

Abstract
Crystal structures of classical cadherins have revealed two dimeric configurations: in the first, N-terminal β-strands of EC1 domains “swap” between partner molecules. The second configuration (the “X-dimer”), also observed for T-cadherin, is mediated by residues near the EC1-2 calcium binding sites, and N-terminal β-strands of partner EC1 domains, though held adjacent, do not swap. Here we show that strand swapping mutants of type I and II classical cadherins form X-dimers. Mutant cadherins impaired for X-dimer formation show no binding in short timeframe surface plasmon resonance assays but in long timeframe experiments, have homophilic binding affinities close to wild-type. Further experiments show that exchange between monomers and dimers is slowed in these mutants. These results reconcile apparently disparate results from prior structural studies, and suggest that X-dimers are binding intermediates that facilitate the formation of strand swapped dimers.
Introduction
Cadherins constitute a large family of cell surface adhesion receptors whose differential binding is important for the development and maintenance of tissue architecture 1. Cadherins have been identified in vertebrate and invertebrate animals and are defined by the presence of extracellular cadherin-like (EC) domains, β-sandwich domains of ~110 amino acids that contain highly conserved calcium binding regions 2,3. Variations in other sequence features and in the number of EC domains group cadherins into numerous families including the “classical cadherins” which include type I and type II subfamilies, desmosomal cadherins, protocadherins, and others 3. The adhesive properties of vertebrate classical cadherins have been most thoroughly studied. These class I transmembrane proteins include an N-terminal signal sequence followed by a pre-domain that must be removed by proteolysis to activate adhesive function; five EC domains; a single transmembrane segment; and a cytoplasmic domain that contains highly conserved binding sites for catenin proteins which provide indirect links to the cytoskeleton 4.
Intensive study of vertebrate classical cadherins has led to the emergence of a widely accepted view that adhesive binding by these proteins occurs via a strand-swapped interface in EC1, supported by data from a number of laboratories. Crystal structures of the whole EC1-EC5 ectodomain from C-cadherin reveal dimerization interactions between paired ectodomains, oriented as if emanating from apposed cells, between the EC1 domains of each molecule 5. Nearly identical interactions have been found in numerous crystallographic studies of adhesive fragments from other type I cadherins 6–8. Similarly, type II cadherin ectodomain fragment structures also reveal binding interfaces that are exclusively formed by elements from the EC1 domain 9. For both type I and type II cadherins these adhesive binding interfaces are formed through β-strand swapping, in which the N-terminal A* strands spatially swap between partner EC1 domains. In type I cadherins the conserved residue Trp2 anchors the swapped strand, whereas type II cadherins contain two conserved anchor residues, Trp2 and Trp4. The positively charged N-termini of both type I and type II classical cadherins form intermolecular salt bridges in the dimeric structures, providing an explanation for the requirement for precise proteolytic processing 6.
A wide array of data using other investigative methods also supports this structural view. Early domain shuffling experiments showed that the adhesive specificity and hence, presumably, the site of adhesive binding of type I cadherins resides in the EC1 domain10–12. Similar behavior was later demonstrated for type II cadherins both in vitro and in vivo 9. Single-particle electron tomography reconstructions of desmosomes from human and mouse skin reveal EC1-EC1 interactions, and the strand-swapped dimeric structure of C-cadherin can be fit directly into these tomograms 13,14. Mutagenesis data also provides support for this model of cadherin adhesion. Mutants that alter elements of the strand swapping interface, including Trp2 in type I cadherins and Trp2 or Trp4 in type II cadherins, and mutations that fill the Trp2 “acceptor pocket” so a tryptophan side chain cannot be accommodated, abolish cadherin adhesive function 15–17. Further, extensions to the mature N-terminus of one or more amino acids, which would prevent salt bridge formation, also abolish adhesive function 6,18,19.
Despite this compelling data in support of the strand swap model of cadherin adhesive binding, some uncertainty has remained for the role of other interfaces which have been observed in high-resolution structures. Two early crystal structures of recombinant E-cadherin EC1-2 fragments, containing small N-terminal extensions derived from the recombinant production method, revealed a non-swapped dimeric association 20,21. In these structures the partner molecules contact one another at a site near the interdomain calcium binding region, such that the dimeric assembly of the elongated molecules resembles the shape of an “X”. For convenience, we refer to this configuration as an “X-dimer”. Once the importance of the strand swapping mechanism became clear, we formed the incorrect opinion that the X-dimer configuration represented a crystal packing artifact 9. However, a recent crystal structure of T-cadherin EC1-2, reported in the accompanying paper22 revealed an X-dimer that superimposes almost perfectly on the previously determined classical cadherin X-dimer structures, and is likely to represent the primary adhesive interface of this atypical cadherin. This prompted us to investigate potential roles for the X-dimer in classical cadherin function. Here we present data from Mus musculus E-cadherin and cadherin-6 showing that the X-dimer configuration, while apparently providing an adhesive binding interface for T-cadherin, functions as a kinetically important intermediate in the dimerization of classical cadherins by facilitating the conformational transition to the mature strand-swapped adhesive form.
Results
Strand swap-incompetent E-cadherin mutants form X-dimers
Two published crystal structures of E-cadherin EC1-2 fragments, with one or two additional amino acids at their N-terminus due to cloning artifacts20,21, revealed protomers arranged in a configuration we refer to here as an ‘X-dimer’. It was previously suggested that the additional amino acids at the N-termini of those constructs could interfere with the normal strand swapping mechanism and allow the alternate X-dimer configuration to be observed 4,6,18. To test this, we produced three mutants designed to inhibit strand swapping in EC1-2 fragments of mouse E-cadherin. W2A should prevent anchoring of the strand exchanged dimer by the Trp2 indole side chain. The AA extension mutant, with an N-terminus extended by two alanines, and E89A mutant should each prevent formation of a salt bridge between the side chain of Glu89 and the amino terminus of the swapped strand that stabilizes strand exchange 5,18,23.
Structures of these mutants were determined at 2.6Å, 2.7 Å and 2.3 Å resolution respectively (Table 1). All three formed almost identical X-shaped dimers that superimpose with a root mean square deviation (rmsd) of <1 Å over all Cα atoms (Fig. 1a). Each dimer is formed from E-cadherin EC1-2 fragments in an approximately parallel alignment with close contacts principally around the EC1-2 linker regions. The EC1 domains are closely aligned with A strands held adjacent, whereas the EC2 domains extend away from each other, making intermolecular contact at their apex. The resulting dimer has an X-shaped appearance and is essentially identical to the previously published dimers of E-cadherin fragments with extra N-terminal residues 20,21, including the specific interactions in the interface (described in the Supplementary Text and Supplementary Figs. 1–4).
Table 1.
