

Abstract
Massively parallel DNA sequencing technologies provide an unprecedented ability to screen entire genomes for genetic changes associated with tumor progression. Here we describe the genomic analyses of four DNA samples from an African-American patient with basal-like breast cancer: peripheral blood, the primary tumor, a brain metastasis, and a xenograft derived from the primary tumor. The metastasis contained two de novo mutations and a large deletion not present in the primary tumor, and was significantly enriched for 20 shared mutations. The xenograft retained all primary tumor mutations, and displayed a mutation enrichment pattern that paralleled the metastasis (16 of 20 genes). Two overlapping large deletions, encompassing CTNNA1, were present in all three tumor samples. The differential mutation frequencies and structural variation patterns in metastasis and xenograft compared to the primary tumor suggest that secondary tumors may arise from a minority of cells within the primary.
Introduction
Basal-like breast cancer is characterized by the absence of estrogen receptor (ER) expression, the lack of ERBB2 gene amplification, and a high mitotic index. The consequent absence of approved targeted therapy options and frequently poor response to standard chemotherapy often result in a rapidly fatal clinical course. The disease also accounts for an elevated percentage of breast cancers in patients with African ancestry1. Clinical progress has been limited by a poor understanding of the genetic events responsible for this tumor subtype and by limited preclinical models to study the disease. Since basal-like breast cancer has a highly unstable genome, a key question is whether the fatal metastatic process is driven by mutations that occur after the tumor cells arrive at the distant site, or whether the primary tumor generates cells with a complete repertoire of somatic mutations required for metastatic growth. The rapid advancement of next generation sequencing technologies allows comprehensive characterization of genomic changes, facilitating the comparison of multiple samples taken from the same patient to address the genetic basis for tumor progression and metastasis.
Results
Case presentation and prior characterization of samples
A 44-year old African-American woman was diagnosed with an ERBB2 negative and ER negative inflammatory breast cancer. She was treated with neoadjuvant dose-dense chemotherapy2, but significant residual tumor was present in the breast and axillary lymph nodes at mastectomy. This indicated chemotherapy resistance and she underwent radiation therapy. Eight months later, she developed a cerebellar metastasis and, despite resection, rapidly succumbed to widely disseminated disease. A transplantable Human-in-Mouse (HIM) xenograft tumor line was generated from a sample of her primary tumor biopsied before treatment3. The xenograft in the mammary fat pad was locally invasive and produced metastatic deposits in lymph nodes and ovaries. Informed consent for full genome sequencing was obtained and DNA samples were prepared from her peripheral blood, primary tumor, brain metastasis, and an early passage xenograft (harvested 101 days after initial engrafting into the mouse host). Application of the PAM50 intrinsic subtype algorithm identified the primary tumor, brain metastasis, and xenograft line as basal-like subtype, with high risk of relapse (ROR) scores4.
Sequence coverage and mutation analysis
Using a paired-end sequencing strategy, we generated 130.7, 124.9, 111.8, and 149.2 billion base pairs of sequence data from genomic DNA derived from blood, primary tumor, brain metastasis, and xenograft samples, respectively, with corresponding haploid coverages of 38.8X, 29.0X, 32.0X, and 23.8X (Supplementary Table 1). These genome-wide coverages were assessed by comparing SNVs detected by MAQ5 with SNPs genotyped using Illumina 1M duo arrays for all tissues excluding the xenograft. Array data from the metastasis were used as a surrogate for monitoring the xenograft SNP coverage and confirmed bi-allelic detection of 98.27%, 96.79%, 96.17%, and 88.77% of the heterozygous array SNPs in the normal, primary tumor, metastasis, and xenograft sequence datasets, respectively (Supplementary Table 1).
The process for selecting somatic mutations is shown in Supplementary Table 2 and is detailed in Supplementary Information. Putative somatic SNVs and indels that overlap with coding sequences, splice sites, and RNA genes were included as “tier 1”. We combined tier 1 sites identified in all three tumor samples and obtained deep read count data for all four samples from Illumina and/or 454 platforms (Supplementary Information). Based on pathology review, the tumor cellularity estimates were 70% for the primary tumor and 90% for both the brain metastasis and xenograft. Utilizing these estimates, we calculated the tumor read counts by proportionally removing the counts derived from the normal tissue reads from the counts obtained from primary tumor and metastasis reads (Supplementary Table 3a). Using the Illumina platform, we also generated 15.6 Gb (4.4X haploid coverage) of sequence data for the NOD/SCID mouse genome used as the host for the xenograft line. The mapping rates of NOD/SCID data to human and mouse C57BL/6 reference sequences were 3.17% and 95.85%, respectively. Since the non-malignant contamination in xenograft is largely from murine cells (which do not significantly affect read mapping), no correction was applied for the xenograft data. Adjusted tumor read counts were used to calculate mutant allele frequencies. Somatic changes were validated by comparing mutant allele frequencies in the three tumor genomes against the germline DNA sample, combined with a manual review of ABI 3730 data from PCR products (Supplementary Information).
In summary, a total of 50 somatic sites, including 28 missense, 11 silent, 2 splice site, 1 RNA, 1 nonsense, 4 insertions, and 3 deletions, were validated in at least one of the three tumor genomes. Of coding point mutations, the observed nonsynonymous:synonymous (NS:SS) ratio of 2.64:1 (29:11) is not significantly different from that expected by chance6 (P = 0.51), suggesting that the majority of coding mutations do not confer a selective advantage to the basal tumor. This is similar to the NS:SS ratio reported in the small-cell lung cancer cell line NCI-H2097, but higher than the ratio reported in the melanoma cell line COLO-8298.
