the intensity of the fundamental C = O stretches. In the MG C = O first overtone spectrum in Figure 2C, the relative intensity of 2ν8∶2ν9 = 1.00∶0.23, whereas in Figure 2D it is 1.00∶0.17. This relative intensity decrease seen for ν9 in Figure 2D is consistent with the decrease in the fundamental C = O, reinforcing the suggestion that the fundamental aldehydic C = O intensity is decreasing because of hydration of the carbonyl to generate diol.
Fig. 2.

Fundamental C = O stretching region of (A) MG and (B) MGD and the first overtone, 2ν8 and 2ν9, of (C) MG and (D) MGD showing a decrease in the relative intensity of ν9 in (B) when compared to (A) and a decrease in the 2ν8 and 2ν9 aldehydic peak in (D) when compared to (C).
OH stretching region of MGD.
In the MGD spectrum (Fig. 3), there are two OH vibrational stretching modes, with the δ denoting MGD vibration, located at 3,505 cm-1 (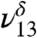 ) and 3,585 cm-1 (
) and 3,585 cm-1 (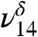 ), in addition to the 3,450 cm-1 peak assigned to 2ν8 and 2ν9. The first OH vibrational stretch at
), in addition to the 3,450 cm-1 peak assigned to 2ν8 and 2ν9. The first OH vibrational stretch at 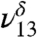 is attributed to the hydrogen-bonded OH of MGD and the
is attributed to the hydrogen-bonded OH of MGD and the 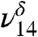 is attributed to the free OH of MGD. Our theoretical frequency puts the
is attributed to the free OH of MGD. Our theoretical frequency puts the 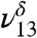 at 3,478 cm-1, which is slightly red shifted compared to the experimental value of 3,505 cm-1. This discrepancy between the theoretically and experimentally derived frequencies is consistent with results from other studies (28, 32–35). The
at 3,478 cm-1, which is slightly red shifted compared to the experimental value of 3,505 cm-1. This discrepancy between the theoretically and experimentally derived frequencies is consistent with results from other studies (28, 32–35). The 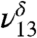 and
and 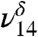 could also have a contribution from tetrol OH stretching vibrations which all fall near to those of MGD, making it difficult to distinguish or model them. In solution, the ratio of diol to tetrol is approximately 60/40 (23). Although this ratio has not been measured for the gas phase, at much lower water concentration in our gas-phase experiments we expect MGD to be favored over the tetrol. The
could also have a contribution from tetrol OH stretching vibrations which all fall near to those of MGD, making it difficult to distinguish or model them. In solution, the ratio of diol to tetrol is approximately 60/40 (23). Although this ratio has not been measured for the gas phase, at much lower water concentration in our gas-phase experiments we expect MGD to be favored over the tetrol. The 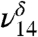 stretch is present in very small amounts even in our lowest relative humidity MG spectrum. The
stretch is present in very small amounts even in our lowest relative humidity MG spectrum. The 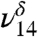 stretch increases in intensity in response to increasing the partial pressure of water in the experiments.
stretch increases in intensity in response to increasing the partial pressure of water in the experiments.
Fig. 3.

The 3,000 cm-1 to 3,650 cm-1 region of (A) MGD and (B) MG, showing the appearance of water clusters (3,100 cm-1 to 3,350 cm-1), the 2ν8 and 2ν9 of C = O, and the 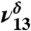 and
and 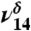 of MGD.
of MGD.
The region from 3,100 to 3,350 cm-1 contains water clusters and hydrated complexes of MG and MGD. The long red shift relative to computations and very broad appearance of these features are consistent with observations and predictions of hydrogen-bonded water clusters (28, 32–34, 36, 37). The broad and overlapping features in this region make it difficult to qualitatively and quantitatively identify these clusters.
Determination of MG, MGD, and Water Partial Pressures.
Based on the spectroscopic identification of MGD, we quantified the amount of MG, MGD, and water present in each spectrum as outlined below. Spectra that were saturated were not used in the calculation of partial pressures, but were useful in making MG and MGD mode assignments.
