

Abstract
Congenital diaphragmatic hernia (CDH) is a common major malformation affecting 1/3000–1/4000 births, which continues to be associated with significant perinatal mortality. Much current research is focused on elucidating the genetics and pathophysiology contributing to CDH to develop more effective therapies. The latest data suggest that many cases of CDH are genetically determined and also indicate that CDH is etiologically heterogeneous. The present review will provide a brief summary of diaphragm development and model organism work most relevant to human CDH and will primarily describe important human phenotypes associated with CDH and also provide recommendations for diagnostic evaluation of a fetus or infant with CDH.
Keywords: CDH+, congenital diaphragmatic hernia, genetic counseling, isolated CDH, non-isolated CDH, syndromic CDH
Overview of congenital diaphragmatic hernia
Congenital diaphragmatic hernia (CDH) is a malformation of the developing diaphragm, a mesodermally derived structure, separating the thoracic and abdominal cavities. Most often, the malformation is an actual ‘hole’ or discontinuity in the diaphragm. Less often, the defect is a thinning or undermuscularization, which is generally referred to as eventration or sac-type CDH. Eventration defects were once considered distinct from cases with a diaphragmatic ‘hole’, but because they have been observed together in the same patient and even in members of the same family, this distinction is not always valid (1–4). Some authorities consider the term CDH too limiting and prefer the designation congenital diaphragmatic defect (5); however, given its widespread use in the medical literature, CDH remains preferred. The consequences of a CDH, be it either a diaphragmatic hole or undermuscularization, are often the same with upward displacement of the abnormal diaphragm and varying degrees of compression of thoracic contents.
CDH embryology
Investigators agree that the diaphragm is a mesodermal structure that forms during weeks 4–10 of human development. There is considerably less agreement regarding the specific components contributing to the mature diaphragm. The long-held view (6) that the diaphragm is ‘fused’ from four separate structures [the septum transversum, the esophageal mesentery, the posterolateral coalescences known as the pleuroperitoneal folds (PPFs), and ingrowth of musculature from the lateral body wall] is now being called into question. More recent work suggests that a non-muscular diaphragmatic anlage first develops (7), with the septum transversum possibly contributing to anterior and central portions of this, and that all diaphragmatic musculature stems from cervical myotome-derived muscle precursors which migrate through the PPFs that appear at approximately day 37 of human embryonic development (Carnegie stage 16) (8). In model organisms, experimentally induced defects in the anlage result in CDH (7).
Type and side of CDH
Approximately 80–85% of diaphragmatic hernias occur on the left side, 10–15% on the right side, while the remainder are bilateral defects (9, 10). Independent of sidedness, most diaphragmatic defects are reported to involve the posterior and lateral aspects of the diaphragm, the so-called posterolateral or Bochdalek CDH. Extremely large defects are sometimes called diaphragm agenesis or aplasia, and it is likely that they represent large Bochdalek hernias rather than a distinct hernia type. Less than 5% of defects are reported in other locations, such as the anterior portion of the diaphragm (e.g. Morgagni hernia) or the central portion of the diaphragm. The fact that most defects are left sided is a robust finding, but the proportion of cases with a posterolateral CDH is not precisely known. This is due to the fact that systematic and precise anatomic localization of diaphragm defects has not been performed in most studies. When Ackerman et al. examined diaphragms from 48 autopsied CDH cases, they found that approximately 25% did not have typical posterolateral defects but rather had central, anterior, unclassifiable, or mixed defect types (11). These observations underscore the need for rigorous and systematic classification of CDH, which will be required for establishing genotype–phenotype correlations in the future. A schematic for collecting this information has been proposed (12).
Pulmonary hypoplasia
Lung hypoplasia with arrest of alveolar development at the mid-canalicular stage is an almost universal finding in CDH and remains one of the major determinants of CDH-associated morbidity and mortality. The lung ipsilateral to the hernia is particularly hypoplastic. This observation stimulated the hypothesis that upward displacement of abdominal contents into the chest mechanically compresses the lung, resulting in pulmonary hypoplasia. Evidence is accumulating from experimental work that pulmonary hypoplasia is bilateral and at least in some models is a primary developmental defect that co-occurs with CDH (13). A ‘dual hit hypothesis’ of pulmonary hypoplasia proposes that the first hit is primary pulmonary underdevelopment and that the second hit is mechanical compression. The combination of two hits accounts for the fact that pulmonary hypoplasia is bilateral but more severe in the lung ipsilateral to the diaphragm defect (14). Additionally, the pulmonary vasculature is underdeveloped with aberrantly muscularized (typically overmuscularized) pulmonary vessels, predisposing to pulmonary hypertension (15). If pulmonary hypertension develops in a neonate with CDH, it can be refractory to treatment and significantly add to morbidity and mortality.
Isolated CDH vs CDH + (non-isolated CDH)
In addition to defining the location and type of CDH, it is important to determine the overall status of the individual in whom the diaphragm defect occurs. In approximately 60% of cases, CDH is the only birth defect, and these cases are classified as having isolated CDH (Fig. 1). Although additional problems commonly coexist with CDH (such as pulmonary hypoplasia, intestinal malrotation, cardiac dextroposition, and left heart hypoplasia; Table 1), these are usually considered part of a CDH sequence and so their presence does not negate designation of a case as having isolated CDH. The remaining approximately 40% of CDH cases are classified as having CDH + (also referred to as complex, non-isolated, or syndromic CDH) due to the presence of additional major malformations in other organ systems, chromosome abnormalities, or single gene disorders (Fig. 1) (9, 10, 16, 17).