Mutation spectrum in basal breast tumor
We investigated the spectrum of DNA sequence changes in this basal tumor and found 55% (22/40) of coding point mutations represent C:G->T:A transitions. A similar frequency of C:G->T:A transitions (56% (18/32)) was observed in the lobular breast tumor recently reported9 (Figure 1a). In addition, 15% (6/40) of coding point mutations representing C:G->A:T transversions were detected in the basal tumor, but none were found in the lobular tumor. The statistical significance of these observations should be explored with the comparative analysis of a larger number of basal and lobular breast tumors. Moreover, the observed C:G->T:A transition frequency is notably higher than those observed in a previous breast cancer study10 (P = 0.027; Figure 1b). A set of extremely high confidence tier 1-4 mutations (somatic score > 55 and average mapping quality > 79) was used to explore the genome-wide mutation spectrum. We found that mutations at A:T bases are significantly expanded in the genome-wide set compared to the coding mutations, especially for A:T->G:C transitions (P = 0.0065). This is consistent with the higher A:T content in non-coding sequences than in coding sequences. Comparison to the whole-genome mutation spectrum reported for the melanoma cell line (COLO-829)8 and a small cell lung cancer cell line (NCI-H209)7 suggests that the tumor genome under study shows no sign of tobacco or ultraviolet influence. We then compared the fraction of the three classes of guanine mutations occurring at CpG dinucleotides in primary tumor, brain metastasis, and xenograft and found that the frequencies of G->A mutations are 27.54%, 27.60%, and 28.05% in each respective tumor, significantly higher than both the genome average of 4.45% (P < 10-10) and the frequency reported in NCI-H209 (P < 10-10; Figure 1c).
Figure 1. Mutational signatures in the basal breast tumor.
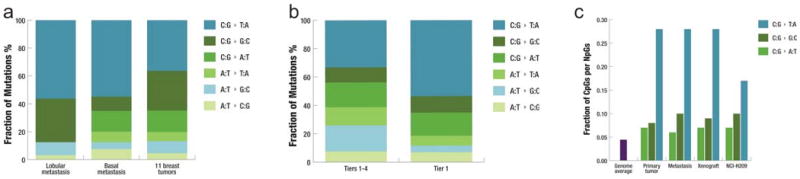
a, Fraction of mutations in each of the transition and transversion categories in the metastasis of a lobular breast tumor9, the metastasis of the basal breast tumor under study, and the 11 breast tumors reported by Wood et al.29 from which 1,104 coding mutations identified in the discovery set were used in the analysis. b, Fraction of mutations in each of the transition and transversion categories in 43 tier1 mutations and 3,204 tier 1-4 mutations in the metastasis under study. c, Fraction of guanine mutations at CpGs in primary tumor, metastasis, xenograft, and NCI-H209 as reported by Pleasance et al.7.
Distribution of mutations among tumors
1. Common mutations detected in three tumor genomes
Of the 50 validated point mutations and small indels, 48 are detectable in all three tumors. We performed a statistical enrichment test that takes the variations of different platforms, experiments, and primer pairs into consideration (Supplementary Information). These 48 sites consist of (1A) 20 sites with relatively comparable frequencies across tumors, (1B) 26 sites significantly enriched (FDR ≤ 0.05) in the metastasis and/or xenograft, and (1C) two sites with significant enrichment (FDR ≤ 0.05) in the primary tumor (Figure 2 and Table 1). The affected genes and the likely consequences of these mutations are summarized in Table 1 and Supplementary Table 3b.
Figure 2. Mutant allele frequency from deep read count data.
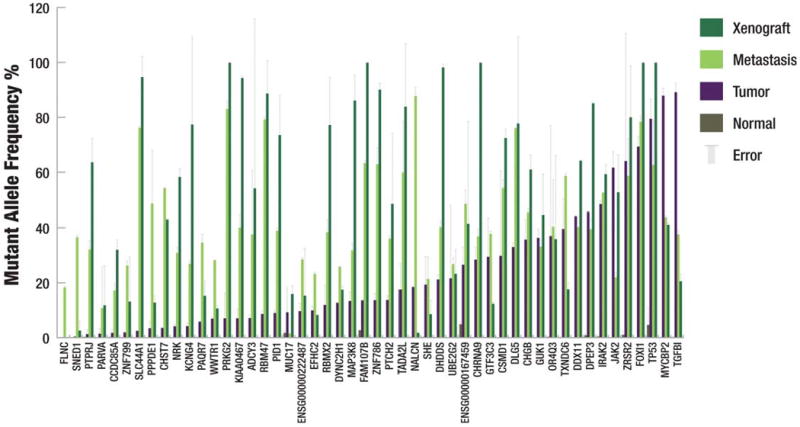
The mutant allele frequency of each somatic mutation is shown. Mutations were validated using both 454 and Illumina sequencing. Each bar represents the average of the frequency yielded by the two technologies for a single primer pair and the error bars represent the standard deviation. Data were considered only if there were at least 200 reads from Illumina sequencing and at least 20 reads from 454 sequencing. If no error bar exists, then data were only available from a single sequencing platform.
Table 1.
Summary of point mutations and small indels identified in the primary tumor, brain metastasis, and xenograft.