Partial pressure of water.
Experimentally observed spectroscopic water lines were compared with high-resolution transmission molecular absorption database (HITRAN) water lines and line strengths to determine the partial pressure of water (Table 2), PH2O, and percent relative humidity, RH%, in each experiment. Three water lines at 1,550; 1,918; and 3,920 cm-1 were isolated and analyzed. Each observed water line was integrated and the H2O number density (N/v) was determined via Eq. 1:
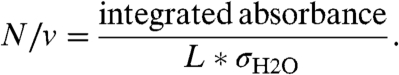 |
[1] |
The path length of the cell, L, was 71 cm and σH2O (cm molecule-1) is the HITRAN line strength for each waterline. The PH2O was determined using the average N/v from the three water lines and converted to %RH using the ideal gas law and the vapor pressure of water.
Table 2.
Partial pressure of water
| Experiment | Int abs 1,550 cm-1 | Int abs 1,918 cm-1 | Int abs 3,920 cm-1 | Avg N/v, molecule cm-3 | PH2O, atm |
| 1 | 0.003 | 0.003 | 0.002 | 1.3 × 1015 | 5.4 × 10-5 |
| 2 | 0.003 | 0.003 | 0.002 | 1.3 × 1015 | 5.4 × 10-5 |
| 3 | 0.003 | 0.003 | 0.002 | 1.3 × 1015 | 5.4 × 10-5 |
| 4 | 0.008 | 0.006 | 0.005 | 3.2 × 1015 | 1.3 × 10-4 |
Partial pressures, PH2O, of water determined from experimental spectra and HITRAN. Integrated absorbance (Int abs) for each experimental water line was taken. Water lines at 1,550; 1,918; and 3,920 cm-1 had the following cross-sections: 2.88 × 1020, 2.93 × 1020, and 2.61 × 1020 cm molecule-1 (55).
Partial pressure of MG.
The partial pressure of MG, PMG, was determined spectroscopically using the integrated absorbance of the MG CH stretch at 2,835 cm-1 (Table 3) and a theoretically calculated MG cross section, σMG, of 1.19 × 10-17 cm molecule-1 (Table 1). The theoretical and literature value for σMG were compared: There is a discrepancy, with the literature value being higher (31). In this work we chose to use the theoretically calculated σMG because of the agreement of the calculated and experimental frequencies and intensities (Table 1). For consistency, the theoretical σMG was used to determine the KP from these experiments. The MG N/v was determined for each experiment using Eq. 1, and the PMG was then determined using the ideal gas law.
Table 3.
Partial pressures of MG
| Experiment | Int abs, cm-1 | N/v, molecule cm-3 | PMG, atm |
| 1 | 26.05 | 3.08 × 1016 | 1.25 × 10-3 |
| 2 | 25.93 | 3.07 × 1016 | 1.24 × 10-3 |
| 3 | 25.66 | 3.04 × 1016 | 1.23 × 10-3 |
| 4 | 33.15 | 3.92 × 1016 | 1.59 × 10-3 |
Partial pressures of MG, PMG, determined from experimental spectra. Integrated absorbance (Int abs) for the MG CH stretch at 2,835 cm-1 was taken. Theoretical calculated σMG of 1.19 × 10-19 cm molecule-1 is used.
Partial pressure of MGD.
The partial pressure of MGD, PMGD, was determined spectroscopically using the MGD OH stretching vibration at 3,585 cm-1 (Table 4). This peak was used because of its presence in each of the spectra containing MGD and its response to water. It is assumed that this peak contains only MGD (although it may also contain a small tetrol contribution) and this feature was integrated to obtain an integrated absorbance. The integrated absorbance along with the theoretically determined cross-section, σOH, of 9.14 × 10-18 cm molecule-1, which is in agreement with a typical alcohol cross-section (34, 38, 39), was used in Eq. 1 to determine MGD N/v. The PMGD was then determined using Eq. 1 and ideal gas law. The experimentally obtained partial pressures are shown in Table 5.
