

Abstract
We previously described a population of regulatory macrophages that produced high levels of IL-10 and low levels of IL-12/23. We now describe and characterize the expression of heparin-binding epidermal growth factor (EGF)-like growth factor (HB-EGF) by these macrophages. HB-EGF has previously been associated with a number of physiological and pathological conditions, including tumor growth and angiogenesis. The induction of HB-EGF in regulatory macrophages is due to new transcription and not to increased mRNA stability. The transcription factor Sp1 is a major factor in HB-EGF production, and knockdown of Sp1 substantially diminishes HB-EGF production. Sp1 was recruited to three sites within the first 2 kb of the HB-EGF promoter following stimulation, and the site located at –83/–54 was required for HB-EGF promoter activity. These regions of the promoter become more accessible to endonuclease activity following macrophage activation, and this accessibility was contingent on activation of the MAPK, ERK. We show that several experimental manipulations that give rise to regulatory macrophages also result in HB-EGF production. These observations indicate that in addition to the secretion of the anti-inflammatory cytokine IL-10, another novel characteristic of regulatory macrophages is the production of angiogenic HB-EGF.
We and others previously described a population of regulatory macrophages that secretes high levels of IL-10 and low levels of IL-12/23 (1). The IL-10 produced by these cells can render macrophages refractory to the activating effects of IFN-γ, and it can bias T cells to produce IL-4 and IL-10 (2, 3). We (1) originally identified these cells by activating macrophages in vitro in the presence of immune complexes (IC).3 Immune complexes interact with macrophage FcγR and initiate a signal transduction cascade that results in the development of regulatory macrophages. Other groups, using different stimuli including cAMP, purinergic receptor ligands, and glucocorticoids have identified macrophages with regulatory characteristics (4). Regulatory macrophages have been identified in parasitic infections and have been shown to contribute to parasite persistence (5). The production of IL-10 from these regulatory macrophages can reverse lethal endotoxemia (6). Recent studies suggest that tumor-associated macrophages and macrophages in atherosclerotic lesions may share characteristics of regulatory macrophages (7–9). Thus, we think that these macrophages may play important roles in a variety of pathological conditions.
Heparin-binding epidermal growth factor (EGF)-like growth factor (HB-EGF) was originally identified in the culture supernatants of the U-937 macrophage-like cell line (10). It was found to be mitogenic for a number of cell types, including fibroblasts, smooth muscle cells, and a number of others. HB-EGF is synthesized as a transmembrane precursor (pro-HBEGF) that can serve as a juxtacrine growth factor (11), and in some species a receptor for diphtheria toxin (12). A number of proteases have been implicated as being responsible for ectodomain shedding, resulting in the formation of soluble HB-EGF (sHB-EGF). These include matrix metalloproteinase (MMP) 3, MMP9, a disintegrin and metalloproteinase (ADAM) 9, ADAM10, ADAM12, and ADAM17 (reviewed in Ref. 13). The resulting C-terminal (membrane-associated) fragment of pro-HB-EGF can contribute to cell cycle progression by translocating to the nucleus and interacting with promyelocytic leukemia zinc finger, the transcriptional repressor of cyclin A (14), or with Bcl6, the transcriptional repressor of cyclin D2 (15). sHB-EGF was shown originally to signal through the EGFR (ErbB1; Ref. 10) but was later demonstrated to also bind ErbB2 and ErbB4 (16, 17).
HB-EGF has been found to play a role in several normal physiological processes, including proper heart (17) and eyelid (18) formation and skin wound healing (19), by inducing keratinocyte migration. It is also associated with a number of pathological conditions. Macrophages, T cells, and vascular smooth muscle cells (SMC) of atherosclerotic plaques have been found to express HBEGF (20, 21). Furthermore, not only is HB-EGF a potent mitogen for SMCs, but it also induces the expression of LOX-1, the receptor for oxidized low-density lipoprotein, by SMCs, potentially aiding in foam cell formation. Furthermore, HB-EGF has recently been shown to be required for low-flow-induced hypertrophic remodeling, further demonstrating a potential role in vascular wall pathology (22).
HB-EGF has also been shown to be an important regulator of tumor growth and angiogenesis. In vitro, HB-EGF has been shown to increase the growth rate of tumor cells and to induce the expression of vascular EGF, and in vivo to strikingly increase angiogenic potential and tumorigenicity (23). Recently, it was shown that HB-EGF may contribute to angiogenesis primarily by driving remodeling of vascular endothelial cells (24). HB-EGF expression was enhanced in many tumors (Ref. 25; reviewed in Ref. 26). HB-EGF can also contribute to chemotherapy resistance (27). Bile acids, which have been implicated as cofactors of colon carcino-genesis, may mediate their activity through the up-regulation and activation of MMP-7, which results in increased shedding of HBEGF and thus proliferation of a human colon cancer cell line (28).
In this study, we describe the induction of HB-EGF by regulatory macrophages and correlate the increased transcription of HBEGF with the activation of two MAPKs, ERK and p38. We show that the activation of ERK results in increased accessibility of the HB-EGF promoter to the transcription factor Sp1, allowing it to initiate transcription.
Materials and Methods
Reagents
The MEK/ERK inhibitor U0126, p38 inhibitor SB203580, and JNK inhibitor II were obtained from Calbiochem (EMD Biosciences) and used in concentrations that were previously optimized for macrophages (29). Actinomycin D, cycloheximide, and N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate (dbcAMP) were purchased from Sigma-Aldrich. Macrophages were pretreated with inhibitors 1 h before stimulation at concentrations given in the figures. ChIP-grade anti-Sp1 and histone H3 Abs were purchased from Upstate Biotechnology. TRIzol reagent and DNase I were purchased from Invitrogen Life Technologies. Klenow enzyme and restriction enzymes were purchased from New England Biolabs. PGE2 was purchased from Caymen Chemical.
Mice
Six- to 8-wk-old BALB/c mice were purchased from Taconic Farms. IL-10−/− mice were purchased from The Jackson Laboratory. Mice were used at 6–8 wk of age as a source of bone marrow-derived Mϕs (BMMϕ). All mice were maintained in high-efficiency particulate air-filtered Thoren units (Thoren Caging Systems) at the University of Maryland (College Park, MD). All procedures were reviewed and approved by the University of Maryland Institutional Animal Care and Use Committee.
Cells and macrophage activation
BMMϕ were prepared as described previously (30). Briefly, bone marrow was flushed from the femurs and tibias of mice, and cells were plated in petri dishes in DMEM-F-12 supplemented with 10% FBS, penicillin/streptomycin, glutamine, and 20% conditioned medium from the supernatants of macrophage-CSF-secreting L929 (LC14) fibroblasts (L cell-conditioned medium). Cells were refed on day 2. Cells were used at 7–10 days for experiments. The RAW264.7 macrophage-like cell line was obtained from the American Type Culture Collection. RAW262.7 cells were maintained in RPMI 1640 supplemented with 10% FBS, penicillin/streptomycin, and glutamine.
Macrophages were activated with LPS or LPS plus ICs, as previously described (2). Briefly, macrophages were harvested from petri dishes and plated into 48-well plates (2 × 105 macrophages) for cytokine measurement and six-well plates (2 × 106 macrophages) for RNA isolation overnight in complete medium without L cell-conditioned medium. The following morning, medium was changed and macrophages were treated with 10 ng/ml Ultra-Pure LPS (Escherichia coli K12; Invivogen) or LPS plus IC. IgG-OVA ICs were made as previously described (2). Briefly, ICs were made by mixing a 10-fold molar excess of rabbit anti-OVA IgG (Cappel) to OVA (Worthington) for 30 min at room temperature. As previously described, any contaminating endotoxin was removed from the anti-OVA IgG before immune complex preparation using EndoClean from BioVitage as described (30). Additionally, macrophages were stimulated with PGE2 (10−8 M) or dbcAMP (100 μM) in the presence or absence of LPS (10 ng/ml).