Fig. 1.
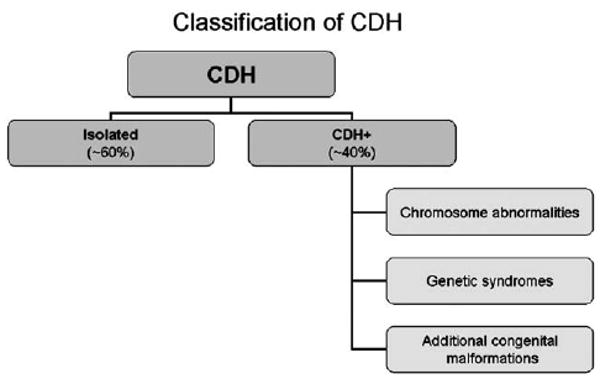
Approach to classification of cases with CDH.
Table 1.
Anomalies frequently found in patients classified as having isolated CDHa
| Pulmonary hypoplasia |
| Malrotation or incomplete rotation of bowel |
| Patent ductus arteriosus |
| Patent foramen ovale |
| Heart ‘hypoplasia’ and/or dextroposition |
| Tricuspid or mitral valve regurgitation |
| Undescended testes |
| Accessory spleen |
CDH, congenital diaphragmatic hernia.
Anomalies that are due to the hemodynamic or mechanical consequences of CDH; therefore, they are part of a CDH sequence rather than independent malformations. Their presence, in and among themselves, does not preclude classifying a case as having isolated CDH.
CDH mortality
Mortality for patients with CDH has declined during recent decades. However, it continues to hover between approximately 30% and 50% in many series and can be even higher among cohorts restricted to prenatally diagnosed cases (17). Recent data emanating from the Congenital Diaphragmatic Hernia Study Group (consisting of 51 tertiary care centers from around the world) reported that overall mortality prior to hospital discharge was 31% (18). A major factor continuing to influence mortality is the ratio of CDH + : isolated cases as mortality remains high for the former but has declined to as low as 10–20% for the latter especially in centers with high volume and particular CDH expertise. A related factor that influences the rate of mortality is referred to as ‘hidden mortality’. This describes overrepresentation of cases at the mild end of the phenotypic spectrum in studies based at referral or tertiary care centers, which can lead to an underestimation of mortality (19). Another factor influencing mortality is hernia sidedness. In most series, patients with right-sided CDH show higher mortality than those with left-sided CDH, and in all series, patients with bilateral CDH show the highest mortality (17).
Timing and method of diagnosis
Approximately 60% of CDH cases are detected prenatally either by ultrasound examination or by fetal magnetic resonance imaging (MRI) scan. These technologies are increasingly accurate for detecting CDH but cannot reliably detect minor anomalies such as facial dysmorphology. Algorithms that predict lung volume at birth are also becoming more accurate and provide useful information for planning the location and method of delivery as well as for prognosticating on morbidity and mortality (20, 21). In approximately 10% of individuals, CDH is diagnosed after the neonatal period, and the defect is either detected incidentally or more commonly is diagnosed during a workup for respiratory symptoms, such as tachypnea, or gastrointestinal symptoms such as abdominal pain, vomiting or constipation (22).
Animal models
Several genetically modified model organisms with abnormalities of the diaphragm have been reported. For example, posterior defects of the diaphragm occur in mice with genetic alterations of Couptf2 (23) and Wt1 genes (24, 25). Anterior and midline defects of the diaphragm are found in Slit3 knockouts (26, 27) and a subset of Gata4 null carrier mice (28), respectively, while muscularization defects are found in the c-Met null model (7) and the Fog2 hypomorph (13). Details on each of these models as well as other transgenic models are provided in several recent excellent reviews (29–31). Bielinska and colleagues suggest that the protein products of currently known CDH-associated genes fall into two major categories: (i) transcription factors (such as Fog2 and GATA-4) or (ii) molecules involved in cell migration, cell–cell signaling or mesodermal patterning (such as Slit3 and Glypican-3). As additional genes are discovered, it will be instructive to learn the gene families and corresponding pathways required for normal diaphragmatic development.
Human CDH
The bulk of human evidence supporting genetic causation of CDH derives from observations in patients with CDH+. The most compelling findings to date include
recurring chromosome abnormalities in unrelated patients revealing CDH hot spots;
single gene disorders containing CDH as part of the core phenotype; genes for many of these disorders are known and provide insight into pathways required for normal diaphragm development;
occurrence of multiplex CDH kindreds.
There is also limited evidence indicating genetic causation in patients with isolated CDH (13).
Isolated (non-syndromic) CDH
In approximately 60% of cases, CDH is isolated, occurring in the absence of additional congenital malformations (Fig. 1).
Associated findings
Labeling a case as having isolated CDH can be misleading because it suggests that the individual is otherwise normal when in fact coexisting problems that affect medical management and cause morbidity are often present. However, these cases are still appropriately classified as isolated because the additional problems listed in Table 1 do not represent independent malformations of other organ systems but rather are hemodynamic or mechanical consequences of the diaphragmatic hernia. The presence of a small septal defect, such as a muscular ventriculoseptal defect (VSD), poses a dilemma for classification because inadequate information exists to know whether the combination of CDH + VSD should be considered as isolated (given that small muscular VSDs often resolve spontaneously) or as CDH+ and therefore impart a different recurrence risk than isolated CDH.