Data collection and refinement statistics
| Ecad EC12 Wild-type | Ecad EC12 AA extension | Ecad EC12 W2A | Ecad EC12 E89A | Ecad EC12 K14E | Ecad EC12 W2AK14E | Ncad EC12 Wild-type | Cad6 EC12 W4A | |
|---|---|---|---|---|---|---|---|---|
| Data Collection | ||||||||
| Space group | C2 | C2 | C2 | C2 | C2 | P1 | C2 | P21 |
| Cell dimensions: a,b,c | 134.71, 42.67, 58.13 | 121,55, 80,17, 72,75 | 120.54, 80.86, 72.52 | 119.63, 77.12, 71.68 | 130.00, 48.54, 54.49 | 45.11, 47.38, 69.90 | 114.66, 86.90, 46.71 | 55,98, 113,56, 67,91 |
| α βγ | 90.00, 111.97, 90.00 | 90,00, 118,79, 90,00 | 90.00, 116.84, 90.00 | 90.00, 115.25, 90.00 | 90.00, 111.63, 90.00 | 70.58, 88.13, 75.50 | 90.00, 97.73, 90.00 | 90,00, 96,01, 90,00 |
| Molecules per asymmetric unit | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 4 |
| Resolution limit (Å) | 2.4 | 2.7 | 2.6 | 2.3 | 2 | 2.5 | 3.2 | 2.82 |
| Unique reflections | 11732 | 13698 | 19548 | 28177 | 19964 | 17683 | 7560 | 20529 |
| Redundancy (Highest resolution shell) | 3.5 (3.1) | 2.6 (2.1) | 3.6 (3.3) | 3.0 (2.8) | 3.2 (1.5) | 2.7 (2.7) | 2.5 (2.1) | 3.3 (2.9) |
| Completeness % (Highest resolution shell) | 95.4 (68.2) | 81.4 (72.8) | 100.0 (99.8) | 99.0 (97.4) | 92.3 (60.9) | 98.6 (97.8) | 90.3 (69.2) | 98.4 (96.4) |
| Average I/σ (I) (Highest resolution shell) | 10.3 (2.5) | 14.0 (3.0) | 12.9 (3.3) | 11.8 (2.5) | 23.4 (4.7) | 8.2 (3.2) | 8.2 (3.9) | 12.9 (2.7) |
| Rmerge (%) (Highest resolution shell) | 8.0 (30.2) | 6.6 (30.4) | 8.6 (38.0) | 9.3 (41.0) | 5.2 (20.0) | 11.1 (40.7) | 11.1 (40.7) | 8.8 (42.6) |
| Refinement | ||||||||
| Rwork (%) | 23.0 | 21.8 | 22.7 | 21.7 | 17.3 | 20.3 | 22.7 | 20.7 |
| Rfree (%) | 28.6 | 26.3 | 26.7 | 27.0 | 21.9 | 24.8 | 29.9 | 28.2 |
| Rmsd bonds (Ǻ) | 0.005 | 0.012 | 0.006 | 0.014 | 0.005 | 0.009 | 0.008 | 0.008 |
| Rmsd angles (°) | 0.885 | 1.343 | 0.995 | 1.429 | 0.938 | 1.170 | 1.164 | 1.051 |
| Protein atoms | 1636 | 3254 | 3196 | 3201 | 1645 | 3176 | 1654 | 6240 |
| Ligand/ion atoms | 4 | 6 | 6 | 6 | 4 | 6 | 3 | 12 |
| Water molecules | 169 | 75 | 157 | 498 | 302 | 169 | 108 | 138 |
| Average B (Ǻ2) protein atoms | 26.5 | 43.3 | 36.9 | 33.6 | 33.0 | 37.6 | 40.7 | 56.1 |
| Average B (Ǻ2) ligand/ion atoms | 24.9 | 31.3 | 23.2 | 21.5 | 48.0 | 36.0 | 33.3 | 42.7 |
| Average B (Ǻ2) water molecules | 27.9 | 33.0 | 34.7 | 37.6 | 46.9 | 35.2 | 26.0 | 46.2 |
Figure 1. Structures of strand-swap mutant and wild-type E-cadherin fragments.

(a) Stereo image showing superposed α-carbon traces of three strand swap interface mutants of E-cadherin EC1-2 that adopt the X-dimer form: W2A (orange), AA-extension (blue) and E89A (green). Trp2 side chains are shown for structures in which the N-terminal strands were resolved (AA extension and E89A mutants). Calcium ions are shown as green spheres. (b) Ribbon diagram showing the strand-swapped dimer formed between symmetry related protomers of wild-type E-cadherin EC1-2 in the crystal. Side chain atoms are shown for Trp2 residues and calcium ions are displayed as green spheres. Green colored protomer is shown in the same orientation as one protomer in the X-dimer structures in panel a to aid comparison.
No strand swapping was observed in the mutant structures, despite close apposition of the A strands. In the W2A mutant, EC1 A-strand residues Asp1-Trp2-Val3-Ile4 were disordered. In the AA-extension and E89A mutants, these residues (except for Asp1) were resolved, showing Trp2 side chains docked into the acceptor pocket of their own protomer (Fig. 1a) as was observed previously in other E-cadherin mutants with extra N-terminal residues 21,24.
We also determined the structure of an EC1-2 fragment of wild-type mouse E-cadherin (Fig. 1b). In contrast to the mutant fragments, the wild-type molecule adopts a strand swapped configuration that is identical to that seen in previously published structures of human and mouse E-cadherin with native N-termini 6,7. In comparison to the X-dimer, the strand swapped dimer has a markedly wider angle between protomers that is maintained by intermolecular contacts involving only the EC1 domain while EC2 and linker regions contribute no buried surface area (Supplementary Table 1).
To determine if X-dimer formation by the E-cadherin mutants could be detected in solution we employed sedimentation equilibrium analytical ultracentrifugation (AUC). The results are summarized in Table 2, and indicate that wild-type protein and AA-extension, W2A and E89A mutants all form homodimers in solution. The measured dissociation constant (KD) for dimerization of the wild-type protein is 98.6 ± 15.5μM from this analysis, in agreement with our previously reported measurements 25. However, all strand-swap mutants dimerized with markedly weaker affinities of at least several hundred micromolar (Table 2), indicating a weak affinity for X-dimer formation. We also prepared a mutant of E-cadherin EC1-2 with a deletion of two N-terminal residues (‘DW-deletion’), which cannot strand swap since the entire swapped structural element is absent. AUC analysis showed this mutant to dimerize with a similar affinity to the W2A and AA-extension mutants (Table 2).
Table 2.
Dissociation constants (KD) for homodimerization of cadherin EC1-2 fragments
| Protein | Description | KD (μM) |
|---|---|---|
| E-cadherin EC1-2 | ||
| Wild type (WT) | Wild type | 98.6 ± 15.5* |
| W2A | strand-swapping mutants | 916 ± 47 |
| AA extension | 811 ± 97 | |
| E89A | 293 ± 11 | |
| DW deletion | 662 ± 28.5 | |
| K14E | X-dimer mutants | 117 ± 8 |
| K14S | 96.0 ± 1.0 | |
| Y142R | 77.4 ± 1.4 | |
| W2A K14E | double interface mutant | (monomer) |
| Cadherin-6 EC1-2 | ||
| Wild type (WT) | Wild type | 3.1 ± 0.1 |
| W4A | strand-swapping mutant | 321 ± 0.5 |
| M188D | X-dimer mutant | 12.6 ± 0.5 |
| W4A M188D | double interface mutant | (monomer) |
Errors indicate data range from two or more experiments
Altogether, our results show that E-cadherin adopts an X-dimer configuration when swapping is prevented, regardless of the mutation used. Further, the X-dimer has a three to ten-fold higher KD than the strand swapped dimer formed by wild-type proteins.
An X-dimer is formed by a swapping-incompetent type II cadherin mutant
To determine whether X-dimer formation is a feature of classical cadherins shared by members of the type II subfamily, we examined a strand swap mutant of cadherin-6. Cadherin-6 is a typical member of the type II classical cadherin subfamily that is >60% identical to cadherin-8 and -11 in its EC1 domain. Previous structural studies have demonstrated that type II cadherins form adhesive dimers via an EC1-EC1 interface in which swapped A* strands are anchored by insertion of the side chains of Trp2 and Trp4 into the opposing hydrophobic cavity 9. Although we were unable to produce crystals of wild-type cadherin-6, its high similarity to other type II cadherins and conservation of Trp2, Trp4, and residues lining the hydrophobic Trp acceptor pocket, strongly suggest that it binds via the same strand exchange mechanism (Supplementary Fig. 3).