A murine aortic smooth muscle cell line, MOVAS, was obtained from the American Type Culture Collection and maintained in DMEM-F-12 medium supplemented with 6% FCS, glutamine, penicillin/streptomycin, and 100 μg/ml G418. G418 was removed before experimentation. Cell proliferation experiments were conducted using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) as described in the manufacturer's instructions.
RNA isolation and real-time PCR
BMMϕs were subject to RNA extraction using TRIzol reagent. The contaminating DNA was then removed by DNase I treatment. ThermoScript RT-PCR system (Invitrogen Life Technologies) was used to generate cDNA from RNA by using oligo(dT)20 primers. Real-time PCR was conducted with the ABI Prism 7700 or Roche LightCycler 480 sequence detection system using iQ SYBR Green Supermix (Bio-Rad Laboratories) following the manufacturer's instructions. Standard PCR analysis was performed using Platinum PCR Supermix (Invitrogen Life Technologies). The following primer pairs were used in this study: HB-EGF (NM_010415), 5′-CAGGACTTGGAAGGGACAGA-3′ and 5′-GGCATTTGCAA GAGGGAGTA-3′; GAPDH (NM_001001303), 5′-TGTTCCTACC CCCAATGTGT-3′ and 5′-GGTCCTCAGTGTAGCCCAAG-3′; IL-10 (NM_010548), 5′-AAGGACCAGCTGGACAACAT-3′ and 5′-TCTCAC CCAGGGAATTCAAA-3′; LOX-1 (NM_138648), 5′-TCATGTGGCAA GAAGCCTAA-3′ and 5′-GCTGAGTAAGGTTCGCTTGG-3′. For data analysis, the comparative threshold cycle (Ct) value for GAPDH was used to normalize loading variations in the real-time PCR reactions. A ΔΔCt value was then obtained by subtracting control ΔCt values from the corresponding experimental ΔCt. The ΔΔCt values were converted to fold difference compared with the control by raising two to the ΔΔCt power.
RNA stability assay
BMMϕs(2 × 106) were stimulated for 2 h with LPS or LPS plus IC before the addition of actinomycin D to a final concentration of 0.5 μg/ml. HBEGF mRNA was subsequently measured by quantitative real-time PCR (QRT-PCR) over the following 2 h.
Immunoprecipitation and Western blot analysis
sHB-EGF was immunoprecipitated using 5 μg of polyclonal goat anti-mouse HB-EGF (M-18; Santa Cruz Biotechnology) per ml of cell culture supernatant. Samples were subjected to SDS-PAGE on 15% resolving gels and transferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were blotted with goat anti-mouse HB-EGF (1/200 dilution) and HRP-conjugated mouse anti-goat IgG secondary Ab (1/10,000) (Santa Cruz). For experiments performed to determine MAPK activation, cells were stimulated and lysed at the indicated times in ice-cold lysis buffer (100 mM Tris (pH 8), 2 mM EDTA, 100 mM NaCl, 1% Triton X-100 containing complete EDTA-free protease inhibitors from Roche Diagnostics, which included 5 mM sodium vanadate, 10 mM sodium fluoride, 10 mM β-glycerophosphate sodium, and 5 mM sodium pyrophosphate). Equal amounts of protein were loaded onto 10% SDS-polyacrylamide gels. Anti-MAPK and anti-phospo-MAPK Abs were purchased from Cell Signaling Technologies. Membranes were developed using ECL Western Blotting Detection Reagents (Amersham Biosciences) according to the manufacturer's instructions.
EMSA
Probes corresponding to potential Sp1-binding sites were generated from the following oligo pairs: consensus, 5′-CTGCGGGGCGGGGCA-3′ and 5′-TCTGCCCCGCCCC-3′; −348/−312, 5′-GGAAGGGGGCGGT GCCGGGCGGGGCGG-3′ and 5′-GGAGCCCCGCCCCGCCCGGCACC GCCCCC-3′;−1277/−1258,5′-AAGTGGGGGTGGGGTG-3′and5′-TCT CCACCCCACCCCC-3′; and −1828/−1809, 5′-CCCCACCCCCACCC CC-3′ and 5′-CCCTGGGGGTGGGGGT-3′. Oligo pairs were annealed by heating to >95°C in a heating block and then allowed to cool to room temperature over several hours. Probes were then radiolabeled using [α-32P]dGTP by the Klenow (fill-in) method. Nuclear extracts were prepared from 1 × 107 RAW264.7 cells as previously described (31). These RAW264.7 macrophages respond similarly to primary macrophages with regard to their HB-EGF induction in response to LPS and LPS plus IC.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were conducted using the ChIP assay kit (Upstate Biotechnology) following the manufacturer's protocol. DNA was sheared using a Cole-Palmer ultrasonic processor (Cole-Parmer Instrument). This resulted in relatively uniform DNA fragment size of ~300 bp (30). The remaining procedures were conducted as previously described (30). HB-EGF (NC_000084) promoter primers used for ChIP analysis are presented in Table I.
Table I.
Schematic diagram and table of primers used for analysis of the HB-EGF promoter

| Amplicon | Primers |
|---|---|
| 1 | 5′-GGTTCCCAGACAGGATCTCA-3′ |
| 5′-AGAAAGAGCTTCAGCATCACC-3′ | |
| 2 | 5′-CTTCCCCGGAGCCTTATTC-3′ |
| 5′-TGAGATCCTGTCTGGGAACC-3′ | |
| 3 | 5′-CCCTACACCCACACTCCAGT-3′ |
| 5′-GAATAAGGCTCCGGGGAAG-3′ | |
| 4 | 5′-CAGATCACCTTTCGCTAGGC-3′ |
| 5′-ACTGGAGTGTGGGTGTAGGG-3′ | |
| 5 | 5′-ACTTCCTTTGCTGTTGCTGG-3′ |
| 5′-GCCTAGCGAAAGGTGATCTG-3′ | |
| 6 | 5′-GTCGAGGTTAGCTGGTTGGA-3′ |
| 5′-CCAGCAACAGCAAAGGAAGT-3′ | |
| 7 | 5′-GTCGAGGTTAGCTGGTTGGA-3′ |
| 5′-CCAGCAACAGCAAAGGAAGT-3′ | |
| 8 | 5′-ATGAAGAGGGGAAGCCAGAT-3′ |
| 5′-TGCTGGGTAGGGAGTTTGAC-3′ | |
| 9 | 5′-CCTCTTCTGACCTCTGTGGG-3′ |
| 5′-ATCTGGCTTCCCCTCTTCAT-3′ | |
| 10 | 5′-ATACTGAGCACCCAAGGTGG-3′ |
| 5′-CCCACAGAGGTCAGAAGAGG-3′ | |
| 11 | 5′-GAGAGCAGGGTGTGAATGGT-3′ |
| 5′-CCACCTTGGGTGCTCAGTAT-3′ | |
| 12 | 5′-GGTTCTCTGCAGTTCTGGGA-3′ |
| 5′-ACCATTCACACCCTGCTCTC-3′ | |
| 13 | 5′-CTTCTTTTTTCTCTTTCTTTGA-3′ |
| 5′-TCCCAGAACTGCAGAGAACC-3′ |
DNase accessibility assay
DNase accessibility assays were performed as previously described (30). Briefly, 1 × 107 BMMϕ grown on 100-mm tissue culture dishes were stimulated with LPS plus IC for the indicated times and then fixed with formaldehyde at a final concentration of 1%. Cells were scraped in cold PBS, washed, and then lysed in ice-cold Nuclei EZ lysis buffer (Sigma-Aldrich). Washed nuclei were resuspended in ice-cold DNase I buffer (100 mM NaCl, 50 mM Tris (pH 8.0), 3 mM MgCl2, 0.15 mM spermine, and 0.5 mM spermidine) supplemented with 1 mM CaCl2. DNase I (Roche Diagnostics) was added and incubated on ice for 1 h. The reaction was stopped by adding DNase stop buffer (10 mM EDTA, 20% SDS, and 0.4 M NaCl). Cross-linking was reversed by incubation at 65°C for ~4 h. Proteinase K and RNase A were added at 37°C overnight. DNA was purified by phenol-chloroform extraction and cold ethanol precipitation.