| Chr | Start | Allele Change | Gene | Amino Acid Change | Mutant Allele Frequency | Copy Number | Enrichment FDR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | T | M | X | T | M | X | M | X | |||||
| 1 | 26062702 | G>A | PAQR7 | p.A72 | 0.17% | 5.78% | 34.55% | 13.70% | 2 | 2 | 2 | 9.00e-4 | 0.011 |
| 1 | 26646672 | G>A | DHDDS | p.R159H | 0.14% | 21.12% | 40.24% | 88.29% | 2 | 2 | 2 | 5.73e-5 | 2.46e-5 |
| 1 | 43684654 | G>A | KIAA0467 | p.G2119R | 0.13% | 7.00% | 40.02% | 84.84% | 2 | 2 | 2 | 0.001 | 5.98e-5 |
| 1 | 45068225 | C>G | PTCH2 | p.W293S | 0.02% | 13.70% | 36.03% | 43.61% | 2 | 2 | 2 | 0.085 | 0.381 |
| 1 | 152723308 | delGCAACTTTTCATT | SHE | p.LPFKG476in_frame_delW | 0.19% | 19.28% | 21.33% | 7.69% | 4.61 | 5.34 | 6.92 | 0.820 | 0.065 |
| 1 | 226395989 | C>A | GUK1 | p.P11Q | 0.01% | 36.29% | 33.12% | 40.17% | 3.66 | 3.96 | 4.64 | 0.374 | 0.365 |
| 1 | 242935580 | A>T | PPPDE1 | p.T151S | 0.12% | 3.39% | 48.57% | 11.47% | 3.45 | 3.71 | 4.38 | 0.012 | 0.063 |
| 2 | 24994872 | G>A | ADCY3 | p.H163 | 0.06% | 7.10% | 37.49% | 48.71% | 3.17 | 3.24 | 3.78 | 0.007 | 0.029 |
| 2 | 56273320 | delG | CCDC85A | p.E161fs | 0.16% | 1.71% | 17.11% | 28.78% | 2.9 | 3.24 | 3.34 | 0.002 | 0.006 |
| 2 | 197349569 | G>C | GTF3C3 | p.R474G | 0.11% | 29.42% | 37.75% | 11.08% | 1.4 | 1.31 | 1.24 | 0.316 | 0.065 |
| 2 | 229835724 | C>T | PID1 | p.S14 | 0.11% | 8.95% | 38.89% | 66.13% | 2 | 2 | 1.36 | 0.001 | 0.166 |
| 2 | 241641282 | C>T | SNED1 | p.T708I | 0.04% | 0.32% | 36.52% | 2.30% | 2 | 2 | 2 | 1.58e-4 | 0.719 |
| 3 | 10236363 | G>T | IRAK2 | e8-1 | 0.38% | 48.37% | 52.69% | 53.33% | 2 | 2 | 2 | 0.156 | 0.762 |
| 3 | 139505123 | G>A | TXNDC6 | p.R221W | 0.29% | 39.50% | 58.62% | 15.81% | 2 | 2 | 2 | 0.039 | 0.012 |
| 3 | 150728323 | A>C | WWTR1 | p.F299V | 0.03% | 6.87% | 28.14% | 9.53% | 2 | 2 | 2 | 1.43e-4 | 0.020 |
| 4 | 40051165 | C>A | CHRNA9 | p.D437E | 0.10% | 28.38% | 36.82% | 90.26% | 2 | 2 | 2 | 0.073 | 9.67e-5 |
| 4 | 40134827 | delG | RBM47 | p.I280fs | 0.05% | 8.62% | 79.15% | 79.74% | 2 | 2 | 2 | 0.030 | 0.124 |
| 4 | 82232630 | C>T | PRKG2 | p.R709 | 0.11% | 6.99% | 82.99% | 91.51% | 2 | 2 | 2 | 0.083 | 0.094 |
| 5 | 135422725 | G>A | TGFBI | p.E576K | 0.17% | 89.09% | 37.58% | 18.45% | 2 | 2 | 2 | 3.34e-6 | 3.23e-5 |
| 5 | 169466048 | C>T | FOXI1 | p.S170F | 0.15% | 69.28% | 78.33% | 93.61% | 2 | 2 | 2 | 0.473 | 0.009 |
| 7 | 100463999 | G>C | MUC17 | p.S861T | 1.69% | 9.22% | 1.46% | 14.43% | 2 | 2.76 | 4.04 | 0.073 | 0.816 |
| 7 | 128284099 | C>T | FLNC | p.N2483 | 0.11% | 0.17% | 18.21% | 0.16% | 2.54 | 2.8 | 2.93 | 0.002 | 0.193 |
| 7 | 148400407 | G>A | ZNF786 | p.F130 | 0.13% | 13.61% | 62.86% | 81.04% | 2.51 | 2.85 | 3.54 | 3.01e-4 | 3.23e-5 |
| 8 | 3232441 | C>A | CSMD1 | p.A409S | 0.04% | 29.75% | 54.22% | 65.18% | 2 | 2 | 2 | 0.355 | 0.141 |
| 8 | 8477326 | C>T | ENSG00000222487 | NULL | 0.11% | 9.61% | 28.43% | 13.74% | 2 | 2 | 2.67 | 0.120 | 0.787 |
| 9 | 5040714 | T>C | JAK2 | p.I166T | 0.09% | 61.63% | 21.93% | 47.40% | 2.83 | 2.67 | 2.84 | 0.246 | 0.999 |
| 9 | 107137789 | G>A | SLC44A1 | p.A132T | 0.08% | 2.59% | 76.14% | 85.31% | 2 | 1.29 | 1.19 | 1.43e-4 | 1.05e-4 |
| 10 | 14603968 | C>T | FAM107B | p.R237Q | 2.65% | 13.53% | 63.25% | 97.88% | 3.7 | 4.04 | 4.76 | 3.29e-6 | 8.54e-8 |
| 10 | 30789749 | C>T | MAP3K8 | p.P461L | 0.11% | 13.33% | 31.72% | 77.47% | 3.44 | 3.71 | 4.21 | 0.002 | 9.67e-5 |
| 10 | 79240899 | G>A | DLG5 | p.D1474 | 0.07% | 32.94% | 76.10% | 74.72% | 2 | 2 | 2 | 6.12e-5 | 0.011 |
| 11 | 12496610 | insATGGAG | PARVA | p.338in_frame_insDG | 0.00% | 1.41% | 10.75% | 10.58% | 2 | 2 | 2 | 0.347 | 0.365 |
| 11 | 48128224 | A>T | PTPRJ | p.K1017N | 0.20% | 1.25% | 32.08% | 57.23% | 2 | 2 | 2.99 | 3.48e-4 | 2.20e-4 |
| 11 | 102687902 | G>A | DYNC2H1 | p.R3867Q | 0.06% | 12.81% | 25.78% | 15.69% | 2 | 2 | 2 | 0.002 | 0.023 |
| 12 | 31122692 | T>G | DDX11 | p.V33G | 0.02% | 44.35% | 40.39% | 57.88% | 1.49 | 1.37 | 1.24 | 0.316 | 0.386 |
| 13 | 76628331 | G>A | MYCBP2 | p.Q2222* | 0.10% | 87.84% | 43.76% | 36.95% | 2 | 2 | 2 | 0.004 | 0.003 |
| 13 | 100688137 | A>T | NALCN | p.D468E | 0.16% | 18.60% | 87.66% | 1.65% | 2 | 2.74 | 2.92 | 0.004 | 0.216 |
| 14 | 19285546 | G>T | OR4Q3 | p.L40 | 0.22% | 36.94% | 40.31% | 32.28% | 2 | 2 | 2 | 0.313 | 0.107 |
| 16 | 66569387 | T>G | DPEP3 | p.R262S | 0.84% | 45.61% | 39.43% | 76.59% | 2 | 2.9 | 3.02 | 0.293 | 6.93e-4 |
| 16 | 82828230 | C>A | KCNG4 | p.G121 | 0.04% | 4.15% | 26.82% | 69.89% | 2.43 | 3.09 | 3.49 | 0.083 | 0.259 |
| 17 | 7519157 | insG | TP53 | p.Q167fs | 4.61% | 79.40% | 62.62% | 97.96% | 2 | 2 | 2 | 0.085 | 0.003 |
| 17 | 32904736 | C>T | TADA2L | p.R339W | 0.12% | 17.49% | 59.92% | 79.47% | 2 | 2 | 2 | 0.002 | 0.002 |
| 19 | 12363315 | G>A | ZNF799 | p.H299 | 0.17% | 2.05% | 26.23% | 11.81% | 2 | 2 | 3.06 | 0.062 | 0.618 |
| 19 | 16006577 | insA | ENSG00000167459 | p.I38fs | 4.82% | 26.53% | 48.47% | 37.74% | 2 | 2 | 3.06 | 0.286 | 0.809 |
| 20 | 5851563 | G>A | CHGB | p.R258Q | 0.14% | 35.64% | 45.50% | 54.87% | 2.57 | 2.86 | 3.64 | 0.057 | 0.005 |