We introduced the mutation W4A into an EC1-2 fragment of mouse cadherin-6 to prevent strand swapping. This mutation has previously been shown to disrupt adhesion mediated by another member of the type II subfamily, VE-cadherin 16. The structure of W4A cadherin-6 was determined at 2.8Å resolution and is shown in Figure 2a. The mutant cadherin-6 shows the same overall fold as observed for other type II cadherins. Each EC domain consists of a seven stranded β-barrel with a short linker connecting EC1 and EC2 that binds three calcium ions. Four cadherin molecules are present in the crystallographic asymmetric unit, arranged into two essentially identical dimers, one of which is shown in the figure. The dimer is X-shaped with protomers held approximately parallel and has an interface centered on the EC1-2 linker region with contribution from regions of EC1 and the apex of EC2. The cadherin-6 W4A dimer superimposes remarkably well on the X-dimer conformation seen in strand swap mutants of E-cadherin (see above and 20,21) and in T-cadherin 22 (see Supplementary Text and Supplementary Figs. 1–4).
Figure 2. Structure of X-dimer formed by strand-swap mutant cadherin-6 and comparison with type II cadherin strand swapped dimer.

(a) X-dimer observed in the crystal structure of a cadherin-6 EC1-2 fragment containing the mutation W4A to prevent strand swapping. One of two identical dimers observed in the crystallographic asymmetric unit is shown. (b) Strand swapped dimer formed by wild-type cadherin-11 reported previously by Patel and colleagues 9 as an example of a type II cadherin strand swapped dimer. The blue colored protomer is oriented as in panel a. Structures are shown as ribbon diagrams with protomers colored orange and blue. Side chains of Trp2 and Trp4 are shown where resolved; calcium ions are shown as green spheres. Dotted lines indicate regions of the structure that were disordered.
For comparison, the structure of a closely-related wild-type type II cadherin fragment (cadherin-11 EC1-2) 9 is shown in Figure 2b. This represents a strand exchanged dimer as expected for wild-type cadherin-6. The swapped interface involves only the EC1 domains and has a wider angle between protomers than the mutant cadherin-6 X-dimer (Supplementary Table 1). The EC1 domains themselves are also at a markedly wider relative angle in the strand exchanged dimer (105° compared to 35° in the X-dimer). Details of the W4A cadherin-6 X-dimer interface are shown in Figure 3a. The interface buries 2496 Ǻ2 and 2692 Ǻ2 of surface area in the two dimers in the crystallographic asymmetric unit, in the same range as the strand swapped dimer which buries 2696 Ǻ2 in cadherin-11 (Supplementary Table 1). The interface can be divided into three main regions. First, the AB loop in EC1 is closely apposed to the BC loop in EC2 of the partner molecule, where the Thr14 side chain forms hydrogen bonds with backbone oxygens of Thr137 and Pro136. Second, the EC1-2 linker regions are in close parallel contact in the dimer, with the Ile99 side chains of both protomers contributing buried hydrophobic surface to the interface. Third, the FG loops at the apex of EC2 of both protomers are apposed. Hydrophobic side chains of Met188, Met192 and Leu195 are buried in this region. Intermolecular hydrogen bonds are also formed between the backbone of Met192 and the Ser196 backbone nitrogen, as well as to Asp101 (backbone oxygen) and Gln103 (side chain) in the linker. It is notable that not all interactions are satisfied on both sides of the dimer, owing to slight asymmetry which may arise from crystal packing forces.
Figure 3. Detailed view of the X-dimer interface formed by mutant cadherin-6.

(a) Ribbon view showing details of the X-dimer interface in cadherin-6 EC1-2 W4A mutant from Fig. 2A. Side chains of residues that contribute 10Å or more of buried surface area to the interface are shown. Intermolecular hydrogen bonds are indicated by dashed lines. Protomers are colored blue and orange. Calcium ions are shown as green spheres. (b) Region of the X-dimer interface involving the A strands in cadherin-6 EC1-2 W4A mutant. Side chains are shown for residues 1 through 10 that comprise the A strand, which is disordered above Asn5 in one protomer (orange). Side chain of mutated residue (Trp4 to Ala) is colored cyan.
As is the case for X-dimers of E-cadherin described above, EC1 domains in the cadherin-6 W4A dimer are oriented with their A strands in close apposition (Fig. 3b). Electron density is observed for the complete A strand of only one protomer in each dimer, whereas the strand is disordered in the other through Met3. No A* strand exchange occurs between protomers; instead the side chain of Trp2 in the ordered A strand is inserted into the hydrophobic core of its own protomer. The mutated residue (W4A, cyan) does not dock into either protomer. Interestingly, while A* strands are not exchanged in X-dimers of either cadherin-6 or E-cadherin, they are positioned close to the partner hydrophobic cavity suggesting that the dimer could in principle help to enable strand swapping.
Sedimentation equilibrium AUC analysis revealed that the cadherin-6 W4A mutant forms homodimers in solution and that the X-dimer, as observed for E-cadherin, has a substantially weaker affinity than the strand swapped dimer formed by wild-type cadherin-6 (Table 2).
X-dimer mutations abolish cadherin binding in SPR assays
Our structural studies established that X-dimers are formed when normal strand swapping is prevented by mutations in type I and type II cadherins. In order to determine whether the X-dimer interface has a functional role in cadherin adhesive interactions, where strand swapping occurs, we introduced mutations designed to prevent X-dimer formation in otherwise wild-type E-cadherin and cadherin-6 EC1-2 fragments.
In E-cadherin, Lys14 which forms an intermolecular salt bridge with Asp138 in the X-dimer interface (Supplementary Fig. 2c) was mutated to glutamic acid (K14E mutant). Reversal of the charge on Lys14 should introduce electrostatic repulsion with Asp138 and disrupt X-dimer formation. By contrast, in the strand exchanged dimer Lys14 is 20Å distant from the partner protomer, solvent exposed, and does not participate directly in binding (see later text).
For cadherin-6, Met188, which is conserved in character in type II cadherins, was changed to an aspartic acid (M188D mutant). The non-polar side chain of Met188 projects into the interior of the X-dimer (Fig. 3a and Supplementary Fig. 2j). Introduction of an unpaired charged side chain was therefore predicted to disrupt X-dimer formation. In the strand swapped dimer structure of cadherin-119 residue 188 is solvent exposed and 7Å from the partner protomer, so the mutation should specifically affect only the X-dimer.
The effects of these mutations on cadherin adhesive interactions were assessed using surface plasmon resonance (SPR) assays. C-terminally biotinylated wild-type and mutant cadherin EC1-2 fragments were captured on neutravidin coated sensor chips, resulting in cadherin surfaces oriented with the functional EC1 domains exposed. Untagged wild-type and mutant cadherin fragments were tested for binding to the cadherin surfaces over a time course of one minute. Since cadherins self-associate, a quantitative SPR analysis is not possible 25 so SPR experiments were employed only to assess relative binding (see Methods).
Wild-type E-cadherin EC1-2 showed specific binding when injected over wild-type E-cadherin EC1-2 captured on the sensor chip (Fig. 4a). In contrast, K14E mutant did not bind to the same surface when tested over the same concentration range (Fig. 4b). In a similar experiment, no binding was detected between immobilized K14E mutant and either wild-type protein or K14E mutant (Fig. 4c, d).
Figure 4. Biophysical characterization of E-cadherin and cadherin-6.

(a-h) SPR binding assays. Purple labels represent the ligand, tethered on the sensor chip surface; green labels represent the analyte, injected in solution. Binding of soluble E-cadherin (a) and E-cadherin K14E (b) to an E-cadherin-immobilized surface. In the same experiment, soluble E-cadherin (c) and E-cadherin K14E (d) were tested for binding over an E-cadherin K14E-immobilized surface. Interaction of soluble cadherin-6 (e) and cadherin-6 M188D (f) was tested over a cadherin-6-immobilized surface. In the same experiment, binding of soluble cadherin-6 (g) and soluble cadherin-6 M188D (h) with immobilized cadherin-6 M188D was also tested. All proteins were tested at a concentration range of 100μM (top trace) to 0.39 μM (bottom trace). (i-j) Sedimentation equilibrium AUC experiments showing similar sedimentation profiles at equilibrium for wild-type and K14E E-cadherin (i) and for wild-type and M188D mutant cadherin-6 (j). Profiles of strand dimer mutants E-cadherin W2A and cadherin-6 W4A are plotted for comparison (yellow traces). Calculated KD values for dimerization of all proteins are displayed in Table 2.