Table 4.
Partial pressures of MGD
| Experiment | Int abs, cm-1 | N/v, molecule cm-3 | PMGD, atm |
| 1 | 0.172 | 2.65 × 1014 | 1.08 × 10-5 |
| 2 | 0.169 | 2.60 × 1014 | 1.06 × 10-5 |
| 3 | 0.171 | 2.63 × 1014 | 1.07 × 10-5 |
| 4 | 0.505 | 7.78 × 1014 | 3.16 × 10-5 |
Partial pressures of MGD, PMGD, determined from experimental spectra. Integrated absorbance (Int abs) for the MGD Fr OH stretch at 3,585 cm-1 was taken. Theoretically calculated σOH of 9.14 × 10-18 cm molecule-1 is used.
Table 5.
Experimental equilibrium constant and gibbs free energy
| Experiment | PMGD, atm | PH2O, atm | PMG, atm | KP | ΔG°, kcal mol-1 |
| 1 | 1.08 × 10-5 | 1.25 × 10-3 | 5.40 × 10-5 | 159 | -3.13 |
| 2 | 1.06 × 10-5 | 1.24 × 10-3 | 5.40 × 10-5 | 157 | -3.12 |
| 3 | 1.07 × 10-5 | 1.23 × 10-3 | 5.40 × 10-5 | 161 | -3.13 |
| 4 | 3.16 × 10-5 | 1.59 × 10-3 | 1.29 × 10-4 | 154 | -2.98 |
Experimentally determined gas-phase water-mediated equilibrium constant, KP, values calculated using Eq. 2 for reaction R1 between MG and MGD and the experimentally determined Gibbs free energy, ΔG°, values calculated using Eq. 3. Temperature was 298 K. KP values have an error of ± 13.0 and ΔG° values have an error of ± 0.05 kcal mol-1.
Water-Mediated Gas-Phase Equilibrium Constant.
Previous MG hydration studies have been primarily performed in the solution phase (21, 22). There have been few thermodynamic studies of the hydration of MG and none performed in the gas phase. As shown in Table 5, under very dry conditions like those used in this study (RH% < 5%), easily detectable quantities of MGD are formed. Assuming that this reaction involves equilibrium R1, and that the tetrol formation is inefficient under conditions like those in our investigation, the equilibrium constant KP can be derived from the partial pressures extracted from our spectra using Eq. 2.
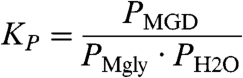 |
[2] |
The KP values for the four experiments (Table 5) fall within the range of 154–161 (± 13.0) and reflect MGD production. The low relative humidity conditions of the experiments did not allow for the water concentration to be varied over a large range and there was little clustering observed by FTIR. As noted previously, there was a discrepancy between the theoretical and literature σMG, which leads to ∼11% discrepancy in the KP values determined using the same spectra, with the theoretical values being slightly higher than the literature KP values.
The Gibbs free energy (ΔG°) was calculated from KP using Eq. 3
| [3] |
Our experimentally determined ΔG° values, which range from -2.98 to -3.10 kcal mol-1 (± 0.051) (Table 5) reflects production of MGD in the gas phase. These values for MGD formation are significantly more favorable than theoretical predictions, even ones made in the aqueous phase (21, 40).
Experimental.
Sample preparation.
MG(CH3COCHO) in aqueous solution (40 wt%) (Sigma-Aldrich) was dried and distilled before use. A 20 mL sample of MG solution was attached to a vacuum line at around 15 m torr and gently heated and stirred at 313 K for ∼12–15 h. Under these conditions, the solution became increasingly viscous. An equimolar amount of P2O5 was added to the sample, which was heated to 393 K and put under vacuum. Two condensers were employed in the distillation, each cooled with cold water, similar to Gurnick et al. (41). A liquid nitrogen bath was used to trap the purified MG. The MG solution was orange-brown in color in contrast to the pure MG, which is bright yellow-green, and MGD, which is colorless. The MG was put under nitrogen and kept frozen so it would remain in its aldehydic form until the experiment. The MGD was prepared in situ, by introducing small amounts of water to the sample in the spectroscopic cell, and allowing MG and water vapor to equilibrate over a period of about 10–30 min.