| 21 | 45015744 | G>A | UBE2G2 | p.I158 | 0.12% | 21.57% | 26.72% | 20.89% | 2 | 2 | 2 | 0.522 | 0.728 |
| X | 15731812 | C>G | ZRSR2 | p.A95G | 1.01% | 64.01% | 58.66% | 72.08% | 2.51 | 2.77 | 2.99 | 0.137 | 0.969 |
| X | 43893087 | C>G | EFHC2 | e15-1 | 0.01% | 9.88% | 23.15% | 7.35% | 2 | 2.68 | 2.82 | 0.114 | 0.381 |
| X | 46318872 | insA | CHST7 | p.T188fs | 0.19% | 3.67% | 54.36% | 38.84% | 2 | 2.68 | 2.82 | 0.073 | 0.058 |
| X | 105040331 | C>A | NRK | p.A681E | 0.12% | 4.08% | 30.84% | 52.45% | 2 | 2 | 2 | 0.085 | 0.017 |
| X | 129374039 | A>G | RBMX2 | p.K169E | 0.30% | 11.88% | 38.36% | 69.46% | 2 | 2.65 | 2.77 | 0.002 | 0.003 |
Gene sets from Ensembl build 54 and Genbank (downloaded in May 2009) were used for annotation of mutations. T: primary tumor; M: metastasis; X: xenograft.
1A. Mutations with comparable frequencies in three tumors
We detected a JAK2 mutation (I166T), residing in the FERM domain, which is different from the previously reported activating mutations in myeloproliferative diseases, often found in the pseudokinase domain11. Screening of an additional 116 breast tumors identified another mutation (R1122P) in the kinase domain of JAK2 from a Luminal B-type breast cancer. A splice site mutation (e8-1) was found in IRAK2. We performed an RT-PCR experiment using RNAs from the brain metastasis and xenograft and found that the first 30 nucleotides of exon 8 (IRAK2, NM_001570) were skipped and an internal exonic AG site was used as a splice acceptor, resulting in an in-frame deletion. A missense mutation (A401S) in CSMD1 was found in all three tumors. Loss of CSMD1 expression is associated with poor survival in invasive ductal breast carcinoma12 and it is frequently deleted in colorectal adenocarcinoma and head/neck carcinomas13. We also identified 3 missense (E608K, T1456R, and Q2204R) and 1 nonsense (Q3005*) mutations in CSMD1 in 4 breast cancers out of 116 screened. A binomial test shows that CSMD1 is significantly mutated in breast cancer (P = 0.022 and FDR = 0.197)(Supplementary Table 4).
1B. Mutations highly enriched in metastasis and/or xenograft
A missense mutation (A681E) in NRK, a protein kinase involved in activating JNK, was found to be present in all three tumors, but at 8- and 13-fold increased allele frequencies in the metastasis and xenograft, respectively (Figure 2 and Table 1). Two somatic mutations (S424C and Q521*) in NRK have been previously reported in breast cancer14. The missense mutation (P461L) identified in the C-terminus of MAP3K8 was present at a roughly 6-fold increase in the xenograft compared to the primary tumor. C-terminal truncation of MAP3K8 has been shown to activate this oncogenic kinase15,16, raising the possibility that this C-terminal substitution (P461L) is an activating mutation.
Another missense mutation (K1017N) in PTPRJ, a protein tyrosine phosphatase, had a mutant allele frequency of 32% in the metastasis and 57% in the xenograft compared to just 1.3% in the primary tumor. This K1017N mutation in PTPRJ is among the most highly enriched mutations in both the metastasis (FDR = 0.00035) and xenograft (FDR = 0.00022). The mutation site is in the juxtamembrane domain (a basic residue motif) and is in close proximity to the tyrosine-protein phosphatase domain (aa 1041-1298). Sacco et al.17 reported that the PTPRJ charged peptide (aa 1013-1024) is responsible for interaction with its substrates, such as ERK1/2. The K1017N mutation found in the basal tumor and the K1016A mutation described in Sacco's report both change a basic residue to a neutral residue, suggesting these two mutations may be functionally similar. A missense mutation (F299V) in WWTR1, assigned as deleterious by SIFT18, was detected at 28% mutant allele frequency in metastasis, but only at 7% and 10% in primary tumor and xenograft, respectively (Figure 2 and Table 1). WWTR1, a 14-3-3 binding protein with a PDZ binding motif, has been shown to modulate mesenchymal stem cell differentiation19. Over-expression of WWTR1 has also been implicated in promoting the migration, invasion, and tumorigenesis of breast cancer cells20.
Another point mutation (R258Q) was identified in CHGB (chromogranin B) encoding a tyrosine-sulfated secretory protein. A SNP at the same position was reported to dbSNP in January 2009 for a Yoruba sample. It was also assigned as a germline site in another African-American with breast cancer when we genotyped this mutation in 112 additional primary tumors and 73 metastatic tumors of various expression classes (Supplementary Information). To investigate this variant further, 84 cancer-free African American women with an average age of 71.2 yrs (low risk for developing breast cancer) and 38 early-onset African American breast cancer patients with an average age of 35.6 yrs were genotyped. The results indicated that 8 out of 84 controls and 3 out of 38 cases carried the variant allele, suggesting this variant is unlikely to be a breast cancer susceptibility allele.