Similar results were observed for the X-dimer inhibiting mutation in cadherin-6. Wild-type cadherin-6 EC1-2 bound to wild-type cadherin-6 EC1-2 tethered on the sensor chip (Fig. 4e). Its M188D mutant, however, failed to bind to the wild-type protein whether it was used as the analyte (in the solution phase) or the ligand (tethered) (Fig. 4f, g). When M188D mutant was used as both the analyte and the ligand a severely diminished binding response of <10 Response Units (RU) was observed (Fig. 4h).
Together the SPR data show that prevention of X-dimer formation by our mutations abolishes binding interactions of E-cadherin and cadherin-6. This is an unexpected finding since cadherin binding is mediated by the strand exchange mechanism which should not be affected directly by these structurally-designed mutations.
Dimerization affinities in AUC are unaffected by X-dimer mutations
In a complementary approach, we used sedimentation equilibrium AUC to measure homodimerization of the X-dimer mutants and wild-type cadherins. In these experiments, the proteins are allowed to establish a monomer-dimer equilibrium in solution throughout a ~20 hour centrifuge run. This is a thermodynamic method which is unaffected by kinetic or hydrodynamic factors 26.
Surprisingly, AUC results for the X-dimer mutants E-cadherin (K14E) and cadherin-6 (M188D) were almost indistinguishable from those obtained from the respective wild-type proteins (Fig. 4i, j). Both dimerize with KD values that are comparable to the non-mutated proteins (Table 2). In comparison, mutations that affect strand swapping had dramatic effects on homodimerization affinities as reported above (Fig. 4i, j; Table 2).
These AUC data indicate that the thermodynamics of cadherin homodimerization are not substantially affected by the X-dimer interface mutations. This is consistent with the observation that the mutated residues lie far from the dimerization interface in the mature strand swapped structures. As a consequence, the dramatic effects of the same mutations in SPR assays cannot be attributed to lowered affinity of the strand swapped dimer. Given the short time scale of SPR experiments in comparison to equilibrium AUC (minutes versus hours) we reasoned that differences in the kinetics of homodimerization between the mutant and wild-type proteins might underlie the differing character of results from these two methods.
Kinetics of monomer-dimer exchange are slowed in X-dimer mutants
To investigate whether dimerization kinetics were altered in the X-dimer mutants we employed sedimentation velocity AUC. Unlike equilibrium AUC, this method does not require that equilibrium between sedimentation and diffusion be established during the run. For this reason the timescale of the experiment is shorter (~45 minutes) and the technique is sensitive to dimerization kinetics. Balbo and colleagues have described theoretically the sedimentation velocity behavior of monomer-dimer equilibria with ‘fast’ or ‘slow’ kinetics 27. Broadly, ‘fast’ equilibria in which substantial exchange between monomer and dimer occurs within the timescale of the experiment will sediment as a single species, representing a mixture of monomers and dimers in rapid exchange. Equilibria with slower kinetics such that little or no exchange occurs during the experiment will exhibit sedimentation of two distinct species representing monomers and dimers that are ‘trapped’ in their current state.
We compared wild-type and X-dimer mutant (K14E) E-cadherin EC1-2 fragments using this method (Fig. 5a, b). Wild-type E-cadherin fragments sedimented as a single species with S ~1.8S (Fig. 5a). In contrast, the X-dimer mutant (K14E, Fig. 5b) sedimented as two distinct species at ~1.5S and ~2.0S. Analysis of a monomeric mutant E-cadherin fragment in which strand swapping and X-dimer formation were prevented (W2A K14E, Table 2) confirmed that the ~1.5S species represents monomer (data not shown). Modeling of sedimentation coefficients using the program HydroPro 28 identified the ~2.0S K14E species as dimeric. Results at lower protein concentrations (43μM) confirm that monomer is more highly represented as predicted; wild-type protein sediments as a single peak in all experiments (Supplementary Fig. 5). Therefore, while the wild-type protein displays sedimentation behavior characteristic of a rapidly exchanging monomer-dimer equilibrium, the X-dimer mutant K14E exhibits a slowly exchanging equilibrium in which very little interconversion between monomers and dimers occurs over the duration of the experiment (approximately 45 minutes).
Figure 5. Sedimentation velocity AUC and size exclusion chromatography of X-dimer mutants.

(a, b) AUC sedimentation velocity profiles of wild-type and X-dimer mutant (K14E) E-cadherin EC1-2 fragments at 86μM protein concentration. Sedimenting species are defined by their sedimentation (S) and frictional coefficients (f/f0). (c-f) Analytical size exclusion chromatography of wild type and X-dimer mutants of E-cadherin EC1-2 (c, d) and cadherin-6 EC1-2 (e, f). Separate monomer and dimer peaks were observed in the mutant samples only. Sample concentrations for E-cadherin were approximately 200μm (blue trace), 100μM (green) and 50μM (red); and for cadherin-6: 40μM (blue) 20μM (green), 10μM (red) and 5μM (orange). Void volume of the column was approximately 7.7mL. Equal fractions of elution were separated by SDS PAGE and Coomassie stained to confirm that all peaks contained the cadherin band at around 23kDa (lower panels).
We confirmed these findings using size exclusion chromatography. Wild-type and K14E mutant E-cadherin EC1-2 fragments were passed through an analytical Superose 12 column under identical conditions at several concentrations spanning the KD for homodimerization. Mirroring the velocity AUC results, wild-type protein eluted as a single peak whereas X-dimer mutant K14E eluted in two distinct peaks representing monomer and dimer (Fig. 5c, d). The position of the wild-type peak was intermediate between the monomer and dimer peaks of K14E and moved towards the monomer size at lower protein concentration, as predicted for a dynamic equilibrium in rapid exchange. The K14E monomer and dimer peaks eluted at the same position at all concentrations of protein, as expected for an slowly exchanging equilibrium, only their relative proportions changed with dimer predominating at the highest concentration tested (approximately 160μM, blue trace in Fig. 5d). Similar experiments with cadherin-6 also showed that wild-type protein eluted as a single peak whereas the X-dimer mutant (M188D) eluted as distinct monomer and dimer peaks (Fig. 5e, f).
Together, the velocity AUC and gel filtration experiments show clear evidence of slowed exchange between monomer and dimer in X-dimer mutants of E-cadherin and cadherin-6.
Structures of X-dimer mutants
To confirm that the X-dimer mutations interfered only with X-interface formation, and did not affect overall cadherin structure, the strand swapped interface, or calcium binding in the EC1-2 linker, we determined the structure of the E-cadherin K14E mutant at 2.0 Ǻ resolution. The structure showed a single cadherin molecule in the asymmetric unit with strand swapped dimers formed between symmetry related protomers (Fig. 6a). The strand swapped dimer is essentially indistinguishable from that found in wild type structures (Fig. 6b 6,7), with rmsd of 1.9Å over 407 Cα atoms aligned with the wild-type structure from Haussinger and colleagues (PDB:1q1p), and 1.1Å when the comparison is restricted to the 32 residues comprising the strand dimer interface. The mutated residue (Glu14, cyan in Fig. 6a) is solvent exposed, far from the dimer interface and its side chain engages neither in inter- nor intra-molecular interactions. Calcium ion binding in the junction between EC1 and EC2 is identical to that seen in structures of wild-type E-cadherin7,20,21, showing no interference from the mutated residue (Fig. 6c). Diffracting crystals of the cadherin-6 X-dimer mutant, M188D, could not be obtained. Nonetheless, the high affinity binding of this mutant in analytical ultracentrifugation experiments (Fig. 4j; Table 2) argues against unintended direct effects on the strand dimer interface.
Figure 6. Structure of E-cadherin EC1-2 K14E mutant.