Clustering of isolated CDH in kindreds
Although the vast majority of individuals have a negative family history, sibling recurrences of isolated CDH are documented (3, 32–39). However, it is not completely possible to exclude the presence of CDH + in some of these cases due to limited physical examination and/or diagnostic workup. Empiric data from several series of consecutively collected cases reveal that sibling clustering of seemingly isolated CDH is low, on the order of 1–2% (5, 40, 41). Therefore, sibling clustering of isolated CDH is a relatively rare occurrence.
Genetic workup
Prenatal diagnosis of isolated CDH either by ultrasound or by MRI scan should prompt a thorough genetics evaluation. In addition to collecting a detailed pregnancy and family history, a standard chromosome analysis and a fetal echocardiogram should be performed. This workup is indicated even in cases with apparently isolated CDH because major as well as minor anomalies can escape prenatal detection. Evaluation of a case first diagnosed postnatally with CDH should be tailored to the findings observed on physical examination; a chromosome analysis may not be required, especially if the patient is non-dysmorphic, appropriately sized for gestational age, and without evidence of other birth defects following appropriate diagnostic surveys.
Genetic counseling
Among the first questions families ask after learning about the diagnosis of CDH are ‘Why did this happen?’ and ‘What caused this?’. The etiology of CDH is likely to be heterogeneous and involve mutations in genes belonging to several pathways necessary for normal diaphragm formation. Although mutations in single genes have been identified as the cause of syndromic forms of CDH (e.g. LRP2 mutations cause Donnai–Barrow syndrome, DBS) (42), only one case with apparently isolated CDH has been reported in association with a de novo mutation in a major gene, FOG2 (13). This finding may represent the ‘tip of the iceberg’. In other words, a high proportion of birth defects associated with reduced survival (such as CDH) could be due to de novo mutations or de novo copy number variants. The low sibling clustering of (apparently) isolated CDH is also consistent with de novo mutational events. However, these empiric data do not exclude polygenic or multifactorial inheritance, but formal proof of an inheritance pattern responsible for the majority of isolated CDH cases is lacking.
Teratogenic animal models of CDH are well described. A consistently reported CDH-associated teratogen in experimental models involves vitamin A deficiency [reviewed in (43)]. Evidence is beginning to accumulate that perturbations in vitamin A metabolism might be associated with human CDH. The most robust evidence comes from cases with CDH+ in that mutations in the receptor for retinol-binding protein (STRA6), as well as mutations in the gene LRP2 encoding a transmembrane receptor that reuptakes numerous compounds including retinol-binding protein, cause Matthew-Wood (MWS) and DBS, respectively (each further discussed below). In terms of isolated CDH, the data are sparse, but a small cohort of newborns with CDH, half of whom had isolated CDH, were found to have decreased levels of vitamin A at birth compared with controls (44). Most recently, an epidemiological study in which the majority of cases had isolated CDH found that self-reporting of decreased retinol intake (among the subset of women who were not vitamin supplement users) was associated with an increased risk of CDH (45).
It is unknown if CDH occurs more commonly in twin pregnancies. When CDH is present in one member of a monozygous (MZ) twin pair, the co-twin is unaffected more often than not, although MZ twins concordant for CDH have also been reported (40). Similar to other common birth defects that often show phenotypic discordance in MZ twins, possible genetic mechanisms for CDH discordance include (i) epigenetic differences, (ii) multifactorial inheritance, and (iii) non-identical genetic constitutions (e.g. twins having copy number differences, or other genomic differences, contributing to their phenotypic differences).
If a fetus is diagnosed prenatally with isolated CDH and the pregnancy is not carried to term, every effort should be made to collect as much information as possible including fetal imaging (e.g. fetal MRI scan and echocardiogram), fetal karyotype, and a postnatal autopsy. A sample of fetal DNA should also be obtained and stored for possible molecular testing. If the results of the diagnostic studies performed are normal, then families should be counseled that the recurrence risk for apparently isolated CDH is approximately 1–2%. This risk of recurrence, extrapolated from available studies on CDH ‘precurrence’, is low but increased at least 20-fold over the CDH occurrence rate in the general population so that subsequent pregnancies should be monitored by fetal imaging (5, 40, 41).
CDH+ (also known as complex, non-isolated, or syndromic CDH)
As many as 40% of CDH cases have additional malformations as part of chromosome aneuploidy, monogenic syndromes, or more commonly, a constellation of major and/or minor anomalies that do not form a currently recognizable pattern or have an identifiable genetic basis (Fig. 1). Selected examples of each of these categories are presented below.
Chromosome aneuploidy associated with CDH
Findings compiled from several studies reveal that, on average, chromosome abnormalities are detected in approximately 10% of CDH cases (46–52). These data derive from routine karyotyping and do not include small genomic deletions or duplications that can be appreciated by the new higher resolution techniques such as array-based comparative genomic hybridization (aCGH). Thus, the actual frequency of chromosome aneuploidy is not known but is likely to be greater than 10%.
Some of the common chromosome abnormalities detected by standard G-banded chromosome analysis include trisomy 18, trisomy 21, and tetrasomy 12p (47, 49, 52–57). Neither the frequency of CDH nor the reason that three 18 chromosomes increase the risk of CDH is currently known. CDH does occur in individuals with trisomy 21 but the association is not that common, and in fact, when a diaphragm defect is present, Morgagni-type hernias predominate (58, 59).