Fourier transformation infrared spectra.
The mid-IR absorption spectra of MG and MGD were measured between 1,000 and 8,000 cm-1 using FTIR spectroscopy at 0.5 cm-1 resolution in a static cell. A Bruker IFSv 66 spectrometer equipped with a globar source, KBr beamsplitter, and mercury cadmium telluride or MCT detector was used for optimal mid-IR absorption spectroscopy. This setup has been described previously (42, 43). The spectroscopic cell was both pressure- and temperature-controlled and was pumped down to approximately 15 m torr and then closed off to the pump. The MG sample was introduced to the cell and water vapor was subsequently added and allowed to equilibrate. All experiments were performed at 298 K.
Theoretical calculations.
The structures and fundamental vibrational mode frequencies of MG and MGD were calculated using the hybrid density functional theory method of B3LYP (44, 45) with the 6 - 31 + G(d,p) basis set (46–50) using the Gaussian 03 program (48). The anharmonic frequencies using the perturbation theory developed by Barone (51) using the B3LYP method is reported along with the intensity calculated by the double harmonic approximation. The optimized structures indicate that trans-MG is the most stable structure of MG, with the cis-MG structure being 4.9 kcal mol-1 higher in energy. Calculations also confirmed that MG would hydrate at the aldehydic group. The theoretical frequencies and intensities along with the experimental frequencies are presented in Table 1.
Conclusion
MG is a known product of VOC oxidation and is prevalent in the atmosphere. In this work, we obtain and assign the gas-phase IR spectrum of MGD and find that the gas-phase hydration of MG is possible at low relative humidity (RH% < 5%). One of the consequences of this gas-phase water-mediated chemistry is a change in the electronic state of the molecule, eliminating the n → π∗ transition of the aldehyde carbonyl which is well known to undergo near-UV photochemistry (52–54). Instead, the OH vibrational chromophore of the diol may react through excitation of the OH vibrational overtone in the near IR to form new products by dehydration, decarboxylation, and decarbonylation, as suggested recently for a number of alcohols and acids (32, 55–58). Using MG and MGD spectral features, we determine the gas-phase water-mediated equilibrium KP. This KP suggests that the gas-phase formation of the diol in the atmosphere is possible and could be expected to affect gas-particle partitioning of MG and its potential to form SOA.
Acknowledgments.
V.V. and J.A. would like to thank B. Ervens and K. Plath for their insightful comments and R. Talukdar and J.Brukholder for sharing unpublished MG spectra. V.V., D.D.H., and J.A. thank the National Science Foundation for support of this work. D.D.H. acknowledges a Cooperative Institute for Research in Environmental Sciences Fellowship. J.A acknowledges a NASA Earth and Space Science Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Solomon S, et al. IPCC, 2007: Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge, UK: Cambridge Univ Press; 2007. [Google Scholar]
- 2.Kanakidou M, et al. Organic aerosol and global climate modelling: A review. Atmos Chem Phys. 2005;5:1053–1123. [Google Scholar]
- 3.De Gouw J, Jimenez JL. Organic aerosols in the Earth’s atmosphere. Environ Sci Technol. 2009;43(20):7614–7618. doi: 10.1021/es9006004. [DOI] [PubMed] [Google Scholar]