(a) Structure of strand swapped dimer formed between symmetry related protomers of E-cadherin EC1-2 K14E, shown in a ribbon representation. The mutation K14E is highlighted in cyan. Calcium ions are shown as green spheres, side chain atoms of Trp2 and of Glu89 are displayed. (b) Superposition of theα-carbon trace of E-cadherin EC1-2 K14E strand swapped dimmer (blue) with that of wild-type E-cadherin (PDB 1q1p, red) reported by Haussinger and colleagues 6. (c) Calcium co-ordination in the K14E EC1-2 junction. Three calcium ions (green) are co-ordinated by residues in EC1, EC2 and the linker region as well as by two water molecules (grey) exactly as observed in the wild-type protein.
The structure of a double mutant of E-cadherin in which both strand swapping and X-dimerization were prevented was also determined (E-cadherin W2A K14E). The 2.5Å structure showed neither strand swapped dimers nor X-dimers in the crystal lattice (Supplementary ig. 6). This structure confirms that the K14E mutation prevents X-dimer formation, even in the high protein concentrations reached in crystallization experiments. In addition, analytical ultracentrifugation experiments with the W2A K14E double mutant confirm that it is monomeric in solution, as is the equivalent double mutant (W4A M188D) of cadherin-6 (Table 2).
Together the E-cadherin mutant structures support the analytical ultracentrifugation data showing that the thermodynamics of strand swapping are unaffected by the K14E mutation and confirm that its sole structural consequence is failure of the X-dimer interface. We tested other cadherin mutants to confirm our interpretation. The X-dimer inhibiting mutation Y142R, was targeted to the EC2-side of the interface. Remarkably, this mutant exhibited biophysical behavior indistinguishable from K14E in SPR, AUC, and size exclusion experiments, strongly supporting our interpretation (Supplementary Fig. 7a-d, Table 2 and data not shown). We also tested a more conservative substitution mutant in which Lys14 was mutated to serine (K14S). This mutant displayed homophilic binding behavior in SPR assays intermediate between wild-type and K14E mutant protein (Supplementary Fig. 7e-g), consistent with the less severe mutation. Similar to other X-dimer mutants, equilibrium analytical ultracentrifugation revealed wild-type binding (Table 2).
X-dimer mutations prevent cadherin-mediated cell aggregation
Having observed that specific mutation of the X-dimer interface abrogated cadherin binding in SPR assays without substantially lowering the affinity of dimerization, we set out to determine if the same mutations would prevent cell aggregation when introduced into full length cadherins expressed on the surface of transfected cells.
Chinese Hamster Ovary (CHO) cells were used for these experiments because they express no endogenous cadherins but acquire the ability to aggregate in a calcium dependent manner when exogenous cadherins are expressed on their surface 29. CHO cells were transfected with either wild-type E-cadherin, E-cadherin with an X-dimer mutation (K14E) or E-cadherin with a strand swapping mutation (W2A). Clonal cell lines of each were established and wild-type and K14E transfectants were shown to express comparable levels of cadherin on the cell surface by immunofluorescent staining of permeabilized (Fig. 7c, g) and non-permeabilized cells (Fig. 7d, h). Surface expression of the W2A mutant was somewhat lower (Fig. 7k, l).
Figure 7. Aggregation of cells transfected with X-dimer mutant E-cadherin.
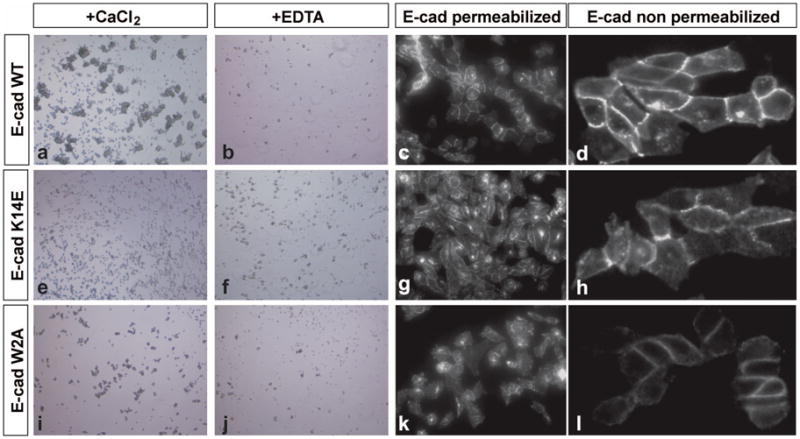
(a) Wild-type E-cadherin expressing CHO cells show robust cell aggregation in a long-term assay whereas E-cadherin K14E (e) and E-cadherin W2A (i) show aggregation similar to that found in the presence of 4mM EDTA (b, f, j). Immunofluorescent staining for the presence of E-cadherin (c, g, k) shows that all constructs are expressed to similar levels and that concentration of E-cadherin towards sites of cell-cell contact can be observed. Surface expression is confirmed by immunofluorescent staining in the absence of permeabilization of the cells (d, h, l)
Cell aggregation of the transfectants was assessed using a long term aggregation assay in which dissociated cells were incubated for 24 hours with constant rotary shaking then viewed under an inverted light microscope 9,30. Cells expressing wild-type E-cadherin showed extensive aggregation in these conditions (Fig. 7a). The aggregation was calcium dependent, becoming undetectable when calcium ions were chelated with EDTA (Fig. 7b).
In contrast, cells expressing the X-dimer mutant (K14E) showed no detectable aggregation under the same conditions (Fig. 7e); results were indistinguishable from assays performed in the absence of calcium (F) or with cells expressing the strand swapping mutant (W2A, panels I, J). These results were surprising, given our structural and biophysical data showing that the K14E mutant forms adhesive strand swapped dimers with an affinity equal to the wild-type protein, albeit with slower kinetics.
We reasoned that the shear forces between cells introduced by rotary shaking in our assay may make aggregation unusually sensitive to slower binding kinetics. For this reason we then used a ‘hanging drop’ method in which cells are allowed to aggregate without shaking in a droplet of culture medium suspended from an inverted cover slide 31. However, even under these conditions, only the wild-type protein mediated cell aggregation whereas cells expressing the X-dimer mutant were indistinguishable from parental CHO cell controls (Supplementary Fig. 8).
Together, the cell aggregation assays suggest that specific mutation of the X-dimer interface renders E-cadherin incapable of mediating cell-cell adhesion sufficient to aggregate cells. In combination with the biophysical data described above this suggests that cadherin-mediated adhesion in a cellular context may be dependent in part on efficient kinetics of adhesive dimer formation.
Discussion
Although the strand swap mechanism of classical cadherin adhesion has been known for some time, certain aspects of binding have remained unclear, in part due to seemingly disparate results from early structural studies. The first crystal structures from a cadherin ectodomain region, the EC1 domain of N-cadherin, revealed a strand-swapped dimer configuration 8. Soon thereafter the structure of a two-domain EC1-2 fragment from E-cadherin was determined 20, yet this structure revealed a different dimer – the configuration that we refer to here as an ‘X-dimer’. – These configurations represent the founding members of two classes of cadherin ectodomain structures, for which examples of each have followed since. Strand swapped dimers have been found in type I cadherin ectodomain structures for E-cadherin, N-cadherin, and C-cadherin, and for the ectodomains of type II cadherins 8, 11, and MN-cadherin 5–9. Prior to the work reported here and in the accompanying paper on T-cadherin, X-dimer configurations had been found for two different crystal forms of EC1-2 constructs of E-cadherin 20,21. Reconciling the existence and potential role of these dimer interfaces is thus essential for a detailed understanding of the molecular mechanisms of cadherin-mediated adhesion.