Two highly informative and thorough reviews provide lists and descriptions of recurring chromosome deletions, duplications, and translocations (54, 57). Analyses contained within these reviews indicate the existence of several chromosome ‘hot spots’ that presumably house genes important for diaphragm development. A complete discussion of chromosome abnormalities in cases with CDH is beyond the scope of this review; instead, an overview of selected ‘hot spots’ already assigned an Online Mendelian Inheritance in Man (OMIM) number is provided.
Tetrasomy 12p (isochromosome 12p or Pallister-Killian syndrome) (OMIM 601803)
Findings in patients with tetrasomy 12p range from mild facial dysmorphology and learning disabilities to multiple malformations, including CDH, that can be incompatible with life. Prenatal vs postnatal diagnosis influences the pattern of anomalies observed, although CDH always occurs in the minority of patients. CDH is present in approximately 10–20% of postnatally diagnosed cases (60, 61) and in 33% of prenatally diagnosed cases (62). The signs triggering consideration of the diagnosis of isochromosome 12p in infants or children can be relatively subtle such as linear streaks of skin hyperpigmentation, sparse hair bitemporally, coarse facies, mental retardation and seizures, while prenatal triggers include the presence of multiple major malformations (such as CDH and brain malformations) along with polyhydramnios, edema, and relative limb shortening.
The genetic basis of tetrasomy 12p accounts for its phenotypic diversity. Affected individuals are tissue mosaics for four copies of the short arm of chromosome 12 by virtue of having the normal chromosome 12 homologues plus a supernumerary isochromosome consisting of two copies of 12p. This isochromosome is rarely recovered from cultured lymphocytes so that a normal stimulated lymphocyte karyotype does not exclude the diagnosis. Demonstration of the supernumerary isochromosome is far more readily accomplished on cultured skin, amniocytes or chorionic villi (62). The current diagnostic standard for isochromosome 12p is a chromosome study on a tissue other than blood. In the future, molecular cytogenetic studies performed on samples derived from lymphocytes, such as fluorescence in situ hybridization (FISH) and aCGH, may become a new diagnostic standard, although at the same time, they may identify patients with very low-level isochromosome 12p mosaicism and create new challenges for clinical interpretation.
The mechanism of isochromosome formation is not fully understood but misdivision of the centromere during or just before maternal meiosis has been suggested (63). It is not currently known which of the approximately 400 genes estimated to reside on chromosome 12p confer risk for CDH.
Del (15)(q26.1-q26.2) (OMIM 142340, DIH1)
There is compelling evidence from more than two dozen cases that loss of the 15q26.1-q26.2 interval significantly increases the risk for abnormal diaphragm formation. Loss of this interval can result from one of several mechanisms such as de novo deletions, unbalanced translocations, or formation of a ring chromosome 15. In addition to CDH, commonly occurring abnormalities include growth retardation, facial dysmorphology, and limb, renal, and cardiovascular malformations (CVMs) (64). Mortality among patients with deletions encompassing this interval remains very high. The facial dysmorphology of patients with del (15)(q26.1-q26.2) is often described as ‘coarse’ accompanied by ‘hypertelorism’. Given overlapping facial appearance with some ‘Fryns syndrome’ patients, perhaps this region houses a locus responsible for a subset of patients falling within this phenotypic spectrum.
The deleted gene (or genes) located in the 15q26 interval that increases the risk for developing CDH is not currently known. The smallest region of deletion overlap is approximately 3.5 million base pairs and may contain as few as half a dozen genes (64,65). A compelling candidate gene within this region is COUP-TFII (chick ovalbumin upstream promoter transcription factor II), encoding a receptor in the steroid/thyroid hormone family. The evidence implicating COUP-TFII in human CDH stems from several sources including the following: COUP-TFII is located in the chromosome 15q26 CDH hot spot (66); CDH occurs in a Couptf2 conditional knockout mouse (23); and COUP-TFII is involved in the retinol pathway (67), a pathway known to be important for normal diaphragm formation. Despite evidence implicating COUP-TFII as a candidate gene, sequencing of several hundred CDH patients to date has yet to reveal a coding region mutation (68–70).
Del (8)(p23.1) (OMIM 222400, DIH2)
Another CDH ‘hot spot’ revealed by cytogenetic and molecular cytogenetic studies is (8)(p23) (47, 53, 55, 71–74). The smallest deletion characterized to date covers approximately 3.5 Mb (73), containing the candidate gene GATA4, known to be important for heart, lung, and diaphragm development (28, 75).
8q23 (OMIM 610187, DIH3)
Cytogenetic rearrangements of this region in patients with CDH are few in number, and none have been characterized at the molecular level (49, 76–78). Despite this, localization of FOG2 to this region and the discovery of a de novo mutation in a patient with fatal but apparently isolated CDH strongly suggest that this is a CDH locus. Further evidence underscoring the importance of FOG2 in human CDH is the presence of pulmonary hypoplasia and diaphragmatic eventration/sac-type hernias in a Fog2 hypomorphic mutant mouse model (13).