HB-EGF promoter reporter construct
Approximately 3 kb of the HB-EGF promoter were cloned using the following primers: forward, 5′-ACCTCGGAGACATGAGCTCTTAC-3′; and reverse, 5′-GCTACCCTCTCAATGAATTAAGCTA-3′. PCR was performed using the Expand Long Template PCR System (Roche Diagnostics). The PCR product was ligated into the pCRII-TOPO vector (Invitrogen) for scale-up. The cloned promoter fragment (−2704/+330, relative to transcriptional start site) was removed using NheI and EcoRV restriction enzymes, gel purified, and then ligated into the pGL4. 19[luc2CP/Neo] luciferase reporter vector (Promega). The −1238/+330 promoter construct was made by KpnI digestion of the −2704/+330 construct, gel purification of the larger fragment, and ligation. The −557/+330 promoter construct was made by digestion of the −2704/+330 construct with Tth111I and NheI restriction enzymes. The larger fragment was then gel purified, and sticky ends were filled in using Klenow enzyme. The resultant fragment was then blunt-end ligated.
Mutation of the Sp1 site in the −557/+330 promoter construct was done using the QuikChange Site-Directed Mutagenesis kit II from Stratagene according to the manufacturer's instructions. The following primers were designed using Stratagene's QuikChange primer design program: sense, 5′-GG GAGGGAAGGGGTAGGTGCCGGTAGG-GTAGGGGCTCCCACTC-3′; and antisense, 5′-GAGTGGGAGCCCCTACCCTACCGGCACC-TACCCC TTCCCTCCC-3′.
Transient transfection and luciferase activity
RAW264.7 cells were transiently transfected using the FuGENE HD transfection reagent according to the manufacturer's instructions. Briefly, 2 μg of plasmid were added to 3 μl of FuGENE diluted in 100 μl of RPMI 1640 and incubated at room temperature for 30 min. Twenty microliters of the resultant transfection complex were added per well of a 48-well plate containing ~2 × 105 RAW cells. Each reporter construct was cotransfected with the pRL-Null Renilla luciferase plasmid at a ratio of 40:1. The next morning, medium was replaced, and cells were stimulated for ~8 h. Luciferase activity was assessed using the Dual-Luciferase Reporter Assay System (Promega). Firefly lucif-erase activity was measured for each HB-EGF reporter construct and normalized using Renilla luciferase activity.
Sp1 knockdown and primary macrophage transfection
Day 6 or day 7 BMMϕs were transfected by nucleofection as previously described (29). Briefly, 5 × 106 BMMϕs were resuspended in 100 μlof cell line kit T solution (Amaxa), and nucleofection was performed using program T20. Cells were transfected with Silencer Select Pre-Designed small interfering RNA(siRNA) to murine Sp1 (Ambion) at a final concentration of 10 or 100 nM, or a scrambled sequence dsRNA. One milliliter of prewarmed medium containing 20% L cell conditioned medium was immediately added to the cuvet, and cells were transferred to tissue culture plates containing preincubated medium. Cells were allowed to recover overnight before medium was replaced. Cells were then incubated for an additional 24 h before Sp1 gene silencing was assessed, and cells were stimulated for the expression of HB-EGF. Sp1 expression was assessed using the primers 5′-CAGTGAGGGAAGAGCCTCAG-3′ and 5′-AGCGCTTCCCACAATATGAC-3′.
Results
The induction of HB-EGF mRNA and protein
We previously demonstrated that macrophages stimulated in the presence of ICs assumed a regulatory phenotype and were able to inhibit a variety of immune responses (3). We performed microarray analysis on these regulatory cells and identified a subset of genes that were overexpressed (Gene Expression Omnibus dataset GDS2041; Ref. 3). One gene, HB-EGF, which was substantially induced in regulatory macrophages was selected for further study. Macrophages stimulated with LPS plus IC synthesized relatively high levels of HB-EGF mRNA (Fig. 1A) compared with unstimulated macrophages (time 0) or with stimulated with LPS alone (Fig. 1A, dashed lines). At the peak of mRNA induction at 90 min, LPS plus IC simulated macrophages expressed 7- to 8-fold more HB-EGF mRNA than cells stimulated with LPS alone, and these elevated levels were maintained for 3 h poststimulation (Fig. 1A).
FIGURE 1.

HB-EGF is induced in BMMϕs in response to LPS and ICs. A, QRT-PCR of HB-EGF expression in BMMϕs after stimulation with LPS (10 ng/ml) alone or LPS plus IgG-OVA ICs. QRT-PCR for HB-EGF mRNA is expressed as a fold change over unstimulated conditions (0 min). B, Western blot for HB-EGF in cell lysates or immunoprecipitated (I.P.) from cell culture supernatants (Sups.) of unstimulated (NS) macrophages (M–) or macrophages stimulated with LPS (10 ng/ml) or LPS plus IC for 16 h. Values are representative of at least three independent experiments. Conditioned medium from IC plus LPS-stimulated macrophages potentiates growth and LOX-1 expression on vascular SMCs. C-E, Macrophage-conditioned medium was generated as described in Materials and Methods from macrophages treated with ICs, LPS, or a combination of LPS plus IC (LPS/IC). C, Murine aortic SMCs (MOVAS) were added to wells of a 96-well plate in DMEM-F-12 medium containing 60% conditioned medium as indicated. Cells were allowed to grow for 48 h, and CellTiter 96 AQueous One Solution Reagents (Promega) were added for 1 h at 37°C. The OD450 was recorded and subtracted from the OD of IC-treated cells. D,1 × 105 MOVAS cells were plated in 96-well plates in DMEM-F-12 medium containing different concentration of IC-, or LPS-, or IC/LPS-conditioned medium. Cells were allowed to grow for 48 h and CellTiter 96 AQueous One Solution Reagents were added as above. E,2 × 106 MOVAS cells were plated in six-well plates in DMEM-F-12 medium containing 60% conditioned medium. Total RNA was isolated after cell culture for 12 h or 24 h, and LOX-1 gene expression was analyzed as described in Materials and Methods. LOX-1 mRNA expression of each sample was normalized to GAPDH mRNA. The normalized LOX-1 mRNA expression of IC-conditioned medium-treated MOVAS cells was arbitrarily set as 1. Each data point represents the mean ± SD of two sets of experiments.
Like other members of the EGF family, HB-EGF is synthesized as a membrane-associated precursor (pro-HB-EGF) that is subsequently cleaved, yielding the active growth factor (32). To determine whether HB-EGF is secreted or retained on the cell surface, macrophages were stimulated for 24 h with LPS or LPS plus IC, and then cell culture supernatants and cell lysates were analyzed by immunoprecipitation using a polyclonal Ab specific for HBEGF. Immunoprecipitated HB-EGF was subjected to SDS-PAGE. A band corresponding to processed sHB-EGF, with a molecular mass of ~20 kDa, was detected in culture supernatants of macrophages stimulated with LPS plus IC at 24 h (Fig. 1B). Macrophages stimulated with LPS alone did not secrete detectable sHBEGF. Furthermore, pro-HB-EGF was not detected in cell lysates from any of the cells. Thus, HB-EGF is synthesized by regulatory macrophages and is rapidly cleaved to yield the soluble secreted form.
Supernatants from stimulated macrophages were added to aortic SMCs, and their growth was measured over a 48-h period. Growth was normalized to cells receiving IC alone. SMCs exposed to LPS plus IC supernatants showed more growth relative to those exposed to supernatants from macrophages stimulated with LPS alone (Fig. 1C). SMC growth was a function of supernatant concentrations, and supernatant concentrations as low as 5 and 10% were adequate to stimulate significant SMC growth (Fig. 1D). Supernatants were also analyzed for their ability to induce low-density lipoprotein receptor mRNA expression on SMCs. Real-time PCR was used to measure LOX-1 mRNA following the addition of supernatants for 12 or 24 h. At both times, LOX-1 mRNA expression was induced by macrophages stimulated with LPS, but higher when supernatants from regulatory macrophages (LPS plus IC) were added (Fig. 1E).