Why do adhesive domains from classical cadherins sometimes form strand-dimer structures and sometimes X-dimers? In their wild type forms, both type I E-cadherin and type II cadherin-6 (by homology with closely related type II cadherins of known structure) adopt strand swap dimer configurations, but our results show that mutations that impair strand swapping in either of these proteins lead to adoption of the X-dimer form. In retrospect, the configurations adopted in previously determined X-dimer structures of E-cadherin can be attributed to the presence of one or two large amino acids – methionine or methionine-arginine – before the natural N-terminus 20,21. Other multidomain fragments of classical cadherins with native N-termini, or with single smaller amino acid substitutions or additions, yielded crystal structures in the strand swapped dimer form 5–7,9. One N-cadherin EC1-2 structure reported by us 17 had a wild-type N-terminus, but revealed a monomeric form in crystals which formed at pH 5.0 in the presence of uranyl acetate. However, we have now re-determined this structure using less harsh crystallization conditions, and found that this molecule also forms a strand swapped dimer (Supplementary Fig. 9).
Experiments reported here, in agreement with prior studies 6, show that the presence of extra N-terminal residues impedes adoption of the strand swapped structure, lowers the affinity of dimerization and yields an X-dimer crystal structure. Similarly, other E-cadherin mutants designed to interfere with strand swapping – W2A (substitution of the Trp2 anchor residue with alanine) and E89A (Glu89 substitution with uncharged residue to prevent a salt bridge that stabilizes swapping) – form X-dimers and have interaction affinities that are weaker than wild-type protein (Table 2). Similar results are found for the type II cadherin-6: a designed strand-swapping mutation targeting one of the tryptophan residues that anchors the swapped strand yields a protein with weaker dimerization affinity that adopts an X-dimer structure. It is remarkable that, despite substantial structural differences in the strand exchanged dimer interfaces in type I and type II cadherins 9, the X-dimer structures formed by their strand swap-incompetent mutants are structurally so similar (Supplementary Text and Supplementary Figs. 1–4).
In a recent NMR study of the EC1 domain of the type II cadherin-8, we discussed two possible mechanisms for the formation of the swapped dimer 32. One involves a pre-existing equilibrium, or selective fit mechanism, where only collisions between monomers with both strands in an “open conformation” 33 lead to dimerization. In the other, monomers in the “closed conformation” form an initial encounter complex and then, subsequently, strand swapping occurs. Although the EC1 fragments used in the NMR study exhibited only very weak dimerization at physiological pH, under conditions of low pH around 3% of the monomers were shown to be in an open conformation suggesting the possibility for a selective fit mechanism, at least for these single domain fragments. However, the data are also consistent with an encounter complex mechanism whereby two monomers would stay bound for a long enough time for strand swapping to occur, a model clearly favored by our current results.
Our data define an encounter complex mechanism where the X-dimer is an intermediate along the path to the formation of a strand swapped dimer (Fig. 8). First, the binding affinity of the X-dimer is lower than that of the swapped dimer, as required by such a model. Second, mutations that compromise X-dimer formation inhibit swapped dimer formation in short time scale experiments, but do not affect the dimerization affinity of the swapped dimer in long time frame experiments. Specifically, the equilibrium AUC data reported here were acquired over the course of ~20 hour centrifuge runs, over which time a stable thermodynamic equilibrium can establish. Remarkably, in these experiments, X-dimer mutants of both E-cadherin (K14E) and cadherin-6 (M188D) behave almost indistinguishably from wild-type protein (Fig. 4i, j), showing that these mutations, as predicted from the structures, do not interfere substantially with the final thermodynamic equilibrium of binding. Similarly, crystallization experiments with the K14E mutant, over the course of days, revealed strand swapped dimers identical to wild-type protein (Fig. 6). However, results from SPR experiments, in which binding must take place over much shorter time periods (~1 minute), reveal no adhesive binding for these mutant proteins (Fig. 4). This difference in behavior between long timescale AUC experiments and short timescale SPR experiments provides strong evidence in support of our hypothesis. Size exclusion chromatography and velocity AUC experiments confirm that the kinetics of monomer-dimer exchange are slowed in these mutants. In either of these methods, wild-type cadherins yield a single species in rapid exchange between monomer and dimer, whereas X-dimer mutants reveal monomer and dimer species in slow exchange (Fig. 5). Together with the equilibrium AUC data these experiments strongly suggest that the X-dimer enhances the kinetics of association and dissociation of the swapped adhesive dimer. A final line of evidence is that the X-dimer structure itself is highly suggestive of its role as an intermediate since the A* strands of partner molecules are positioned adjacent to one another as if poised to swap. Moreover, although the X-dimer affinities are relatively weak they are sub-millimolar and the buried interfacial area is substantial. Thus, it is not unreasonable to expect that the X-dimer has a long enough lifetime to allow the two A* strands to swap prior to dissociation of the complex. In a recent single molecule optical study, published while this manuscript was under review, Sivasankar and colleagues found that E-cadherin EC1-5 ectodomains containing the W2A mutation were able to form weak Ca2+-dependent encounter complexes in which EC1 domains are in close proximity 34. These findings are in close agreement with the data presented here, which identify the X-dimer as the encounter complex.
Figure 8. Two-step model of classical cadherin binding.

A parsimonious model is suggested by the data presented here. Cadherin monomers first associate with fast kinetics in an X-dimer configuration. In the X-dimer, strand swapping regions of the paired EC1 domains are positioned adjacent to one another, enabling the slower transition to the mature strand-swapped dimer state.
It is thought that tissue patterning mediated by cadherins depends, at least in part, on differential binding between different cadherin family members 35–38. The work presented here suggests that, in principle, such specificity could arise either from thermodynamic differences in binding energy, or from differences in binding kinetics. Cell aggregation assays (Fig. 7) show that cell adhesion, under the conditions we tested, critically depends on X-dimer function. Therefore, cadherins with mismatched X-dimer interfaces might not be expected to dimerize by strand swapping, providing a potential means for specificity. However we recently demonstrated that heterophilic binding between N- and E-cadherin is measurable in SPR assays 25. Since these short time-scale assays are very sensitive to failure of X-dimer formation (see above), the findings effectively rule out the existence of strict selectivity at this level. Nonetheless, the functional roles of kinetic and thermodynamic processes in the binding specificity of cadherins in vivo remain to be determined.
The discovery of a possible intermediate in the kinetic pathway leading to strand swapping in classical cadherins, which also appears to mediate adhesion in T-cadherin, suggests a potential role for the X-dimer interface in the evolution of strand swapping in cadherins. Since strand swapping that is rapid enough to drive cell adhesion appears to require the positioning function of an X-dimer form, it is likely that the existence of this interface was a prerequisite for the evolution of strand swapped dimers. Furthermore, it is possible that some extant branches of the cadherin family that diverged from the classical cadherins before strand swapping arose may utilize the X-dimer interface as their primary adhesive mechanism.
T-cadherin is unlikely to represent a “pre swapping” cadherin. Although T-cadherin is a family outlier in that it is “truncated” by the absence of trans-membrane and cytoplasmic domains, it is a close relative of classical cadherins. Phylogenetic analysis and conserved exon boundary positions suggest that classical and T-cadherins diverged relatively recently subsequent to a gene duplication event 3,39. When amino acid sequence identities over the entire ectodomain are compared, T-cadherins are more similar to type I classical cadherins than are the type II or desmosomal cadherins. Since these sub-families are known or are predicted to swap, respectively, this implies that T-cadherin may have branched from the classical cadherins after the emergence of strand swapping and divergence of the type I, type II, and desmosomal cadherins. Thus T-cadherin may have lost its strand swap binding function and reverted to the intermediate X-dimer configuration.
In several branches of the cadherin superfamily the sequence determinants thought to be required for strand swapping are not conserved, including a shortened A strand, tryptophan at position 2, and a glutamic acid that forms a salt bridge with the amino terminus of the swapped strand 2. Non-classical cadherins such as the clustered and non-clustered protocadherins and all invertebrate cadherins do not possess these features in their EC1 domains, which are more closely related to classical cadherin domains EC2-5. Consequently, the dimerization mechanisms of these cadherins remain unknown. The possibility remains that some of these cadherins may utilize X-dimers to achieve their adhesive function.