Three validated indels were enriched in the metastasis and/or xenograft. One was the 1-bp insertion in exon 4 of the TP53 gene, which creates a frameshift mutation (Q167fs) in the DNA binding domain and results in a truncated protein. We found the TP53 mutation significantly enriched in the xenograft, while present at a relatively constant frequency in primary tumor and metastasis (Figure 2 and Table 1).
1C. Mutations enriched in the primary tumor
A nonsense mutation (Q2222*) in MYCBP2 and a missense mutation (E576K) in TGFBI, both found in all three tumors, had higher mutant allele frequencies in the primary tumor (88% for MYCBP2 and 89% for TGFBI) than in the metastasis (44% for MYCBP2 and 38% for TGFBI) or the xenograft (37% for MYCBP2 and 18% for TGFBI) (Figure 2 and Table 1).
2. De novo mutations identified in the metastasis
Two de novo mutations were discovered in the metastatic tumor, neither of which was detected in the primary or xenograft tumor genomes. One was a missense mutation (T708I) in SNED1, with a mutant allele frequency of 37%. The other was a silent mutation (N2483) in FLNC with a mutant allele frequency of 18% (Figure 2 and Table 1). Since the xenograft line, without these two mutations, exhibits metastatic lesions in ovarian, lymphoid, and subcutaneous tissue (data not shown), it is unlikely that these mutated genes are essential to the metastatic process.
Elevated copy number alterations in metastasis and xenograft
The cnvHMM algorithm (unpublished) was applied to the aligned sequence reads to detect regions of copy number alterations in all three tumors. Using pathology-based purity estimates for the primary tumor and brain metastasis, we calculated the read depth contributed from the tumor cells alone and then computed the copy number for all genomic positions. Read depth correction was not applied to the xenograft, as stated earlier. We subsequently compared the copy number data from all three tumors with those from peripheral blood, to identify genomic segments with significant copy number alterations (CNAs) (Supplementary Information). A total of 516.5 Mb, 640.4 Mb, and 754.5 Mb were amplified, while 342.5 Mb, 383.1 Mb, and 562.5 Mb were deleted, in primary tumor, metastasis, and xenograft, respectively (Supplementary Table 5-7). Moreover, 96.11% and 93.98% of CNA sequences in the primary tumor also were found in CNA segments in the metastasis and xenograft respectively, suggesting most primary tumor CNAs are preserved during disease progression and engraftment. On the other hand, only 80.65% of metastasis and 61.29% of xenograft CNA sequences overlap with primary tumor CNAs. Furthermore, 155 regions with focal copy number segments (≤ 2Mbp) were detected in the primary tumor, but only 101 and 97 regions in the metastasis and xenograft (Supplementary Table 8-10). Our result also shows that 111 (average span = 745,183 bp) and 99 (average span = 799,395 bp) focal copy number segments (≤ 2Mbp) in the primary tumor overlap with broader copy number segments in the metastasis (average span = 2,245,546 bp) and xenograft (average span = 3,565,456 bp), suggesting possible expansion of primary focal regions or selection of new adjacent events during disease progression and in the mouse host. Sequence depth-based copy number analysis shows overall the highest concordance with other platforms, including the array CGH and Illumina SNP array, and also provided the highest concordance of copy number (correlation coefficients: 0.89-0.97) between primary tumor, metastasis, and xenograft(Supplementary Table 11).
Common and unique structural variations in three tumors
We used BreakDancer 21 to detect structural variants (SV) in sequencing data from paired end libraries (Supplementary Table 12) and applied a set of thresholds to identify putative somatic structural events.
1. Deletions, insertions, and inversions
Breakpoint-containing contigs from the three tumor samples, that were not present in the matched normal genome, were successfully assembled for 137 deletions, 15 insertions, and 38 inversions using the TIGRA assembler (unpublished), suggesting they were putative somatic events. We then re-mapped individual reads to these assembled contigs to screen out germline SVs and to confirm somatic SVs (Supplementary Information), resulting in the detection of 59 deletions and 18 inversions. PCR primers were designed successfully to validate 73 out of 77 putative SV events and the resulting amplicons were sequenced by either the Roche454 or ABI 3730 platform. Subsequently, 28 deletions and 6 inversions were validated as somatic events (Table 2). Among them, a 46,462 bp heterozygous deletion in FBXW7 removes the last 10 exons and a portion of the first exon of NM_018315, likely inactivating FBXW7. FBXW7 targets Cyclin E and mTOR for ubiquitin-mediated degradation22,23. Numerous cancer-associated mutations in FBXW7 have been previously reported, and loss of FBXW7 function causes chromosomal instability and tumorigenesis24. Two overlapping deletions (538,467 bp and 515,465 bp in length) on chromosome 5, affecting CTNNA1 along with LRRTM2, MATR3, SNORA74A, and SIL1 also were validated. This result is consistent with the detection of a focal copy number deletion encompassing this region in both metastasis (copy number = 0.65) and xenograft (copy number = 0.03) (Figure 3 and Supplementary Table 9 and 10). Careful examination of this region in the aligned sequence reads for the primary tumor confirms the existence of copy number deletion. Loss of CTNNA1 was shown to result in global loss of cell adhesion in human breast cancer cells25 and increased in vitro tumorigenic characteristics26, suggesting this bi-allelic deletion has functional importance. A 109,563 bp heterozygous deletion on chromosome 8 was assembled and validated in all three tumors. This event removed three exons of NRG1, which encodes a peptide growth factor that binds to ERBB3 and ERBB4. Interestingly, a 26,919 bp deletion in MECR was only identified, assembled, and validated in the metastasis, suggesting its de novo nature in this sample.
Table 2.