Induction of HB-EGF by various regulatory macrophage populations
HB-EGF expression was examined in a variety of regulatory macrophage populations that were induced by stimuli other than ICs. The readout used to show the induction of regulatory macrophages was high IL-10 production. In addition to ICs, macrophages were stimulated with PGE2 or dbcAMP in combination with LPS. Previous work demonstrated that a combination of two stimuli was required to induce regulatory macrophages (2). Stimulation of macrophages with LPS in the presence of ICs (Fig. 2A), PGE2 (Fig. 2B), or dbcAMP (Fig. 2C) enhanced the production of both IL-10 (Fig. 2, left) and HB-EGF (Fig. 2, right). None of the three stimuli alone induced HB-EGF production (Fig. 2, solid lines). Thus, HB-EGF is produced by regulatory macrophages, and like IL-10 it requires two stimuli for induction.
FIGURE 2.

Regulatory macrophages express HB-EGF. BMMϕs were stimulated with ICs (A), 10−8M PGE2 (B), or 100 μM dbcAMP (C) in the presence or absence of LPS (10 ng/ml). IL-10 (left) and HB-EGF (right) mRNA was measured at 0, 30, 60, 90, 120, 150, and 180 min by QRT-PCR and expressed relative to the maximum level of HB-EGF mRNA during the stimulation conditions. Values are representative of at least three independent experiments.
Sp1 binds to the HB-EGF promoter in situ and in vitro
The robust induction of HB-EGF mRNA in regulatory macrophages prompted us to determine which transcription factors might play a role in HB-EGF transcription. Preliminary promoter analysis using Transfac (default 85% cutoff; http://www.gene-regulation.com/pub/databases.html and Ref. 33) revealed three potential Sp1 binding sites within the first 2 kb of the HB-EGF promoter. EMSAs were performed to determine whether the predicted promoter elements could be bound by Sp1. For these assays, the macrophage-like RAW264.7 cell line was used. These cells respond similarly to primary macrophages in their HB-EGF induction, following stimulation with LPS or LPS plus IC (Supplemental Fig. 1).4
Nuclear extracts were mixed with a −86/−48 probe containing the proximal Sp1-binding site. Nuclear extracts bound to this probe (Fig. 3A, *), and this binding was competed for by increasing concentrations (10–50×) of either a cold consensus Sp1 oligo or the cold HB-EGF probe itself. A supershift analysis using mAbs to Sp1 was performed, to demonstrate that Sp1 specifically bound to this oligo (Fig. 3A, arrow). An irrelevant Ab (α-H3) failed to cause a supershift. Similar studies were performed with probes corresponding to the other two Sp1-binding sites (−1566/−1548 and −1015/−996). In all cases, nuclear extracts bound to these probes in a manner that was competed by cold consensus or HB-EGF-specific probes and supershifted by Ab to Sp1 (Supplemental Fig. 2).
FIGURE 3.
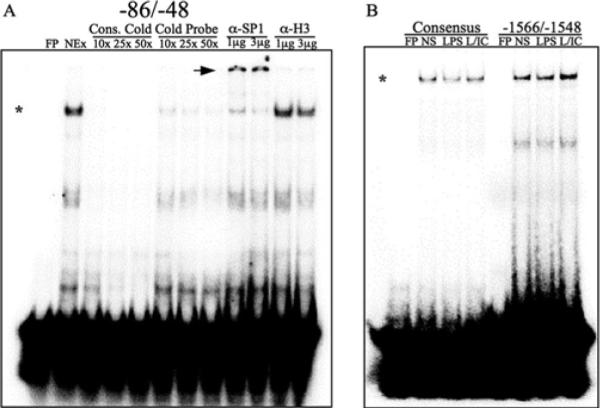
Sp1 binds to portions the HB-EGF promoter in vitro. A, 32P-labeled double-stranded oligomers corresponding to a −86/−48 fragment of the HB-EGF promoter were incubated on ice for 1 h with nuclear extracts (NEx) from LPS plus IC-stimulated (45 min) RAW264.7 cells. Binding was competed with increasing concentrations (10–50×) of cold unlabeled probe or cold consensus probe. DNA-Sp1 protein complexes were supershifted by incubating nuclear extracts with 1 or 3 μg of Ab against Sp1 or histone H3 for 30 min on ice before incubation with labeled probe. *, Sp1-DNA complexes; arrowheads, supershifted Sp1-DNA complexes. B, Labeled double-stranded oligomers corresponding to −1566/−1548 of the HB-EGF promoter were incubated with nuclear extracts from unstimulated (NS), LPS, or LPS plus IC-stimulated (L/IC) RAW264.7 cells. FP, Free probe; α-, anti-.
Despite the substantial induction of HB-EGF expression following stimulation of macrophages with LPS plus IC, to our surprise there were no detectable differences in the amount of Sp1 binding that occurred when nuclear extracts from unstimulated cells, or cells stimulated with LPS or LPS plus IC were used. All three of the probes containing Sp1 binding sites bound equal amounts of Sp1 regardless of the macrophage stimulation condition (Fig. 3B and data not shown). Thus, all macrophage nuclear extracts contained Sp1 that was competent to bind to consensus and HB-EGF-specific probes.
A ChIP assay was performed to determine whether the three Sp1-binding sites identified by EMSA also bound Sp1 in situ in live cells. BMMϕs were stimulated with LPS plus IC and then processed for ChIP analysis using an anti-Sp1 Ab. An analysis of the first 2000 bp of the HB-EGF promoter (−2000/+292) using 13 different primer pairs (Table I) revealed three Sp1-binding regions, mapping to amplicons 3, 8, and 11 (Fig. 4A), corresponding to the three predicted Sp1-binding sites. A kinetic analysis of these regions revealed a rapid, although transient binding of Sp1 which peaked at ~45 min (Fig. 4B, amplicons 3, 8, and 11). As a control, an upstream region (−2000/−1849) of the HB-EGF promoter failed to efficiently recruit Sp1 (Fig. 4B, amplicon 13).
FIGURE 4.
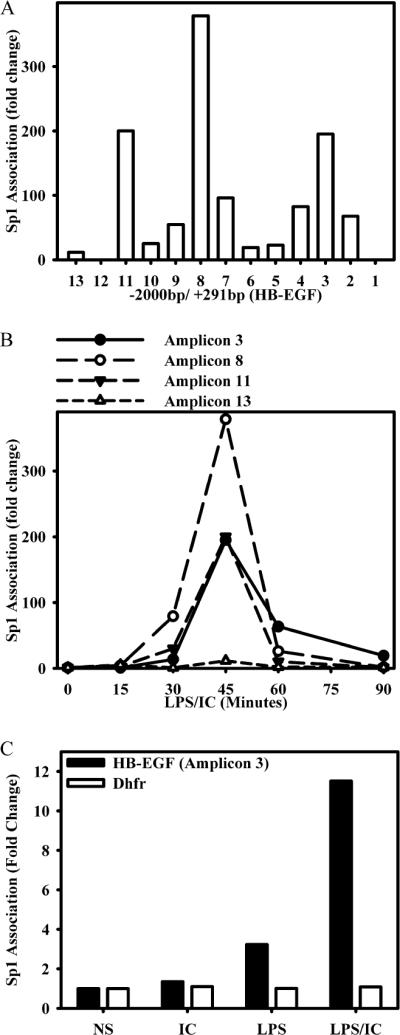
Sp1 is recruited to the HB-EGF promoter in situ by BMMϕs after stimulation with LPS plus IC. A, BMMϕs were stimulated with ICs and 10 ng/ml LPS (LPS/IC) for 0 and 45 min. Cross-linked chromatin fragments were immunoprecipitated with anti-Sp1 Ab. Immunoprecipitated DNA was purified and examined for the presence of HB-EGF promoter sequences corresponding to each of the 13 amplicons with the corresponding primers listed in Table I as measured by QRT-PCR. The data were normalized to inputs at each time point and plotted as fold changes relative to the data at 0 min. B, BMMϕ were stimulated with immune complexes (IC) and 10 ng/ml LPS (IC/LPS) for 0, 15, 30, 45, 60, and 90 min and processed for ChIP as in A. DNA was purified and examined for the presence of HB-EGF promoter sequences corresponding to amplicons 3, 8, 11, and 13 by QRT-PCR. C, BMMϕs were stimulated with ICs, LPS, or LPS/IC for 45 min or left unstimulated (NS) and processed for ChIP as in A. DNA was purified and examined for the presence of the HB-EGF promoter sequence corresponding to amplicon 3 and the Sp1 site within the promoter of the housekeeping gene dihydrofolate reductase (Dhfr). Values are representative of at least two independent experiments.