Methods
Crystallization and structure determination
We expressed and purified proteins as described in Supplementary Methods, and grew crystals using the vapor diffusion method by mixing 0.6μL protein solution with 0.6μL reservoir solution at 20°C. Crystallization conditions were: 0.2M Tris-Cl pH 8.0, 0.1M ammonium chloride, 6% (w/v) PEG 8000, 1.5mM dithiothreitol (Ecad EC1-2); 1.3M ammonium sulfate, 0.1M Tris-Cl pH 8.5, 15% (v/v) glycerol (Ecad EC1-2 W2A, Ecad EC1-2 AA); 0.25M ammonium sulfate, 0.1M MES pH 6.5, 26% (w/v) PEG 5000 monomethylether (Ecad EC1-2 E89A); 0.1M MES pH 6.5, 0.75M sodium sulfate (Ecad EC1-2 K14E); 1.2M ammonium sulfate, 0.1M Tris-Cl pH 8.5, 15% (v/v) glycerol (Ecad EC1-2 W2A K14E); 0.1M HEPES pH 7.5, 17% (v/v) isopropanol, 12% (w/v) PEG 4000, 9mM CaCl2 (cad-6 EC1-2 W4A) and 0.1M Tris-Cl pH 7.1, 30% (w/v) PEG 3350 (Ncad EC1-2). We cryoprotected crystals by transfer to mother liquor supplemented with 30% (v/v) glycerol and flash froze them at 100K.
We collected data on single frozen crystals at 0.979Å wavelength at the X4A and X4C beam lines of the National Synchrotron Light Source, Brookhaven National Laboratory. We solved structures by molecular replacement using as a search model the structure of mouse E-cadherin EC1-2 (PDB code 1edh, chain A) for all E-cadherin structures; mouse N-cadherin EC1-2 (PDB code 1ncj) for Ncad EC1-2; and mouse cadherin-11 EC1-2 (PDB code 2a4e) modified with the program Chainsaw 40 for cad-6 EC1-2 W4A. Refinement proceeded by manual building in Coot 41 and automated refinement in Refmac 42 and CNS 43. Data collection and Refinement statistics are summarized in Table 1. Ramachandran plot statistics (% favored/allowed/disallowed) for the final structural models were: 97.6/2.4/0 (Ecad EC1-2); 97.4/2.6/0 (Ecad EC1-2 AA); 97.1/2.9/0 (Ecad EC1-2 W2A); 97.8/2.2/0 (Ecad EC1-2 E89A); 91.4/8.6/0 (Ecad EC1-2 K14E); 97.3/2.7/0 (Ecad EC1-2 W2A K14E); 89.7/10.3/0 (Ncad EC1-2) and 95.7/4.3/0 (cad-6 EC1-2 W4A).
Surface Plasmon Resonance (SPR)
We performed experiments using a Biacore T100 biosensor with a Series S CM4 sensor chip (GE Healthcare). We immobilized neutravidin over all flow cells in HEPES-Buffered-Saline (HBS, 10mM HEPES, pH 7.4, 150mM NaCl) at 32°C. A mixture of 200mM N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC) and 50mM N-hydroxysuccinimide (NHS) was injected over the flow cells for 7 minutes at 20 μL min−1. Immunopure neutravidin (Thermo Scientific) dissolved in 10 mM sodium acetate pH 4.5, was injected over the activated surfaces for 7 minutes at 20 μL min−1. We blocked using a 7-minute pulse of 1.0M ethanolamine-HCl, pH 8.5 at the same flow rate, giving immobilization levels of 9,000–13,000 RU.
We performed biotinylated-protein captures and binding analysis at 25oC in a running −1 BSA and buffer of 20mM Tris-HCl, pH 8.0, 150mM NaCl, 3mM CaCl2, 0.25mg mL 0.005% (v/v) Tween 20. E-cadherin EC1-2 and its K14E mutant were captured over flow cells 1 and 3 respectively, using a 50-fold dilution of each protein in running buffer, and injecting consecutive 10-s pulses at 20 μL min−1 until approximately 500 Response Units (RU) of each protein was captured onto their respective flow cells. Using this approach, the same protein injection can flow consecutively over flow cells 1, 2 and 3. In a separate experiment, we captured wild type cadherin-6 EC1-2 and its mutant M188D using the same protocol. In both experiments, nothing was captured over flow cell 2, which was also immobilized with neutravidin, to serve as a reference.
We prepared analyte proteins in running buffer at a concentration range of 100–0.39μM using a two-fold concentration series. The association rate was monitored at 50μL min−1 for 60s, followed by a 60s dissociation phase. After each binding cycle, buffer was injected at the same flow rate for 60s to wash out the flow path and minimize sample carryover. We injected samples in order of increasing concentration and we repeated each concentration series at least twice for each protein. Every third protein sample, a buffer injection was performed for double referencing of binding responses. The first 10 injections in each experiment contained running buffer only, to allow for instrument equilibration. We removed these initial 10 injections from the analysis. Binding data was processed using Scrubber 2.0 (BioLogic).
As discussed in a previous publication 25, determination of rate and equilibrium constants from SPR data is not possible for cadherins because they self-associate both in the bulk flow and on the chip surface rendering such analyses unreliable. Therefore SPR analysis is used here only as a comparative binding assay.
Analytical ultracentrifugation
We performed analytical ultracentrifugation (AUC) experiments using a Beckman XLA/I ultracentrifuge, with a Ti50An or Ti60An rotor. For sedimentation equilibrium experiments, we dialyzed all proteins overnight at 4oC in 10mM Tris-HCl, pH 8.0, 150mM NaCl, 3mM CaCl2, with 3mM TCEP added for E-cadherin samples. We loaded 120μL of each protein at a concentration of 10, 20, or 30μM into a cell with a 12 mm six-channel centerpiece and sapphire windows. We collected data at 25oC at 280nm and 660nm, spinning at 23400g for 20 hours after which four scans (1 per hour) were collected. Speed was increased to 35200g for 10 hours, then to 49100g for 10 hours, with four hourly scans taken after each period. This protocol gives 72 scans for each protein. We calculated buffer density and v-bar using SednTerp. (Alliance Protein Laboratories), and analyzed the scans using HeteroAnalysis 1.1.0.28 (http://www.biotech/uconn.edu/auf/). We fitted data from all concentrations, speeds and both detection systems globally by non-linear regression to a monomer/dimer equilibrium model to calculate the KD for each homodimer. All values were determined from at least two independent experiments.
For sedimentation velocity experiments, we loaded 400μL of each protein at 43 or 86μM and collected data at 6°C at 45000rpm, at 660nm. Scans were collected every third minute, with a total 340 scans collected per sample. Qualitatively similar results were observed at 25°C for wild-type and mutant proteins. We edited and processed data using SedFit software (https://sedfitsedphat.nibib.nih.gov).
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants R01 GM062270 (L.S.), R01 GM30518 (B.H.), U54 CA121852 (B.H. and L. S.) and by NSF grant MCB-0416708 (B.H.). B.H. is an investigator of the Howard Hughes Medical Institute. X-ray data were acquired at the X4A and X4C beamlines of the National Synchrotron Light Source, Brookhaven National Laboratory; the X4 beamlines are operated by the New York Structural Biology Center.
Footnotes
Accession codes
Protein Data Bank: Coordinates for mouse E-cadherin EC1-2 wild-type, W2A, AA-extension, E89A, K14E and W2A K14E, mouse N-cadherin EC1-2 and for mouse cadherin-6 W4A, have been deposited with accession codes XXXX, XXXX, XXXX, XXXX, XXXX, XXXX, XXXX, and XXXX, respectively.