Del (4)(p16) (OMIM 194190, Wolf–Hirschhorn syndrome)
Patients with Wolf–Hirschhorn syndrome (WHS) can have large 4p deletions detectable by standard karyotyping or smaller ‘microdeletions’ detectable only by FISH or aCGH. Although CDH is a relatively rare part of the WHS phenotype, it has been reported in more than a dozen patients in association with variable-sized deletions (49, 56, 70, 79–82). The smallest deletion characterized at the molecular level is 2.6 Mb pairs (79), and molecular characterization of additional cases is needed to define the CDH critical region.
+ der (22) t(11;22)(q23;q11) (OMIM 609029) and trisomy 22
Cases of CDH, occurring with additional anomalies, have been reported in patients with trisomy 22 as well as in patients with ‘partial trisomy 22q’ (83–87). The latter most commonly arises following 3:1 meiotic non-disjunction from a t(11;22) (q23.3; q1 1.2) balanced translocation carrier parent. Thus, trisomy for one or more genes in this interval appears to confer risk for developing CDH.
Del (1)(q41-q42.12)
Although not yet assigned an OMIM number, evidence is accumulating that loss of one or more genes in this interval predisposes to CDH plus additional anomalies (70, 88–91). Screening of approximately 10,000 patients using targeted aCGH identified 7 patients harboring a deletion of this interval, 2 of whom had CDH (92). Both cases had previously normal routine karyotypes, the first published as a long-term Fryns syndrome survivor by Van Hove et al. (93) and the second published and characterized by Kantarci et al. (88). Combining results from all available cases, the smallest region of overlap is approximately 1.2 Mb pairs. Several genes map to this region, but Shaffer et al. suggest that deletion of the gene dispatched 1 (DISP1) is the prime candidate gene, given its interaction with sonic hedgehog (92). Our group has just identified a de novo mosaic DISP1 variant in a highly conserved residue in a karyotypically normal CDH+ patient (94).
Known monogenic syndromes associated with CDH
A dozen or so syndromes have a well-documented association with CDH; several of these disorders are listed in Table 2 and are described in greater detail below. Other syndromes have CDH as an occasional finding; examples of these are Apert, Beckwith–Weidemann, CHARGE, Coffin–Siris, Goltz, Perlman, and Swyer syndromes. For further discussion of these phenotypes, the reader is referred to Pober et al. (95). Important reasons for recognizing monogenic syndromes that include CDH are proper genetic and recurrence risk counseling, mutation testing if available, guidelines for prenatal evaluation, and potential insight into pathways responsible for CDH that in turn could inform developmental mechanisms and even therapy.
Table 2.
Monogenic syndromes in which CDH prominently features and description of several monogenic syndromes, further elaborated on in the text, in which CDH commonly occurs
| Syndrome | OMIM/inheritance | Locus/gene | Additional prominent features |
|---|---|---|---|
| Cornelia de Lange | 122470/AD 300560/XL |
5p13.1/NIPBL Xp11.2/SMC1A |
Growth retardation, micromelia, facial dysmorphology, mental retardation, and hirsutism |
| Craniofrontonasal dysplasia | 304110/XL | Xq12/EFNB1 | Craniosynostosis, hypertelorism, bifid nasal tip, musculoskeletal abnormalities, and longitudinal grooves on nails. Females more severely affected than males |
| Donnai–Barrow syndrome | 222448/AR | 2q24.3-2q31.1/LRP2 | Hypertelorism, high myopia, agenesis of corpus callosum, sensorineural hearing loss, low-molecular-weight proteinuria, and developmental delay |
| Fryns syndromea | 229850/UNK | UNK | Coarse face, distal digital hypoplasia, anomalies of the central nervous, cardiovascular, and/or genitourinary systems |
| Matthew-Wood syndrome (PDAC or PMD syndrome) | 601186/AR | 15q24.1/STRA6 | Micro- or anophthalmia, pulmonary hypoplasia, and cardiovascular and genitourinary malformations |
| Multiple vertebral segmentation defects (spondylocostal dysostosis and Jarcho–Levin syndrome) | 277300/AR (milder apparently AD form exists as well) | 19q13/DLL3 (most commonly mutated gene to date) | Short stature, hemivertebrae, fused vertebrae, rib anomalies, and crab-like thorax in some subtypes |
| Simpson–Golabi–Behmel | 312870/XL | Xq26/GPC3 | Overgrowth, coarse facies, musculoskeletal and limb anomalies (e.g. polydactyly and brachydactyly), renal anomalies, and risk for embryonal tumors |
| WT1-opathies (Denys–Drash, Frasier, and Meacham syndromes) | 194080, 136690, and 608978/AD | 11p13/WT1 | Overlapping disorders. All have ambiguous genitalia but renal anomalies and risk for embryonal tumors typify Denys–Drash and Frasier syndromes, while cardiovascular malformations are found in Meacham syndrome |
AD, autosomal dominant; AR, autosomal recessive; PDAC, Pulmonary hypoplasia/agenesis, Diaphragmatic hernia/eventration,
Anophthalmia/microphthalmia, and Cardiac defects; PMD, Pulmonary agenesis, microphthalmia, and diaphragmatic defect; UNK, unknown; XL, X-linked.
Etiologic heterogeneity is likely (due to chromosome microdeletions and AR phenocopies).
Cornelia de Lange syndrome (OMIM 122470 and 300560)
Cornelia de Lange syndrome (CdLS) is a phenotypically striking and well-characterized multiple malformation syndrome whose cardinal features include growth retardation, hirsutism, mental retardation, and upper limb anomalies (ranging from subtle micromelia to dramatic oligodactyly) plus a characteristic dysmorphic facial appearance (96). CDH has been reported in several cases and in fact may be as prevalent as 5–10% (97). Prenatal detection of CDH and upper limb defects should always raise suspicion about the diagnosis of CdLS (98).