Additionally, a ChIP analysis comparing relative Sp1 association with the HB-EGF promoter after stimulation with IC alone, LPS alone, and LPS plus IC was performed (Fig. 4C). Sp1 association was not detected after the addition of ICs alone, and it was only modestly increased following stimulation with LPS alone. In contrast, there was robust recruitment of Sp1 to the HB-EGF promoter after stimulation with LPS plus IC. Thus, there was a clear distinction between the results obtained with ChIP and those obtained with EMSA. Resting cells did not exhibit significant Sp1 ChIP activity (Fig. 4B, time 0), whereas by EMSA their nuclei clearly contained Sp1 that was fully competent to bind DNA (Fig. 3B).
As a control for these studies, the binding of Sp1 to an Sp1-binding site within the promoter of the housekeeping gene dihydrofolate reductase (Dhfr) was analyzed. Dhfr mRNA levels were unchanged by these stimulation conditions (Supplemental Fig. 3), and the binding of Sp1 to Dhfr by ChIP was unaffected by any of these stimulation conditions (Fig. 4C).
Sp1 is required for full expression of HB-EGF
To directly determine whether Sp1 regulates the expression of HB-EGF, siRNA specific to Sp1 was transfected into primary BMMϕs. Knockdown of Sp1 mRNA expression was measured by real-time PCR, 48 h after transfection, and demonstrated a dose-dependent decrease in Sp1 mRNA following transfection with Sp1-specific siRNA (Fig. 5A). Parallel wells of transfected macrophages were stimulated with LPS plus IC for 2 h, and the expression of HB-EGF was measured. HB-EGF mRNA levels were diminished by ~60–70% when transfected with 10 and 100 nM Sp1-specific siRNA, but not by nonspecific scrambled siRNA (Fig. 5B).
FIGURE 5.
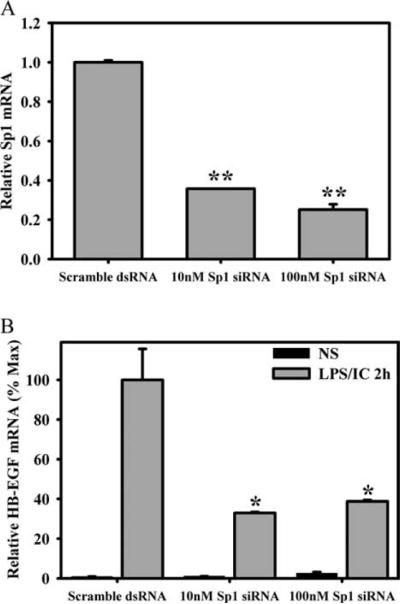
Sp1 is required for full expression of HB-EGF. Day 6 or 7 primary macrophages were transfected with Sp1 siRNA (final concentration, 10 or 100 nM) or scrambled sequence dsRNA by Amaxa Nucleofection for 48 h. Sp1 expression levels were assessed without stimulation (A), and expression levels for HB-EGF were assessed without stimulation and after stimulation with LPS plus IC for 2 h (B) by QRT-PCR. Bars, mean ± SD. *, p <0.001. Values are representative of three independent experiments.
Activity of an HB-EGF reporter construct in response to stimulation
To address which of the three Sp1-containing promoter elements was necessary for the transcription of HB-EGF, reporter plasmids containing portions of the HB-EGF promoter were transfected into RAW264.7 cells. Three HB-EGF promoter reporter plasmids were constructed, including the first ~2700 bases of the HB-EGF promoter (−2704/+330), as well as two truncations (−1238/+330 and −557/+330) (Fig. 6A). The −2707/+330 plasmid contains three potential Sp1-binding sites, whereas the −1230/+330 and the −557/+330 plasmid contained two and one binding site, respectively. Luciferase activity was unchanged following stimulation of RAW cells transfected with empty pGL4.19 vector (Fig. 6A). Transfection of the −2704/+330 plasmid resulted in only minor increases over the level of activity of the empty vector. However, truncation of the promoter (to −1238) strongly enhanced the activity of the promoter upon stimulation (Fig. 6A). The most severely truncated HB-EGF promoter analyzed (−557) displayed similarly increased levels of luciferase activity upon stimulation (Fig. 6A). Both of these vectors (−1238 and −557) responded equally well to stimulation with either LPS alone or LPS plus IC. Thus, the luciferase assay did not accurately reflect actual HB-EGF mRNA induction. HB-EGF production required a combination of LPS plus IC, whereas luciferase activity was maximally induced by LPS alone.
FIGURE 6.

Luciferase activity of RAW264.7 cells transfected with wild-type (WT) and mutant HB-EGF reporter vectors. A, pGL4.19 luciferase reporter constructs containing −2704/+330, −1238/+330, −557/+330, or none of the HB-EGF promoter were transfected into RAW264.7 cells overnight and then stimulated with LPS (1 or 10 ng/ml) or LPS plus IC for 8 h. Each sample was measured for firefly luciferase activity and normalized to Renilla luciferase activity (transfected at a ratio of 40:1). Values are represented as fold changes relative to unstimulated cells transfected with the empty vector. B, Schematic of the mutations made in the Sp1 site contained in the −557/+330 vector. The central GC of the three conserved core GGGCGG sequences was mutated to TA by site-directed mutagenesis. RAW264.7 cells were transfected with the empty vector or the −557/+330 HB-EGF reporter vector containing the wild-type or mutant Sp1-binding site overnight and then stimulated with LPS (10 ng/ml) plus IC for 8 h. Samples were analyzed as described in A and represented as fold changes from unstimulated cells transfected with the empty vector. Bars, mean ± SD. *, p < 0.001 comparing medium or LPS plus IC conditions from wild-type to mutant −557/+330-transfected cells. Values are representative of at least three independent experiments.
Because both the −1230/+330 and the −557/+330 promoter plasmids responded similarly to stimulation with LPS plus IC, we investigated whether the Sp1-binding site located within −83/−54 was responsible for the response to LPS plus IC. This region actually contains three potential Sp1-binding sites in tandem (Fig. 6B). To assess the importance of this region, we used site-directed mutagenesis to modify two nucleotides of the conserved core binding site of GGGCGG to GGTAGG. Transfection of the −557/+330 led to a constitutive enhancement in luciferase activity that was enhanced upon stimulation (Fig. 6B). However, both the constitutive and induced activities were nearly completely nullified by mutating these sites. Thus, this site binds Sp1, and this binding can activate the HB-EGF promoter.
Other groups have reported that alterations in HB-EGF mRNA stability can contribute to HB-EGF expression (34). To analyze mRNA stability, macrophages were stimulated with LPS or LPS plus IC for 2 h, and then 0.5 μg/ml actinomycin D was added. HB-EGF mRNA levels were measured by QRT-PCR for 2 h (Supplemental Fig. 4). Although macrophages stimulated with LPS plus IC had higher levels of HB-EGF mRNA, the rate at which the mRNA was degraded was similar in both conditions. Therefore, the high degree of HB-EGF mRNA induction in regulatory macrophages was not due to alterations in mRNA stability.