Validated structural variations.
| Type | Tumor source | Chromosome A | Breakpoint A | Orientation A | Chromosome B | Breakpoint B | Orientation B | Event size | Gene |
|---|---|---|---|---|---|---|---|---|---|
| Translocation | T,M,X | 1 | 245548334 | minus | 2 | 64855174 | plus | ZNF496 | |
| Translocation | T,M | 1 | 245548342 | Plus | 6 | 144243130 | plus | ZNF496, C6orf94 | |
| Translocation | T,M,X | 2 | 64855565 | Plus | 6 | 144243118 | minus | C6orf94 | |
| Translocation | T,M,X | 2 | 165126335 | plus | 16 | 4537866 | plus | GRB14 | |
| Translocation | T,M,X | 4 | 188855443 | plus | 9 | 139022260 | plus | ABCA2 | |
| Translocation | T,M,X | 12 | 10874022 | plus | 14 | 99382256 | minus | EML1 | |
| Translocation | T,M | 19 | 17188977 | minus | 3 | 188010735 | plus | USE1 | |
| Inversion | T,M,X | 1 | 35703682 | 1 | 35732148 | 28465 | KIAA0319L | ||
| Inversion | T,M,X | 1 | 95919529 | 1 | 95920940 | 1410 | |||
| Inversion | T,M,X | 1 | 204459097 | 1 | 204461297 | 2200 | |||
| Inversion | T,M,X | 1 | 204459547 | 1 | 204460581 | 1033 | |||
| Inversion | T,M,X | 4 | 177886041 | 4 | 177890171 | 4129 | VEGFC | ||
| Inversion | T,M,X | 19 | 17800861 | 19 | 17801858 | 996 | JAK3 | ||
| Deletion | M | 1 | 29389213 | 1 | 29416133 | 26919 | MECR | ||
| Deletion | T,M,X | 1 | 76496719 | 1 | 76496797 | 79 | ST6GALNAC3 | ||
| Deletion | T,M,X | 1 | 88291885 | 1 | 88292292 | 406 | |||
| Deletion | T,M,X | 2 | 18629189 | 2 | 19196656 | 567466 | NT5C1B | ||
| Deletion | T,M,X | 2 | 64853205 | 2 | 65010694 | 157488 | |||
| Deletion | T,M,X | 2 | 128745303 | 2 | 128898612 | 153308 | HS6ST1 | ||
| Deletion | T,M,X | 4 | 1203395 | 4 | 1265560 | 62164 | CTBP1 | ||
| Deletion | T,M,X | 4 | 135737399 | 4 | 135738718 | 1318 | |||
| Deletion | T,M,X | 4 | 147221480 | 4 | 147294628 | 73147 | AK057233 | ||
| Deletion | T,M,X | 4 | 153446894 | 4 | 153493357 | 46462 | FBXW7 | ||
| Deletion | T,M,X | 5 | 15572469 | 5 | 15572649 | 179 | FBXL7 | ||
| Deletion | T,M,X | 5 | 130743604 | 5 | 130743718 | 113 | CDC42SE2 | ||
| Deletion | T,M,X | 5 | 138131495 | 5 | 138669963 | 538467 | CTNNA1, LRRTM2, MATR3, SNORA74A, SIL1 | ||
| Deletion | T,M,X | 5 | 138141753 | 5 | 138657219 | 515465 | CTNNA1, LRRTM2, MATR3, SNORA74A, SIL1 | ||
| Deletion | T,M,X | 6 | 39689264 | 6 | 39689652 | 387 | KIF6 | ||
| Deletion | T,M,X | 7 | 999743 | 7 | 999984 | 240 | |||
| Deletion | T,M,X | 7 | 135419232 | 7 | 135419453 | 220 | |||
| Deletion | T,M,X | 8 | 32597100 | 8 | 32706664 | 109563 | NRG1 | ||
| Deletion | T,M,X | 8 | 116552846 | 8 | 116634665 | 81818 | TRPS1 | ||
| Deletion | T,M,X | 8 | 136595795 | 8 | 136596285 | 489 | KHDRBS3 | ||
| Deletion | T,M,X | 9 | 2746534 | 9 | 2746735 | 200 | |||
| Deletion | T,M,X | 10 | 77142378 | 10 | 77142881 | 502 | C10orf11 | ||
| Deletion | T,M,X | 11 | 115974418 | 11 | 115974688 | 269 | |||
| Deletion | T,M,X | 11 | 125479377 | 11 | 125479744 | 366 | |||
| Deletion | T,M,X | 17 | 24451601 | 17 | 24475255 | 23653 | MYO18A | ||
| Deletion | T,M,X | 17 | 73733446 | 17 | 73733547 | 100 | BIRC5 | ||
| Deletion | T,M,X | 18 | 46765510 | 18 | 46768017 | 2507 | ELAC1 | ||
| Deletion | T,M,X | X | 149511547 | X | 149548642 | 37094 | MTM1 |
T: primary tumor; M: metastasis; X: xenograft.
Figure 3. Two overlapping CTNNA1 deletions on chromosome 5 in three tumors.
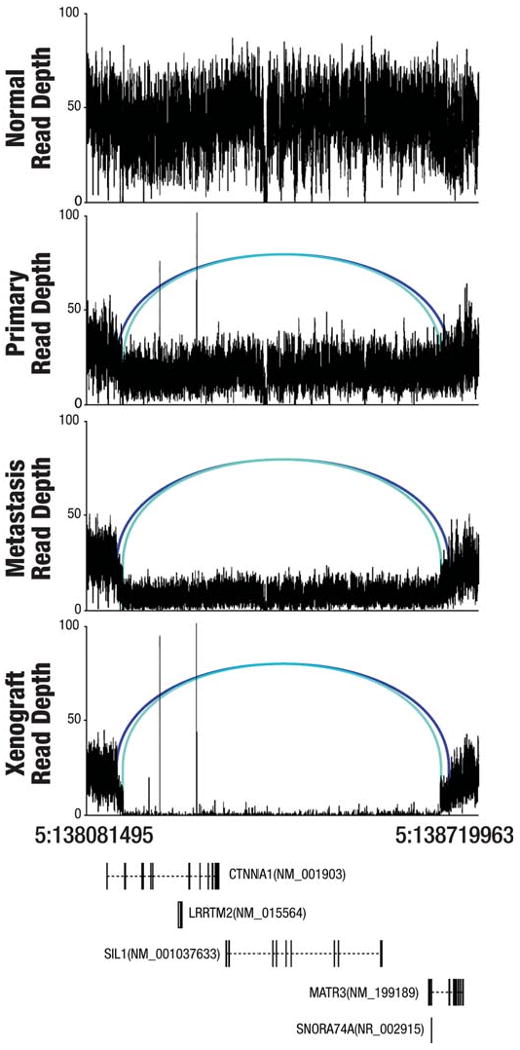
A graph of sequence depths, read pairs, and genes in a 638,468 bp region containing two overlapping deletions. The top four panels display the read depths at each base and the reads within the region whose mates mapped at an abnormal distance are displayed as blue bars, with matched pairs connected by arcs. Two different shades of blue indicate the two separate allelic deletion events (538,467 bp and 515,465 bp in length). The bottom panel displays genes annotated in this genomic region.