The vast majority of cases are caused by de novo dominant mutations predominantly in the gene Nipped-B-Like (NIPBL) present in approximately 50% of CdLS patients (96). More rarely, mutations in the X-linked structural maintenance of chromosomes 1A (SMC1A) gene cause CdLS. Genotype–phenotype correlations for CDH do not yet exist.
Craniofrontonasal syndrome (OMIM 304110)
Craniofrontonasal syndrome (CFNS) is an X-linked disorder whose prominent findings involve, although are not restricted to, the craniofacial region. Paradoxically, in this X-linked disorder, females are more severely affected than males and typically manifest coronal craniosynostosis, hypertelorism, a broad nasal bridge with a broad bifid nasal tip, facial asymmetry, frontal bossing plus a variety of skeletal abnormalities including thorax and clavicular abnormalities, scoliosis and longitudinally grooved fingernails. Males however are more likely to have hypertelorism or telecanthus, short stature, and either no or only mild musculoskeletal abnormalities (99). CDH has been reported in several CFNS patients, including both males and females [reviewed in (100)]. Interestingly, the mutations found in two CFNS females who had CDH were truncating rather than missense mutations (101).
CFNS is caused by mutations in the ephrin-B1 gene (EFNB1) coding for a transmembrane protein that functions both as a ligand and as a receptor. The reason that females are more severely affected than males is not fully understand. One hypothesis posits that mutation carrier females fail to preferentially inactivate their mutant X chromosome, so they are ephrin-B1 mosaics. Thus, carrier females have cellular patches of ephrin expression surrounded by cellular patches of no expression, resulting in abnormal cellular signaling (so-called ‘cellular interference’) (102). As predicted by the ‘cellular interference’ model, mutant males are more mildly affected than females because complete lack of ephrin-B1 expression should be better tolerated than patchy ephrin-B1 expression. Males compensate for the absence of ephrin-B1 by utilizing other ephrin ligands (103, 104).
Donnai–Barrow syndrome/faciooculoacousticorenal syndrome (OMIM 222448)
Initially reported as separate entities, recent work shows that DBS and faciooculoacousticorenal syndrome (FOAR) are the same autosomal recessive disorder caused by mutations in the low-density lipoprotein receptor-related protein 2 gene (LRP2). LRP2 encodes the protein megalin, a transmembrane receptor important for endocytosis and reuptake of numerous ligands (42).
DBS was first reported by Donnai and Barrow in 1993 (105). Based on their work as well as findings from subsequent cases (42, 106, 107), invariant features of the DBS phenotype include high myopia, hypertelorism, complete or partial agenesis of the corpus callosum, sensorineural hearing loss, facial dysmorphology, and developmental delay. Approximately half of the patients clinically diagnosed with DBS also have CDH and/or omphalocele, while coloboma is less commonly seen. Features distinguishing patients diagnosed clinically with FOAR have been macrocephaly (but not agenesis of the corpus callosum), proteinuria, and infrequent occurrence of CDH (107–109). However, it is now known that proteinuria, in fact a characteristic pattern of low-molecular-weight proteinuria, is present in all DBS/FOAR patients. The human phenotype resulting from LRP2 mutations resembles that of the Lrp2 (e.g. megalin) knockout mouse, although the mouse abnormalities of microophthalmia and mild holoprosencephaly are not recapitulated in humans (110). The mechanism of action by which absent or dysfunctional megalin protein causes the characteristic pattern of structural birth defects seen in DBS/FOAR is not known; possible mechanisms include disruption of megalin–sonic hedgehog interactions or failure to endocytose critical ligands, such as retinol-binding protein required for normal development.
Mutations in LRP2 are scattered throughout the gene without evidence of a mutation ‘hot spot’. In light of this and the fact that the gene is large containing 79 exons, clinical suspicion about the diagnosis of DBS/FOAR should be bolstered by quantitative (not dipstick) urine protein determination to look for low-molecular-weight proteinuria. If proteinuria is present, then urine electrophoresis to look for the characteristic pattern of low molecular moieties should be obtained, before proceeding to LRP2 sequencing (42).
In seven of eight mutation-proven DBS/FOAR cases, each parent carried a mutant allele as expected for a disorder with autosomal recessive inheritance. However, one DBS/FOAR patient, born to non-consanguineous parents, was homozygous for an LRP2 mutant allele that was present only on the paternal haplotype. Microsatellite markers showed that the proband had uniparental isodisomy for paternal chromosome 2 (111). Consequently, the DBS/FOAR recurrence risk for this couple was reduced from 25% to virtually negligible.
Fryns syndrome phenotype
The clinical diagnosis most commonly made when CDH occurs in the presence of additional malformations is that of Fryns syndrome. However, it is not likely that all patients diagnosed as such have the same entity, indicating that etiologic heterogeneity exists in this group. It may be more appropriate at this juncture to describe a Fryns syndrome phenotype that encompasses clinically overlapping subsets of patients with (i) autosomal recessive Fryns syndrome (OMIM 229850), (ii) patients with chromosomal microdeletion and microduplication phenocopies, and (iii) patients with autosomal recessive phenocopies due to mutations in other genes. The responsible gene (or genes) for any of these subsets is not currently known.