Role of MAPK activation in HB-EGF induction
We previously demonstrated that immune complexes could activate ERK (29). We therefore wished to investigate whether ERK and p38 were involved in the induction of HB-EGF by regulatory macrophages. As a first step, macrophages were stimulated with LPS alone or in combination with ICs, PGE2, or dbcAMP (Fig. 7). The activation of ERK and p38 were examined by Western blot analysis using Abs specific to the phosphorylated forms of ERK and p38 (Fig. 7A) and quantitated by densitometry analysis (Fig. 7B). Stimulation with LPS alone resulted in a modest level of ERK and p38 activation, but the combination of LPS plus any of the three second stimuli resulted in enhanced early MAPK activation (Fig. 7B). The inhibition of either ERK or p38 resulted in a profound decrease in HB-EGF induction by any of the three stimuli (Fig. 7C). Thus, HB-EGF induction depends on the activation of two of the major MAPKs, ERK and p38, and inhibiting either of them results in decreased HB-EGF expression.
FIGURE 7.

MAPKactivation following stimulation with LPS plus IC. A, Macrophages were stimulated with 10 ng/ml LPS alone or LPS plus IC, PGE2, or dbcAMP (cAMP). After 5 and 20 min, macrophage lysates were prepared for Western blotting analysis using mAbs to phosphorylated ERK or p38. Blots were normalized with Ab to total ERK and p38. B, ERK and p38 phosphorylation at 5 min poststimulation was quantitated using ImageQuant software. The phosphorylation of MAPKs following LPS stimulation was arbitrarily set as 1, and the data were expressed as fold induction. C, Quantitative PCR measurements of HB-EGF mRNA in macrophages stimulated with LPS plus IC, PGE2, or dbcAMP for 2 h in the presence or absence of MAPK inhibitors U0126 or SB203580. All real-time PCR results are normalized to GAPDH and are expressed as percent HB-EGF mRNA.
To examine the extent to which HB-EGF expression was dependent on the MAPKs, macrophages were pretreated with increasing concentrations (0–4 μM) of the MEK1,2 inhibitor (U0126), the p38 inhibitor (SB203580), or the JNK inhibitor II before stimulation with LPS in the presence of ICs. HB-EGF mRNA was measured by QRT-PCR 2 h after stimulation. HBEGF mRNA production was inhibited in a dose-dependent manner by pretreating macrophages with either the ERK inhibitor or the p38 inhibitor (Fig. 8A). As a control, the JNK inhibitor only minimally decreased HB-EGF production when used at similar concentrations (Fig. 8A). This concentration of JNK inhibitor was sufficient to significantly inhibit the secretion of TNF-α by macrophages (data not shown). The inhibition of ERK activation by U0126 exerted a similar dose-dependent inhibition of HB-EGF production regardless of the stimulation conditions used to induce HB-EGF (Fig. 8B). Thus, HB-EGF can be induced by a variety of stimuli and in all cases this induction depends on the activation of ERK.
FIGURE 8.
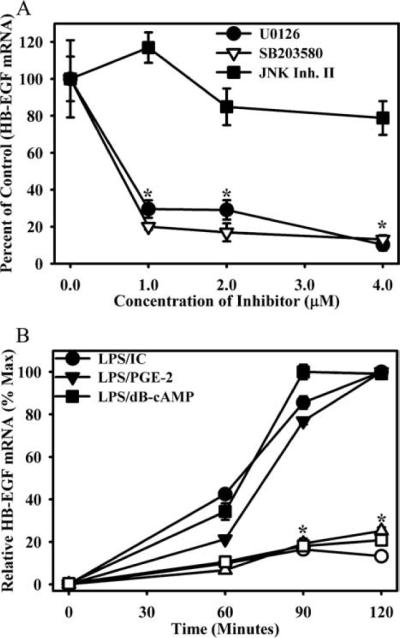
HB-EGF induction requires the activity of ERK1/2 and p38. A, BMMϕs were pretreated with increasing concentrations of the inhibitors of the ERK1/2 pathway (U0126), p38 (SB203580), or JNK (JNK Inhibitor (Inh.) II). BMMϕs were then stimulated with LPS (10 ng/ml) + IC for 2 h. HB-EGF mRNA was measured by QRT-PCR and expressed relative to vehicle controls. Values are representative of at least three independent experiments. Symbols, mean ± SD. B, BMMϕs were pretreated with 5 μM U0126 (open symbols) or vehicle (closed symbols) for 1 h and then stimulated with LPS plus IC, PGE2, or dbcAMP for 60, 90, or 120 min. HB-EGF mRNA was measured by QRT-PCR and expressed relative to the maximum level of HB-EGF mRNA during the stimulation conditions. *, p < 0.01 as compared with vehicle control.
Enhanced HB-EGF promoter accessibility in response to LPS plus IC
We hypothesized that HB-EGF gene expression was regulated at the level of chromatin, and dependent on locus accessibility. This would explain the seemingly discordant data that were obtained with EMSA and luciferase assays, relative to ChIP. We hypothesized that assays depending on extrachromosomal DNA elements (EMSA, luciferase) did not require the combination of LPS plus IC for maximal stimulation. We examined the sensitivity of DNA to cleavage by DNase before and after stimulation of macrophages with LPS plus IC. Chromatin sensitivity to DNase was measured at amplicons 3, 8, and 11 and compared with a region of the TdT gene, which is not expressed in macrophages (35). At 0 min (unstimulated cells), all of these regions were relatively resistant to DNase cleavage (Fig. 9). Within minutes of stimulation, all three regions studied showed a substantial increase in sensitivity to DNase digestion. Although each region showed slightly different kinetics, the extent of the accessibility increase was comparable in all three regions (Fig. 9). This was not the case for the TdT gene, which showed essentially no changes in accessibility over this period of observation (Fig. 9). These data are consistent with the hypothesis that although Sp1 is constitutively found in the nucleus in a form that is able to bind to its cognate sequences (see EMSA in Fig. 3), this binding does not occur in unstimulated cells. Changes in HB-EGF promoter accessibility enable Sp1 to associate with the HB-EGF promoter following stimulation.
FIGURE 9.
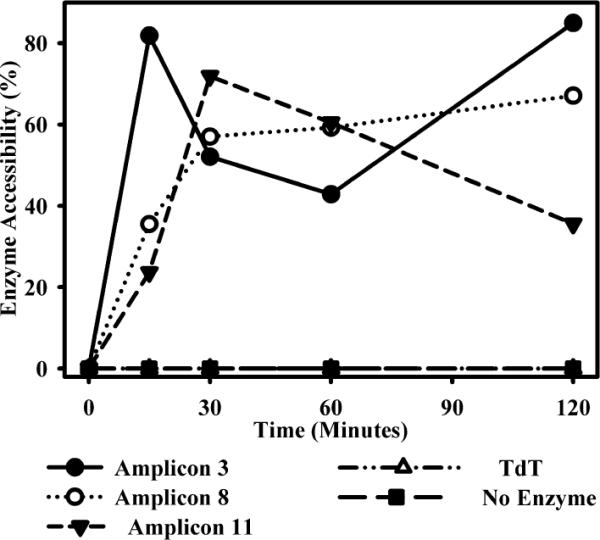
Changes in DNase I accessibility at the HB-EGF promoter. BMMϕs were stimulated with LPS plus IC for 0, 15, 30, 60, or 120 min and then fixed with paraformaldehyde. Nuclei were isolated and treated with DNase for 1 h on ice. DNA was then purified and subject to QRT-PCR. Data are presented as a percent of DNase accessibility to amplicons 3, 8, and 11 as normalized to undigested DNA and then to unstimulated control. The lymphocyte-specific TdT gene is present as a negative control for changes in DNase sensitivity. Values are representative of at least three independent experiments.
Requirement of ERK activity for efficient Sp1 recruitment and promoter accessibility
To determine whether ERK activity was required for efficient Sp1 recruitment to the HB-EGF promoter, macrophages were pretreated with the ERK inhibitor U0126, and Sp1 recruitment to the HB-EGF promoter that was measured by ChIP (Fig. 10A). Similar to the observations made in Fig. 4, stimulation of macrophages with LPS plus IC resulted in a strong recruitment of Sp1 to all three regions of the HB-EGF promoter at 45 min (amplicons 3, 8, and 13). Blocking the ERK pathway with U0126 completely diminished Sp1 binding at all three of these sites (Fig. 10A).