2. Translocations
Of the 112 assembled putative translocations, 34 passed manual review using Pairoscope graphs (unpublished), and 19 with an assembly score greater than our experimentally-supported cutoff of 10 were included in Supplementary Table 13. Seven translocations were experimentally validated (Table 2). One validated translocation t(4;9)(188855443;139022258), assembled in all three tumors, involved an LTR from the ERVL-MaLR family on chromosome 4 and ABCA2 on chromosome 9. The translocation removes the final exon of the ABCA2 gene (NM_001606). Two other validated translocations, identified in all three tumors, are t(1;2)(245548338;64855172) and t(2;6)(64855607;144243116) (Supplementary Figure 1). Noticeably, the breakpoints on chromosome 2 for these two translocations are only separated by 393 bp in a TcMar-Tigger repeat. The chromosome 1 breakpoint of t(1;2)(245548338;64855172) is in intron 5 of NM_032752 in ZNF496. We expect the translation of ZNF496 to continue through exon 5 into intron 5 due to lack of a splice acceptor site. On the other hand, t(2;6)(64855565;144243116) involves FAM164B on chromosome 6 and the translocation contig retains 3 exons of XM_928657. We have also validated t(1:6)(245548342;144243110) (not detected by BreakDancer), whose breakpoints are only 4 bp and 6 bp away from the breakpoints identified on chromosomes 1 and 6 for t(1;2)(245548338;64855172) and t(2;6)(64855607;144243116), respectively (Supplementary Figure 1). This translocation is found in both the primary tumor and the metastasis, but apparently is lost in the xenograft (Supplementary Figure 1 and 4). Sequencing of two PCR products generated using two primer pairs from chromosomes 1 and 6 demonstrated the presence of two forms of genomic fusions: one includes chromosomes 1 and 6 and the other includes chromosomes 1, 2, and 6. The former is only present in the primary tumor and the metastasis.
Figure 4. Circos plots for the primary tumor, metastasis, and xenograft genomes.
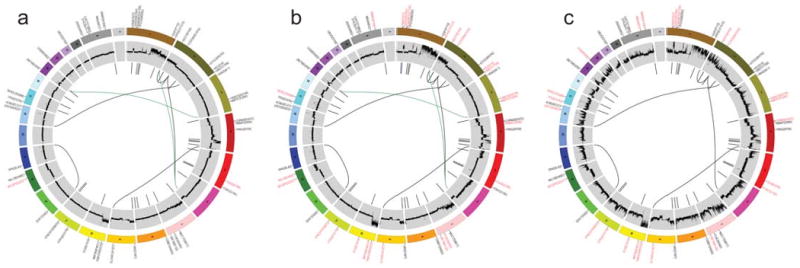
Circos30 plots display the validated tier 1 somatic mutations, DNA copy number, and validated structural rearrangements in the primary tumor (a), metastasis (b), and xenograft (c). Mutations enriched in the primary tumor (a) are labeled in red and mutations enriched in the metastasis or xenograft are in red (b and c). Mutations and the large deletion unique to the metastasis are in blue (b). Translocations only present in primary tumor and metastasis are in green. All shared events are in black. The copy number difference between the tumor and normal is shown (scale: -4 to 4). No purity-based copy number corrections were used for plotting.
Discussion
Our comprehensive analysis of this sample set identified 50 novel somatic point mutations and small indels in coding sequences, RNA genes, and splice sites as well as 28 large deletions, 6 inversions and 7 translocations. In terms of functional annotation, a hierarchy can be suggested. The first level includes somatic changes likely to be functional, such as the small indel in TP53, the large heterozygous deletion in FBXW7, and the bi-allelic deletion in CTNNA1. The second level consists of non-synonymous mutations in genes previously noted to be targeted for somatic mutation in cancer or found to be recurrently mutated in this study, although the exact mutations are novel and their functional importance requires further investigation (JAK2, PTCH2, CSMD1, and NRK). The third level contains mutations known to be related to signal transduction in the malignant cells and/or found to be enriched during disease progression (MAP3K8, PTPRJ, and WWTR1). The final level, by far the largest group, awaits the acquisition of new data. Analysis of germline variants for over 500 classic tumor suppressor genes and oncogenes27 identified a large number of SNPs, none of which were unequivocal hereditary breast cancer susceptibility alleles (data not shown).
The wide range of mutant allele frequencies suggests considerable genetic heterogeneity in the cellular population at the primary site. The mutation frequency range narrowed in brain metastasis and xenograft, suggesting the metastatic and transplantation processes selected for cells utilizing a distinct subset of the primary tumor mutation repertoire. The overlap between the mutation frequency changes seen in the metastatic and xenograft samples argues that genomic progression during xenograft formation is similar to that during metastasis. Moreover, it suggests that the changes were not therapy-related, since the xenograft was established prior to any treatment. GO annotation of enriched mutations suggests that transcription factor activity is possibly selected in xenograft (Supplementary Table 14). In contrast to our observation of only two new tier 1 mutations at the metastatic site, sequencing of an indolent metastatic lobular breast tumor showed that the great majority of the mutations detected were completely novel when compared to the primary tumor9. However, in this instance, the metastatic process evolved over nine years, as opposed to less than one year in the case we describe here. Another difference relative to the lobular cancer genome, where no structural variants were validated, was that paired-end sequencing detected 41 structural variations within this basal-like tumor genome. Our study of a primary tumor-metastasis-xenograft trio therefore demonstrates that, while additional somatic mutations, copy number alterations, and structural variations do occur during the clinical course of the disease, most of the original mutations and structural variants present in the primary tumor are propagated. The preservation of all primary mutations in the xenograft suggests that early passage xenograft lines are valid for functional and therapeutic studies. However, the altered mutation frequency and elevated degree of copy number alterations suggest caution when interpreting the results of such experiments.
In conclusion, the first completed basal-like breast cancer genome is highly complex, as would be anticipated from a tumor-type associated with chromosomal instability and DNA repair defects. Indeed this cancer genome, in comparison with the two AML cases published recently by our group27,28, revealed a 3-4 fold increase in high confidence SNVs genome-wide, suggesting a much greater background mutation rate. Future studies should extend our analysis approach of primary, metastatic, and normal tissue trios and include affected individuals with diverse geographic origins to produce a complete catalog of recurrent somatic and inherited variants associated with the development of this common malignancy.