Common features found in patients classified as having the Fryns phenotype can be grouped into five categories (112) adapted from Fryns (113):
Diaphragmatic defect: any type of diaphragmatic abnormality including typical CDH, an eventration, or sac-type hernia. Diaphragm defects are extremely common although not a universal finding in patients diagnosed with Fryns syndrome.
Pulmonary hypoplasia: pulmonary hypoplasia almost always accompanies CDH and so this may not represent an independent diagnostic criterion. However, pulmonary hypoplasia was reported in at least one case without diaphragmatic abnormality on autopsy (114).
Facial dysmorphology: coarsened face, flattened nasal bridge, hypertelorism, and nonspecific ear anomalies (113).
Distal digital hypoplasia: small nails and/or distal phalanges.
Characteristic pattern of additional anomalies: numerous malformations have been reported in patients diagnosed with Fryns syndrome including but not limited to CVMs (such as septal and conotruncal defects), renal anomalies (such as cystic dysplasia), and brain malformations (e.g. agenesis of the corpus callosum and Dandy–Walker malformation).
The occurrence of a similarly affected sibling counts as a sixth criterion. Lin et al. propose that patients with classic (or narrowly defined) Fryns syndrome have at least four of the six findings, while those with only three findings can be considered as having broadly defined Fryns syndrome (112). The validity of this dichotomous classification schema is not currently known and will remain unknown until the criteria are evaluated prospectively and/or genetic mutations responsible for some cases falling within the Fryns phenotypic spectrum are identified.
Prior to counseling autosomal recessive risk of recurrence to a couple who has conceived a child with Fryns syndrome, aCGH should be performed on a DNA sample from the child (74, 88). Detection of a microdeletion or microduplication alters the recurrence risk and also permits specific genetic testing in subsequent pregnancies.
Matthew-Wood syndrome (Spear syndrome, PDAC or PMD syndrome) (OMIM 601186)
MWS is a rare autosomal recessive disorder with a striking phenotype, most often including microophthalmia or anophthalmia, pulmonary hypoplasia or agenesis, and diaphragmatic defects. Chitayat et al. suggest that the acronym PDAC (Pulmonary hypoplasia/agenesis, Diaphragmatic hernia/eventration, Anophthalmia/microphthalmia, and Cardiac defects) replace the eponyms historically used to refer to this phenotype (115). Mutations in STRA6 (a member of the stimulated by retinoic acid gene family) have been found in some, but not all, cases with this constellation of anomalies (116). STRA6 encodes for the transmembrane retinol-binding protein receptor that functions in cellular uptake of retinol. Mutations in this important developmental gene interfere with morphogenesis of multiple organs, possibly through disruption of normal retinoic acid signaling.
Multiple vertebral segmentation defects (OMIM 277300)
CDH has been reported in more than a dozen cases in conjunction with disorders of vertebral segmentation (variously called spondylocostal dysostosis, spondylothoracic dysostosis, and Jarcho–Levin syndrome) (117–120). The distinction among these disorders continues to be debated, but it is important for the clinician to recognize their association with CDH (121). In common, these disorders show multiple contiguous vertebral abnormalities (such as butterfly and hemivertebrae), rib anomalies (such as absent or fused ribs), and shortened trunk (122, 123).
To date, mutations have been identified in several genes [Delta-like 3 (DLL3), lunatic fringe (LFNG), and mesoderm posterior 2 (MESP2) (123)] in the Notch signaling pathway, which plays an important role in body planning and somite formation.
Simpson–Golabi–Behmel syndrome (OMIM 312870)
There are reports of CDH occurring in this X-linked disorder although far more common manifestations are prenatal and postnatal overgrowth, coarse facial features with hypertelorism, skeletal anomalies (polydactyly, brachydactyly, and syndactyly), renal anomalies (cystic dysplastic kidneys), cardiovascular anomalies, and supernumerary nipples (124, 125). The range of intellectual outcomes is broad extending from normal intelligence to mental retardation. There is a significant risk for occurrence of embryonal tumors that requires monitoring particularly for the development of Wilms tumor and hepatocellular carcinoma (126).
Loss-of-function mutations in the gene glypican-3 (GPC3) are responsible for Simpson–Golabi– Behmel syndrome, but the mechanism by which this predisposes to CDH remains unknown.
‘WT1-opathies' [Denys–Drash syndrome (OMIM 194080), Frasier syndrome (OMIM 136690), and Meacham syndrome (OMIM 608978)]
De novo mutations in the WT1 gene have been identified in several cases diagnosed clinically with Denys–Drash, Frasier, and Meacham syndromes (127, 128). These allelic disorders share the presence of genital abnormalities (e.g. ambiguous genitalia and male pseudohermaphroditism) but also have distinct differences in that complex CVMs occur in Meacham syndrome, while progressive renal failure and risk for embryonal tumors typify the other two conditions. CDH has been reported in all three entities, albeit infrequently in Frasier and Denys–Drash syndromes (128–132). In mouse models, Wt1 is expressed in the developing structures of the diaphragm including the septum transversum and the PPFs, so it is not surprising that some Wt1 knockout mice manifest a typical posterolateral Bochdalek-type hernia (24, 25, 133).
CDH with other congenital malformations that do not constitute a recognizable pattern
Among the approximately 40% of cases with CDH +, a chromosomal or syndromic diagnosis is established in the minority. Thus, the majority of these cases show a non-diagnostic pattern of anomalies, with the most common coexisting malformations involving the cardiovascular, central nervous, and musculoskeletal systems. These patterns are presumably non-random, but instead reflect disruption of genes whose products function in all the affected organ systems.