FIGURE 10.
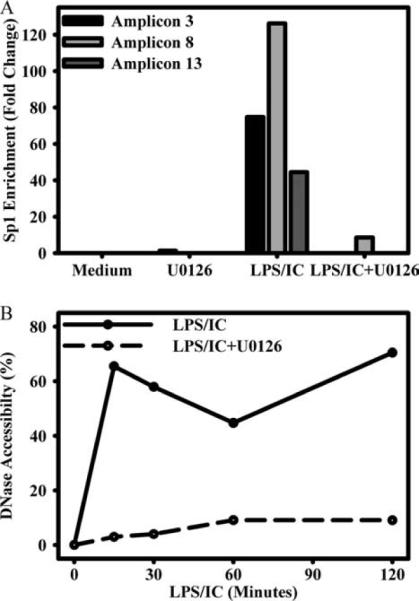
ERK-dependent alterations at the HB-EGF promoter. A, BMMϕs were treated with medium alone or 5 μM U0126 for 1 h and then left unstimulated or stimulated with LPS (10 ng/ml) plus IC for 45 min. Sp1 association was measured as in Fig. 4 and is expressed as fold enrichment from unstimulated vehicle control for amplicons 3, 8, and 13, each of which contain Sp1-binding sites. B, BMMϕs were pretreated with U0126 or vehicle control as in A, then stimulated for 0, 15, 30, 60, or 120 min, then measured for changes in DNase accessibility as in Fig. 9. DNase accessibility is presented as a percentage of digested DNA as normalized to unstimulated and undigested controls. Values are representative of at least two independent experiments.
Blocking the ERK pathway also prevented stimulation-dependent increases in DNA accessibility at the HB-EGF promoter. Similar to Fig. 9, when macrophages were treated with LPS plus IC, the HB-EGF promoter became more accessible to DNase activity (amplicon 3; Fig. 10B). Inhibition of the ERK pathway with U0126, however, greatly diminished the stimulus-dependent increase in accessibility at the HB-EGF promoter (Fig. 10B). Thus, both Sp1 association and DNase accessibility were dependent on the activation of ERK.
Discussion
We have previously described a population of regulatory macrophages that are associated with a variety of immunosuppressive conditions. The most consistent feature of these regulatory macrophages is a reciprocal alteration in the production of IL-10 and IL-12/23. Unlike classically activated macrophages, regulatory macrophages make high levels of IL-10 and little or no IL-12/23. These regulatory cells have been assigned different names, depending on the conditions under which they were generated or the tissue in which they were identified (36). In tumors, a population of macrophages with a regulatory phenotype has been considered a harbinger of a poor outcome and metastasis (37). A population of macrophages with characteristics of regulatory macrophages has also been identified in atherosclerosis (9). We previously demonstrated that the expansion of a regulatory population of macrophages correlated with disease progression in leishmaniasis (5). The macrophages in these diseases have well-described immunosuppressive activities, most of which can be assigned to IL-10 production. We now propose that these regulatory macrophages coexpress a second novel activity that allows them to remodel blood vessels. In this work, we demonstrate that the expression of HB-EGF on regulatory macrophages may help to explain the atherogenic and angiogenic potential of these macrophages.
We also show here that two of the three major MAPKs, p38 and ERK, are required for HB-EGF production. Blocking either of them inhibits transcription. We also show that the coordination of two signals is required for the development of regulatory macrophages and HB-EGF induction. Signal 1 involves the binding of a stimulus such as LPS to TLRs to activate transcription factors. This first signal depends on p38 to activate the transcription factors to initiate transcription. Signal 2 involves the activation of ERK, and in the present work we describe three different stimuli that can activate ERK when added to macrophages. These stimuli include ICs, PGs, and dbcAMP. We show that ERK activation makes the HB-EGF promoter more accessible to the transcription factor Sp1.
These conclusions were reached after studies to analyze the binding of Sp1 to the HB-EGF promoter yielded very different results, depending on the assays that were used. The EMSA assays indicated that Sp1 is resident in the nucleus of unstimulated cells and fully competent to bind elements in the HB-EGF promoter in the absence of stimulation. This constitutive binding to unstimulated cells, however, was not observed by ChIP analysis, and it was not reflected in HB-EGF mRNA production. A potential explanation for this difference is that the HBEGF promoter was inaccessible to transcription factors in unstimulated cells. Increased accessibility due to chromatin alterations would also explain the discordant luciferase data in which LPS alone induced as much luciferase activity as did LPS plus IC. Regulation at the level of chromatin accessibility would not be obvious during EMSA or luciferase assays, where naked DNA was used as the probe or readout. This increased accessibility of the HB-EGF promoter following stimulation was confirmed by DNase accessibility assays. After stimulation, the HB-EGF promoter became more accessible to DNase cleavage, whereas the accessibility of a control gene went unaltered. This increased accessibility did not occur when ERK was inhibited. Thus, the two signals required for HB-EGF production activate different MAPKs and both MAPKs are required for HB-EGF transcription.
By EMSA assays, we show that Sp1 can bind to three positions along the HB-EGF promoter. Luciferase reporter assays were performed to determine which site was most closely associated with transcriptional activation. The binding of Sp1 to the 3′-most site, located adjacent to the transcriptional start site, appeared to be required for the activity of the HB-EGF promoter. This site was sufficient to induce maximal luciferase activity. Surprisingly, an analysis of an extended promoter, including the 5′-most site, not only failed to contribute to transcription but also substantially diminished luciferase activity. The logical interpretation of these results is that a repressor element was located within this site. Studies are ongoing to examine this site more carefully.
We examined two other conditions in which regulatory macrophages were induced, and we demonstrate that HB-EGF production was regulated in an ERK dependant fashion in all three populations. PGE2 has been previously shown to enhance the endotoxin-driven production of IL-10 from macrophages and monocytes (38). We show that HB-EGF production is also enhanced under these conditions. Similar to what we observed for ICs, PGE2 induced no HB-EGF on its own, but rather synergized with LPS to produce HB-EGF (Fig. 2). The same observation was made when macrophages were stimulated in the presence of dbcAMP and LPS. In both cases, HB-EGF was induced, and this induction was substantially inhibited by the addition of the MEK inhibitor, U0126. Thus, the activation of ERK in macrophages results in a phenotype that is quite distinct from classically activated macrophages and results in macrophages that are not only immunosuppressive but also angiogenic and atherogenic.
A link between IL-10 production and angiogenesis was initially established in studies of tumor-associated macrophages (39). Tumor-associated macrophages are a rich source of IL-10 (40) and tumor-promoting growth factors (39). Elevated expression of HB-EGF has been found in many human tumors, and high levels have been found to correlate with poor prognosis (26). In vitro and in vivo studies indicate that the expression of HB-EGF in the developing tumor microenvironment can contribute to angiogenesis, and thus to metastasis (23). In this work, we provide a molecular mechanism to explain the coexpression of these two activities in regulatory macrophages and show that both activities are dependent on the activation of ERK. These findings suggest that the inhibition of ERK may prevent both the immunosuppressive and the angiogenic activities of these macrophages.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant AI49383.
Footnotes
Disclosures The authors have no financial conflict of interest.
Abbreviations used in this paper: IC, immune complex; EGF, epidermal growth factor; HB-EGF, heparin-binding EGF-like growth factor; pro-HBEGF, HB-EGF transmembrane precursor; sHB-EGF, soluble HB-EGF; MMP, matrix metalloproteinase; ADAM, a disintegrin and metalloproteinase; SMC, smooth muscle cell; BMMϕ, bone marrow-derived macrophage; dbcAMP, N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate; QRT-PCR, quantitative real-time PCR; ChIP, chromatin immunoprecipitation; siRNA, small interfering RNA.
The on-line version of this article contains supplemental material.