Methods Summary
Illumina reads from peripheral blood, primary tumor, metastasis, and xenograft were aligned to NCBI build36 using MAQ5 and coverage levels were defined by comparison of SNPs identified by Illumina 1M duo arrays to SNVs called by MAQ. Somatic mutations were identified using our in-house programs glfSomatic and a modified version of the Samtools indel caller (http://samtools.sourceforge.net/). Putative variants were manually reviewed and then validated by Illumina, 3730 or 454 sequencing. Structural variations were identified using BreakDancer21, manually reviewed and validated by a combination of localized Illumina read assembly, PCR and either 3730 or 454 sequencing. A complete description of the materials and methods used to generate this data set and results is provided in the Supplementary Information.
Supplementary Material
Acknowledgments
We thank the many members of The Genome Center and Siteman Cancer Center at Washington University in St. Louis for support. This work was funded by grants to R.K.W. (Richard K. Wilson) from Washington University in St. Louis and the National Human Genome Research Institute (NHGRI U54 HG003079), and grants to M.J.E. (Matthew J. Ellis) from the National Cancer Institute (NCI 1 U01 CA114722-01), the Susan G Komen Breast Cancer Foundation (BCTR0707808), and the Fashion Footwear Charitable Foundation, Inc. NCI U10 CA076001 and a Breast Cancer Research Foundation grant awarded to the American College of Surgeons Oncology Group supported the acquisition of samples for recurrence testing. The tissue procurement core was supported by an NCI core grant to the Siteman Cancer Center (NCI 3P50 CA68438). The Human and Mouse Linked Evaluation of Tumors Core was supported by the Institute of Clinical and Translational Sciences at Washington University (CTSA grant UL1 RR024992). Lastly, we thank Illumina, Inc. for their support and role in the Washington University Cancer Genome Initiative.
Footnotes
Author Contributions: E.R.M., L.D., R.S.F., M.J.E., T.J.L., and R.K.W. designed the experiments. L. D. and M.J.E. led data analysis. L.D., D.E.L., K.C., J.W.W., C.C.H., M.D.M., D.C.K., Q.Z., H.S., J.K., L.C., L.L., M.C.W., N.D.D., D.S., D.M.T., J.L.I., P.J.G., J.S.H., W. S., G.M.W., and Y.T. performed data analysis. D.E.L., C.C.H., J.W.W., J.F.M., and L.D. prepared figures and tables. R.S.F., L.L.F., R.M.A., J.H., K.D.D., C.C.F., K.A.P., J.S.R., V.J.M., L.C., S.D.M., T.L.V., E.A., K.D., S.D., T.G., L.L., R.C., J.E.S., D.P.L., M.E.W., M.C., G.E., M.D.M., D.M.T., J.L.I., and P.J.G. performed laboratory experiments. S.L. and M.J.E. created the xenograft line. M.J.E., M.W., and R.A. provided samples. D.J.D., S.M.S., A.F.D., G.E.S., C.S.P., J.M.E., J.B.P., B.J.O., J.T.L., F.D., A.E.H., M.D.O., and K.E.B. provided informatics support. L.D., M. J. E., E. R. M., and R. K. W. wrote the manuscript. T.J.L., D.E.L, M.C.W., D.C.K., and C.M.P. critically read and commented on the manuscript.
References
- 1.Carey LA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 2.Citron ML, et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21(8):1431–1439. doi: 10.1200/JCO.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 3.Kuperwasser C, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101(14):4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18(11):1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bardelli A, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300(5621):949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- 7.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 463(7278):184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 463(7278):191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461(7265):809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 10.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 11.Mullighan CG, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106(23):9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamal M, et al. Loss of CSMD1 expression is associated with high tumour grade and poor survival in invasive ductal breast carcinoma. Breast Cancer Res Treat. 2009 doi: 10.1007/s10549-009-0500-4. [DOI] [PubMed] [Google Scholar]
- 13.Toomes C, et al. The presence of multiple regions of homozygous deletion at the CSMD1 locus in oral squamous cell carcinoma question the role of CSMD1 in head and neck carcinogenesis. Genes Chromosomes Cancer. 2003;37(2):132–140. doi: 10.1002/gcc.10191. [DOI] [PubMed] [Google Scholar]
- 14.Stephens P, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37(6):590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 15.Gandara ML, Lopez P, Hernando R, Castano JG, Alemany S. The COOH-terminal domain of wild-type Cot regulates its stability and kinase specific activity. Mol Cell Biol. 2003;23(20):7377–7390. doi: 10.1128/MCB.23.20.7377-7390.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark AM, Reynolds SH, Anderson M, Wiest JS. Mutational activation of the MAP3K8 protooncogene in lung cancer. Genes Chromosomes Cancer. 2004;41(2):99–108. doi: 10.1002/gcc.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacco F, et al. Tumor suppressor density-enhanced phosphatase-1 (DEP-1) inhibits the RAS pathway by direct dephosphorylation of ERK1/2 kinases. J Biol Chem. 2009;284(33):22048–22058. doi: 10.1074/jbc.M109.002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong JH, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309(5737):1074–1078. doi: 10.1126/science.1110955. [DOI] [PubMed] [Google Scholar]
- 20.Chan SW, et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68(8):2592–2598. doi: 10.1158/0008-5472.CAN-07-2696. [DOI] [PubMed] [Google Scholar]
- 21.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Koepp DM. Fbw7 isoform interaction contributes to cyclin E proteolysis. Mol Cancer Res. 2006;4(12):935–943. doi: 10.1158/1541-7786.MCR-06-0253. [DOI] [PubMed] [Google Scholar]
- 23.Mao JH, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321(5895):1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8(2):83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 25.Bajpai S, Feng Y, Krishnamurthy R, Longmore GD, Wirtz D. Loss of alpha-catenin decreases the strength of single E-cadherin bonds between human cancer cells. J Biol Chem. 2009;284(27):18252–18259. doi: 10.1074/jbc.M109.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plumb CL, et al. Modulation of the tumor suppressor protein alpha-catenin by ischemic microenvironment. Am J Pathol. 2009;175(4):1662–1674. doi: 10.2353/ajpath.2009.090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 30.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.