- 4.Rincon AG, Guzman MI, Hoffmann MR, Colussi AJ. Optical absorptivity versus molecular composition of model organic aerosol matter. J Phys Chem A. 2009;113(39):10512–10520. doi: 10.1021/jp904644n. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Q, et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys Res Lett. 2007;34(13):L13801. [Google Scholar]
- 6.Heald CL, et al. A large organic aerosol source in the free troposphere missing from current models. Geophys Res Lett. 2005;32(18):L18809. [Google Scholar]
- 7.Volkamer R, et al. Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected. Geophys Res Lett. 2006;33(17):L17811. [Google Scholar]
- 8.Finlayson-Pitts BJ. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols. Phys Chem Chem Phys. 2009;11(36):7760–7779. doi: 10.1039/b906540g. [DOI] [PubMed] [Google Scholar]
- 9.Fast J, et al. Evaluating simulated primary anthropogenic and biomass burning organic aerosols during MILAGRO: Implications for assessing treatments of secondary organic aerosols. Atmos Chem Phys. 2009;9(16):6191–6215. [Google Scholar]
- 10.De Gouw JA, et al. Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002. J Geophys Res-Atmos. 2005;110(D16):D16305. [Google Scholar]
- 11.Ervens B, Feingold G, Frost GJ, Kreidenweis SM. A modeling study of aqueous production of dicarboxylic acids: 1. Chemical pathways and speciated organic mass production. J Geophys Res-Atmos. 2004;109(D15):D15205. [Google Scholar]
- 12.Tervahattu H, Juhanoja J, Kupiainen K. Identification of an organic coating on marine aerosol particles by TOF-SIMS. J Geophys Res-Atmos. 2002;107(D16):1–7. [Google Scholar]
- 13.Veres P, et al. Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for the measurement of gas-phase organic acids in the atmosphere. Int J Mass Spectrom. 2008;274(1-3):48–55. [Google Scholar]
- 14.Altieri KE, et al. Oligomers formed through in-cloud methylglyoxal reactions: Chemical composition, properties, and mechanisms investigated by ultra-high resolution FT-ICR mass spectrometry. Atmos Environ. 2008;42(7):1476–1490. [Google Scholar]
- 15.Ervens B, et al. Secondary organic aerosol yields from cloud-processing of isoprene oxidation products. Geophys Res Lett. 2008;35(2):L02816. [Google Scholar]
- 16.Carlton AG, et al. Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments. Atmos Environ. 2007;41(35):7588–7602. [Google Scholar]
- 17.Warneck P. In-cloud chemistry opens pathway to the formation of oxalic acid in the marine atmosphere. Atmos Environ. 2003;37(17):2423–2427. [Google Scholar]
- 18.Fu TM, et al. Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J Geophys Res-Atmos. 2008;113(D15):D15303. [Google Scholar]
- 19.Jang MS, Kamens RM. Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene. Environ Sci Technol. 2001;35(18):3626–3639. doi: 10.1021/es010676+. [DOI] [PubMed] [Google Scholar]
- 20.Tuazon EC, Macleod H, Atkinson R, Carter WPL. Alpha-dicarbonyl yields from the NOX-air photooxidations of a series of aromatic-hydrocarbons in air. Environ Sci Technol. 1986;20(4):383–387. doi: 10.1021/es00146a010. [DOI] [PubMed] [Google Scholar]
- 21.Krizner HE, De Haan DO, Kua J. Thermodynamics and kinetics of methylglyoxal dimer formation: A computational study. J Phys Chem A. 2009;113(25):6994–7001. doi: 10.1021/jp903213k. [DOI] [PubMed] [Google Scholar]
- 22.Betterton EA, Hoffmann MR. Kinetics, mechanism, and thermodynamics of the reversable-reaction of methylglyoxal (CH3COCHO) with S(IV) J Phys Chem. 1987;91(11):3011–3020. [Google Scholar]