Author contributions
OH, XJ, FB, and KJC determined and refined all the crystal structures; FB, OH, and JB produced all the wild-type and mutant proteins; PK performed and analyzed the SPR experiments; JV performed bioinformatic analysis; GA performed the AUC experiments; SRP performed the cell aggregation and immunofluorescence studies; OH, BH and LS designed experiments, analyzed data, and wrote the manuscript.
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Takeichi M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol. 1995;7:619–27. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 2.Posy S, Shapiro L, Honig B. Sequence and structural determinants of strand swapping in cadherin domains: do all cadherins bind through the same adhesive interface? J Mol Biol. 2008;378:952–66. doi: 10.1016/j.jmb.2008.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nollet F, Kools P, van RF. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Mol Biol. 2000;299:551–72. doi: 10.1006/jmbi.2000.3777. [DOI] [PubMed] [Google Scholar]
- 4.Patel SD, Chen CP, Bahna F, Honig B, Shapiro L. Cadherin-mediated cell-cell adhesion: sticking together as a family. Curr Opin Struct Biol. 2003;13:690–8. doi: 10.1016/j.sbi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Boggon T, et al. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–13. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 6.Haussinger D, et al. Proteolytic E-cadherin activation followed by solution NMR and X-ray crystallography. EMBO J. 2004;23:1699–708. doi: 10.1038/sj.emboj.7600192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parisini E, Higgins JM, Liu JH, Brenner MB, Wang JH. The Crystal Structure of Human E-cadherin Domains 1 and 2, and Comparison with other Cadherins in the Context of Adhesion Mechanism. J Mol Biol. 2007;373:401–11. doi: 10.1016/j.jmb.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shapiro L, et al. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–37. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 9.Patel S, et al. Type II cadherin ectodomain structures: implications for classical cadherin specificity. Cell. 2006;124:1255–68. doi: 10.1016/j.cell.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 10.Nose A, Nagafuchi A, Takeichi M. Expressed recombinant cadherins mediate cell sorting in model systems. Cell. 1988;54:993–1001. doi: 10.1016/0092-8674(88)90114-6. [DOI] [PubMed] [Google Scholar]
- 11.Nose A, Tsuji K, Takeichi M. Localization of specificity determining sites in cadherin cell adhesion molecules. Cell. 1990;61:147–55. doi: 10.1016/0092-8674(90)90222-z. [DOI] [PubMed] [Google Scholar]
- 12.Shan WS, Koch A, Murray J, Colman DR, Shapiro L. The adhesive binding site of cadherins revisited. Biophys Chem. 1999;82:157–63. doi: 10.1016/s0301-4622(99)00115-5. [DOI] [PubMed] [Google Scholar]
- 13.Al-Amoudi A, Frangakis AS. Structural studies on desmosomes. Biochem Soc Trans. 2008;36:181–7. doi: 10.1042/BST0360181. [DOI] [PubMed] [Google Scholar]
- 14.He W, Cowin P, Stokes D. Untangling desmosomal knots with electron tomography. Science. 2003;302:109–13. doi: 10.1126/science.1086957. [DOI] [PubMed] [Google Scholar]
- 15.Kitagawa M, et al. Mutation analysis of cadherin-4 reveals amino acid residues of EC1 important for the structure and function. Biochem Biophys Res Commun. 2000;271:358–63. doi: 10.1006/bbrc.2000.2636. [DOI] [PubMed] [Google Scholar]
- 16.May C, et al. Identification of a transiently exposed VE-cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood. 2005;105:4337–44. doi: 10.1182/blood-2005-01-0010. [DOI] [PubMed] [Google Scholar]
- 17.Tamura K, Shan W, Hendrickson W, Colman D, Shapiro L. Structure-function analysis of cell adhesion by neural (N-) cadherin. Neuron. 1998;20:1153–63. doi: 10.1016/s0896-6273(00)80496-1. [DOI] [PubMed] [Google Scholar]
- 18.Harrison O, Corps E, Kilshaw P. Cadherin adhesion depends on a salt bridge at the N-terminus. J Cell Sci. 2005;118:4123–30. doi: 10.1242/jcs.02539. [DOI] [PubMed] [Google Scholar]
- 19.Ozawa M, Kemler R. Correct proteolytic cleavage is required for the cell adhesive function of uvomorulin. J Cell Biol. 1990;111:1645–50. doi: 10.1083/jcb.111.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–4. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- 21.Pertz O, et al. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–47. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciatto C, et al. T-cadherin structures reveal a novel adhesive binding mechanism. Nature Structural and Molecular Biology. 2009 doi: 10.1038/nsmb.1781. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laur OY, Klingelhofer J, Troyanovsky RB, Troyanovsky SM. Both the dimerization and immunochemical properties of E-cadherin EC1 domain depend on Trp(156) residue. Arch Biochem Biophys. 2002;400:141–7. doi: 10.1006/abbi.2002.2774. [DOI] [PubMed] [Google Scholar]
- 24.Schubert W, et al. Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell. 2002;111:825–36. doi: 10.1016/s0092-8674(02)01136-4. [DOI] [PubMed] [Google Scholar]
- 25.Katsamba P, et al. Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0905349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balbo A, Brown PH, Braswell EH, Schuck P. Measuring protein-protein interactions by equilibrium sedimentation. Curr Protoc Immunol. 2007;Chapter 18(Unit 18):8. doi: 10.1002/0471142735.im1808s79. [DOI] [PubMed] [Google Scholar]
- 27.Brown PH, Balbo A, Schuck P. Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr Protoc Immunol. 2008;Chapter 18(Unit 18):15. doi: 10.1002/0471142735.im1815s81. [DOI] [PubMed] [Google Scholar]
- 28.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–30. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niessen C, Gumbiner B. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J Cell Biol. 2002;156:389–399. doi: 10.1083/jcb.200108040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimoyama Y, Tsujimoto G, Kitajima M, Natori M. Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. Biochem J. 2000;349:159–67. doi: 10.1042/0264-6021:3490159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kii I, Amizuka N, Shimomura J, Saga Y, Kudo A. Cell-cell interaction mediated by cadherin-11 directly regulates the differentiation of mesenchymal cells into the cells of the osteo-lineage and the chondro-lineage. J Bone Miner Res. 2004;19:1840–9. doi: 10.1359/JBMR.040812. [DOI] [PubMed] [Google Scholar]
- 32.Miloushev VZ, et al. Dynamic properties of a type II cadherin adhesive domain: implications for the mechanism of strand-swapping of classical cadherins. Structure. 2008;16:1195–205. doi: 10.1016/j.str.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C, Posy S, Ben-Shaul A, Shapiro L, Honig B. Specificity of cell-cell adhesion by classical cadherins: Critical role for low-affinity dimerization through beta-strand swapping. Proc Natl Acad Sci U S A. 2005;102:8531–6. doi: 10.1073/pnas.0503319102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivasankar S, Zhang Y, Nelson WJ, Chu S. Characterizing the initial encounter complex in cadherin adhesion. Structure. 2009;17:1075–81. doi: 10.1016/j.str.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duguay D, Foty R, Steinberg M. Cadherin-mediated cell adhesion and tissue segregation: qualitative and quantitative determinants. Dev Biol. 2003;253:309–23. doi: 10.1016/s0012-1606(02)00016-7. [DOI] [PubMed] [Google Scholar]
- 36.Steinberg MS. On the Mechanism of Tissue Reconstruction by Dissociated Cells, Iii. Free Energy Relations and the Reorganization of Fused, Heteronomic Tissue Fragments. Proc Natl Acad Sci U S A. 1962;48:1769–76. doi: 10.1073/pnas.48.10.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinberg MS. Differential adhesion in morphogenesis: a modern view. Curr Opin Genet Dev. 2007;17:281–6. doi: 10.1016/j.gde.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 38.Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem. 1990;59:237–52. doi: 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- 39.Hulpiau P, van Roy F. Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol. 2009;41:349–69. doi: 10.1016/j.biocel.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 40.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. Journal of Applied Crystallography. 2008;41:641–643. [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.