References
- 1.Sutterwala FS, Noel GJ, Salgame P, Mosser DM. Reversal of proinflammatory responses by ligating the macrophage Fcγ receptor type I. J. Exp. Med. 1998;188:217–222. doi: 10.1084/jem.188.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson CF, Mosser DM. Cutting edge: biasing immune responses by directing antigen to macrophage Fcγ receptors. J. Immunol. 2002;168:3697–3701. doi: 10.4049/jimmunol.168.8.3697. [DOI] [PubMed] [Google Scholar]
- 3.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J. Leukocyte Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosser DM. The many faces of macrophage activation. J. Leukocyte Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 5.Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J. Exp. Med. 2005;201:747–754. doi: 10.1084/jem.20041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerber JS, Mosser DM. Reversing lipopolysaccharide toxicity by ligating the macrophage Fcγ receptors. J. Immunol. 2001;166:6861–6868. doi: 10.4049/jimmunol.166.11.6861. [DOI] [PubMed] [Google Scholar]
- 7.Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 8.Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, Bottazzi B, Colombo MP, Mantovani A, Sica A. p50 nuclear factor-κB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432–11440. doi: 10.1158/0008-5472.CAN-06-1867. [DOI] [PubMed] [Google Scholar]
- 9.Mallat Z, Heymes C, Ohan J, Faggin E, Leseche G, Tedgui A. Expression of interleukin-10 in advanced human atherosclerotic plaques: relation to inducible nitric oxide synthase expression and cell death. Arterioscler. Thromb. Vasc. Biol. 1999;19:611–616. doi: 10.1161/01.atv.19.3.611. [DOI] [PubMed] [Google Scholar]
- 10.Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science. 1991;251:936–939. doi: 10.1126/science.1840698. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Mosser DM, Zhang X. Activation of the MAPK, ERK, following Leishmania amazonensis infection of macrophages. J. Immunol. 2007;178:1077–1085. doi: 10.4049/jimmunol.178.2.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69:1051–1061. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- 13.Nishi E, Klagsbrun M. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a mediator of multiple physiological and pathological pathways. Growth Factors. 2004;22:253–260. doi: 10.1080/08977190400008448. [DOI] [PubMed] [Google Scholar]
- 14.Nanba D, Mammoto A, Hashimoto K, Higashiyama S. Proteolytic release of the carboxy-terminal fragment of proHB-EGF causes nuclear export of PLZF. J. Cell Biol. 2003;163:489–502. doi: 10.1083/jcb.200303017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinugasa Y, Hieda M, Hori M, Higashiyama S. The carboxyl-terminal fragment of pro-HB-EGF reverses Bcl6-mediated gene repression. J. Biol. Chem. 2007;282:14797–14806. doi: 10.1074/jbc.M611036200. [DOI] [PubMed] [Google Scholar]
- 16.Elenius K, Paul S, Allison G, Sun J, Klagsbrun M. Activation of HER4 by heparin-binding EGF-like growth factor stimulates chemotaxis but not proliferation. EMBO J. 1997;16:1268–1278. doi: 10.1093/emboj/16.6.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G, et al. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc. Natl. Acad. Sci. USA. 2003;100:3221–3226. doi: 10.1073/pnas.0537588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mine N, Iwamoto R, Mekada E. HB-EGF promotes epithelial cell migration in eyelid development. Development. 2005;132:4317–4326. doi: 10.1242/dev.02030. [DOI] [PubMed] [Google Scholar]
- 19.Shirakata Y, Kimura R, Nanba D, Iwamoto R, Tokumaru S, Morimoto C, Yokota K, Nakamura M, Sayama K, Mekada E, Higashiyama S, Hashimoto K. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. J. Cell Sci. 2005;118:2363–2370. doi: 10.1242/jcs.02346. [DOI] [PubMed] [Google Scholar]
- 20.Miyagawa J, Higashiyama S, Kawata S, Inui Y, Tamura S, Yamamoto K, Nishida M, Nakamura T, Yamashita S, Matsuzawa Y, et al. Localization of heparin-binding EGF-like growth factor in the smooth muscle cells and macrophages of human atherosclerotic plaques. J. Clin. Invest. 1995;95:404–411. doi: 10.1172/JCI117669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peoples GE, Blotnick S, Takahashi K, Freeman MR, Klagsbrun M, Eberlein TJ. T lymphocytes that infiltrate tumors and atherosclerotic plaques produce heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor: a potential pathologic role. Proc. Natl. Acad. Sci. USA. 1995;92:6547–6551. doi: 10.1073/pnas.92.14.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Sunnarborg SW, McNaughton KK, Johns TG, Lee DC, Faber JE. Heparin-binding epidermal growth factor-like growth factor signaling in flow-induced arterial remodeling. Circ. Res. 2008;102:1275–1285. doi: 10.1161/CIRCRESAHA.108.171728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ongusaha PP, Kwak JC, Zwible AJ, Macip S, Higashiyama S, Taniguchi N, Fang L, Lee SW. HB-EGF is a potent inducer of tumor growth and angiogenesis. Cancer Res. 2004;64:5283–5290. doi: 10.1158/0008-5472.CAN-04-0925. [DOI] [PubMed] [Google Scholar]
- 24.Mehta VB, Besner GE. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth Factors. 2007;25:253–263. doi: 10.1080/08977190701773070. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka Y, Miyamoto S, Suzuki SO, Oki E, Yagi H, Sonoda K, Yamazaki A, Mizushima H, Maehara Y, Mekada E, Nakano H. Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin. Cancer Res. 2005;11:4783–4792. doi: 10.1158/1078-0432.CCR-04-1426. [DOI] [PubMed] [Google Scholar]
- 26.Miyamoto S, Yagi H, Yotsumoto F, Kawarabayashi T, Mekada E. Heparin-binding epidermal growth factor-like growth factor as a novel targeting molecule for cancer therapy. Cancer Sci. 2006;97:341–347. doi: 10.1111/j.1349-7006.2006.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Liu R, Lee SW, Sloss CM, Couget J, Cusack JC. Heparin-binding EGF-like growth factor is an early response gene to chemo-therapy and contributes to chemotherapy resistance. Oncogene. 2007;26:2006–2016. doi: 10.1038/sj.onc.1209999. [DOI] [PubMed] [Google Scholar]
- 28.Cheng K, Xie G, Raufman JP. Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem. Pharmacol. 2007;73:1001–1012. doi: 10.1016/j.bcp.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucas M, Zhang X, Prasanna V, Mosser DM. ERK activation following macrophage FcγR ligation leads to chromatin modifications at the IL-10 locus. J. Immunol. 2005;175:469–477. doi: 10.4049/jimmunol.175.1.469. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Edwards JP, Mosser DM. Dynamic and transient remodeling of the macrophage IL-10 promoter during transcription. J. Immunol. 2006;177:1282–1288. doi: 10.4049/jimmunol.177.2.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao S, Zhang X, Edwards JP, Mosser DM. NF-κB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 2006;281:26041–26050. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.no M, Raab G, Lau K, Abraham JA, Klagsbrun M. Purification and characterization of transmembrane forms of heparin-binding EGF-like growth factor. J. Biol. Chem. 1994;269:31315–31321. [PubMed] [Google Scholar]
- 33.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorensen BS, Ornskov D, Nexo E. The chemotherapeutic agent VP16 increases the stability of HB-EGF mRNA by a mechanism involving the 3′-UTR. Exp. Cell Res. 2006;312:3651–3658. doi: 10.1016/j.yexcr.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Zhou L, Nazarian AA, Smale ST. Interleukin-10 inhibits inter-leukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol. Cell Biol. 2004;24:2385–2396. doi: 10.1128/MCB.24.6.2385-2396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67:5064–5066. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- 38.Strassmann G, Patil-Koota V, Finkelman F, Fong M, Kambayashi T. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J. Exp. Med. 1994;180:2365–2370. doi: 10.1084/jem.180.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 40.Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme DJ, Mantovani A. Autocrine production of IL-10 mediates defective IL-12 production and NF-κB activation in tumor-associated macrophages. J. Immunol. 2000;164:762–767. doi: 10.4049/jimmunol.164.2.762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.