- 23.Nemet I, Vikic-Topic D, Varga-Defterdarovic L. Spectroscopic studies of methylglyoxal in water and dimethylsulfoxide. Bioorg Chem. 2004;32(6):560–570. doi: 10.1016/j.bioorg.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 24.Loeffler KW, Koehler CA, Paul NM, De Haan DO. Oligomer formation in evaporating aqueous glyoxal and methyl glyoxal solutions. Environ Sci Technol. 2006;40(20):6318–6323. doi: 10.1021/es060810w. [DOI] [PubMed] [Google Scholar]
- 25.Bell RP, Rand MH, Wynnejones KMA. Kinetics of the hydartion of acetaldehyde. T Faraday Soc. 52(8):1093–1102. [Google Scholar]
- 26.Buschmann HJ, Dutkiewicz E, Knoche W. The reversable hydration of carbonyl-compounds in aqueous-solution 2. The kinetics of the keto gem-diol transition. Ber Bunsen Phys Chem. 1982;86(2):129–134. [Google Scholar]
- 27.Wolfe S, Kim CK, Yang K, Weinberg N, Shi Z. Hydration of the carbonyl group—A theoretical-study of the cooperative mechanism. J Am Chem Soc. 1995;117(15):4240–4260. [Google Scholar]
- 28.Plath KL, et al. Gas-phase vibrational spectra of glyoxylic acid and its gem diol monohydrate Implications for atmospheric chemistry. React Kinet Catal L. 96(2):209–224. [Google Scholar]
- 29.Bao L, et al. Gas/particle partioning of low-molecular-weight diacarboxylic acids at the suburban site in Saitama, Japan. Atmos Environ. 2009 in press. [Google Scholar]
- 30.Matsunaga S, Mochida M, Kawamura K. Variation on the atmospheric concentrations of biogenic carbonyl compounds and their removal processes in the northern forest at Moshiri, Hokkaido Island in Japan. J Geophys Res-Atmos. 2004;109(D4):D04302. [Google Scholar]
- 31.Zhou SM, et al. Atmospheric Chemistry of Acetylacetone. Environ Sci Technol. 2008;42(21):7905–7910. doi: 10.1021/es8010282. [DOI] [PubMed] [Google Scholar]
- 32.Havey DK, Feierabend KJ, Vaida V. Vapor-phase vibrational spectrum of glycolic acid, CH2OHCOOH, in the region 2000–8500 cm-1. J Phys Chem A. 2004;108(42):9069–9073. [Google Scholar]
- 33.Plath KL, Takahashi K, Skodje RT, Vaida V. Fundamental and Overtone vibrational spectra of gas-phase pyruvic acid. J Phys Chem A. 2009;113(26):7294–7303. doi: 10.1021/jp810687t. [DOI] [PubMed] [Google Scholar]
- 34.Vaida V, Feierabend KJ, Rontu N, Takahashi K. Sunlight-initiated photochemistry: Excited vibrational states of atmospheric chromophores. Int J Photoenergy. 2008;2008:1–13. [Google Scholar]
- 35.Vaida V. Spectroscopy of photoreactive systems: Implications for atmospheric chemistry. J Phys Chem A. 2009;113(1):5–18. doi: 10.1021/jp806365r. [DOI] [PubMed] [Google Scholar]
- 36.Vaida V, Daniel JS, Kjaergaard HG, Goss LM, Tuck AF. Atmospheric absorption of near infrared and visible solar radiation by the hydrogen bonded water dimer. Q J Roy Meteor Soc. 2001;127(575):1627–1643. [Google Scholar]
- 37.Vaida V, Kjaergaard HG, Feierabend KJ. Hydrated complexes: Relevance to atmospheric chemistry and climate. Int Rev Phys Chem. 2003;22(1):203–219. [Google Scholar]
- 38.Lange KR, Wells NP, Plegge KS, Phillips JA. Integrated intensities of O-H stretching bands: Fundamentals and overtones in vapor-phase alcohols and acids. J Phys Chem A. 2001;105(14):3481–3486. [Google Scholar]
- 39.Miller BJ, Kjaergaard HG, Hattori K, Ishiuchi S, Fujii M. The most stable conformer of benzyl alcohol. Chem Phys Lett. 2008;466(1-3):21–26. [Google Scholar]
- 40.Barsanti KC, Pankow JF. Thermodynamics of the formation of atmospheric organic particulate matter by accretion reactions - 2 Dialdehydes, methylglyoxal, and diketones. Atmos Environ. 39(35):6597–6607. [Google Scholar]
- 41.Gurnick M, Chaiken J, Benson T, McDonald JD. Vibrational and rotational spectroscopy of the 1st electronically allowed transition of alpha-dicarbonyls. J Chem Phys. 1981;74(1):99–105. [Google Scholar]
- 42.Rontu N, Vaida V. Vibrational spectroscopy of perfluoroproplonic acid in the region between 1000 and 11000 cm-1. J Mol Spectrosc. 2006;237(1):19–26. [Google Scholar]
- 43.Dunn ME, Shields GC, Takahashi K, Skodje RT, Vaida V. Experimental and theoretical study of the OH vibrational spectra and overtone chemistry of gas-phase vinylacetic acid. J Phys Chem A. 2008;112(41):10226–10235. doi: 10.1021/jp805746t. [DOI] [PubMed] [Google Scholar]
- 44.Becke AD. Density-functional thermochemistry 3. The role of exact exchange. J Chem Phys. 1993;98(7):5648–5652. [Google Scholar]
- 45.Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys Rev B. 1988;37(2):785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 46.Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PV. Efficient diffuse function-augmented basis-sets for anion calculations 3. The 3 - 21 + G basis set for 1st-row elements, LI-F. J Comput Chem. 4(3):294–301. [Google Scholar]
- 47.Frisch MJ, Pople JA, Binkley JS. Self-consistent molecular-orbital methods. 25. Supplementary functions for Gaussian-basis sets. J Chem Phys. 1984;80(7):3265–3269. [Google Scholar]
- 48.Frisch MJ, et al. Gaussian03. C.02 Ed. Wallingford, CT: Gaussian Inc; 2004. [Google Scholar]
- 49.Krishnan R, Binkley JS, Seeger R, Pople JA. Self-consistent molecular-orbital methods. 20. basis set for correlated wave-functions. J Chem Phys. 1980;72(1):650–654. [Google Scholar]
- 50.McLean AD, Chandler GS. Contracted Gaussian-basis sets for molecular calculations. 1. Second row atoms, Z = 11–18. J Chem Phys. 1980;72(10):5639–5648. [Google Scholar]
- 51.Barone V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J Chem Phys. 2005;122(1):014108. doi: 10.1063/1.1824881. [DOI] [PubMed] [Google Scholar]
- 52.Chen YQ, Wang WJ, Zhu L. Wavelength-dependent photolysis of methylglyoxal in the 290–440 nm region. J Phys Chem A. 2000;104(47):11126–11131. [Google Scholar]
- 53.Staffelbach TA, Orlando JJ, Tyndall GS, Calvert JG. The UV-visable absorption-spectrum and photolysis quantum yields of methylglyoxal. J Geophys Res-Atmos. 1995;100(d7):14189–14198. [Google Scholar]
- 54.Meller R, Raber W, Crowley JN, Jenkin ME, Moortgat GK. The UV-visable absorption-spectrum of methylglyoxal. J Photoch Photobio A. 1991;62(2):163–171. [Google Scholar]
- 55.Takahashi K, Plath KL, Skodje RT, Vaida V. Dynamics of vibrational overtone excited pyruvic acid in the gas phase: Line broadening through hydrogen-atom chattering. J Phys Chem A. 2008;112(32):7321–7331. doi: 10.1021/jp803225c. [DOI] [PubMed] [Google Scholar]
- 56.Donaldson DJ, Tuck AF, Vaida V. Atmospheric photochemistry via vibrational overtone absorption. Chem Rev. 2003;103(12):4717–4729. doi: 10.1021/cr0206519. [DOI] [PubMed] [Google Scholar]
- 57.Mills MJ, et al. Photolysis of sulfuric acid vapor by visible light as a source of the polar stratospheric CN layer. J Geophys Res-Atmos. 2005;110(D8):D08201. [Google Scholar]
- 58.Vaida V, Kjaergaard HG, Hintze PE, Donaldson DJ. Photolysis of sulfuric acid vapor by visible solar radiation. Science. 2003;299(5612):1566–1568. doi: 10.1126/science.1079297. [DOI] [PubMed] [Google Scholar]