CDH and CVMs
After excluding syndromic cases, CVMs coexist with CDH in approximately 10–15% of cases (134) (Note that the frequency of CVMs coexisting with CDH doubles or triples if one neglects to exclude syndromic cases.) (9, 50, 52, 135–138). All types of CVMs are found in association with CDH, and efforts to identify a particular CVM– CDH pattern have been unrevealing. For example, an excess of conotruncal malformations has been suggested in some studies but has not been substantiated in others. The pitfall of diagnosing (or ‘overdiagnosing’) hypoplastic left heart syndrome in the setting of CDH is further discussed by Lin et al. (134).
CDH and central nervous system abnormalities
Central nervous system anomalies, particularly neural tube defects and hydrocephalus are present in 5–10% of non-syndromic CDH cases (9, 41, 139). The basis for the association with neural tube defects is not known although problems of schisis-fusion have been suggested (140).
CDH and limb abnormalities
Limb defects co-occur in approximately 10% of non-syndromic CDH cases. Although different defects such as syndactyly and polydactyly have been reported, the coexistence of CDH with limb reduction defects has received the most scrutiny (10, 141) due to speculation that this unique combination of malformations will reveal candidate genes responsible for both defects.
Genetic workup
Given the considerable etiologic heterogeneity found in patients with CDH +, it is not possible to provide a single or uniform approach to the diagnostic evaluation of these cases. The initial workup is as described above for cases with isolated CDH but is then tailored to the existing abnormalities in a given case.
Genetics evaluation of a patient with CDH + begins with a routine chromosome analysis (amniocentesis or chorionic villus sampling for prenatally detected cases and lymphocyte-stimulated blood culture for postnatally detected cases). When a patient's phenotype is suggestive of isochromosome 12p, then a chromosome study on a non-lymphocyte-derived tissue is required.
Several recent reports describe CDH + cases with prior normal 46,XX or 46,XY routine karyotypes but abnormalities detected on aCGH (64, 74, 88). The yield of aCGH in this setting is not currently known, but application of this technology should be considered in prenatally diagnosed cases of CDH (due to limitations in detecting all major malformations, dysmorphology, and minor anomalies). aCGH should also be considered in postnatally evaluated patients who have growth retardation, facial dysmorphology, and/or major and minor anomalies in conjunction with CDH and for whom a diagnosis is not established. As the number of CDH chromosome hot spots continues to grow, targeted arrays that densely cover these hot spots are being developed.
Because causal genes have been identified for several CDH + syndromes, specific mutation analysis should be part of the diagnostic evaluation when patients fitting these phenotypes are being evaluated. Given the continued high mortality of CDH, especially CDH +, banking of patient DNA should be suggested to the family. DNA banking is further discussed in the Gene-Reviews Congenital Diaphragmatic Hernia Overview (95).
Challenging counseling dilemmas can arise for CDH + cases in which a genetic syndrome or chromosome abnormality cannot currently be identified. The common coexistence of CDH and congenital heart disease, described earlier, is an example in point. In approximately half of the cases, the combined presence of both these malformations is part of a genetic syndrome (e.g. Fryns syndrome) or a chromosome abnormality [e.g. del (15)(q26.1-q26.2)]. However, in the other half of the cases, a unifying etiology is not recognized even after appropriate diagnostic evaluation. It seems likely that the proportion of diagnosed cases will continue to grow over time with advances in molecular cytogenetic techniques and gene identification for CDH-associated syndromes. For the undiagnosed CDH + cases, no empiric data are currently available to guide recurrence risk counseling. Until such data exist, it seems prudent to provide multiple congenital anomaly (MCA) recurrence risk counseling to parents whose child has CDH plus other major malformations such as complex congenital heart disease. Counseling the parents of a child with CDH and a muscular VSD is particularly challenging. Assuming that neither parent has a history of a VSD, the conservative approach is to provide an MCA recurrence risk of <5% until more information from family studies and from natural history studies on small muscular VSDs become available.
Pathways important for diaphragm development
As reviewed above, progress is being made in the genetic dissection of human CDH. For example, critical chromosome hot spots and specific genes responsible for several CDH-associated monogenic syndromes have been identified, and each new gene contributes to knowledge about diaphragm development. Rather than looking at genes in isolation, it is important to consider them as potential windows into developmental pathways required to support normal diaphragm formation (68). Recognizing pathways in their entirety may lead to more rapid treatment advances than recognizing only a single member of the pathway.
Current evidence suggests that an intact vitamin A pathway is needed to support normal diaphragm formation (142). Lines of evidence that perturbations in this pathway confer risk for CDH are multifold and were presented above. Suffice it to say that data come from both model organism work [dietary deficiency (143, 144), transgenic (145), and teratogenic (43)] as well as from human studies [epidemiologic (44, 45) and human malformation syndromes (42, 116)]. An important next step is to assess whether vitamin A pathway disturbances contribute to the most common form of CDH, namely isolated (nonsyndromic) CDH. Two potential directions for further study include analyses of high-density single nucleotide polymorphism arrays to assess whether variations in vitamin A pathway genes occur more commonly in CDH patients than controls and implementation of prospective studies to measure vitamin A levels throughout gestation. Similar approaches may prove useful for identifying and characterizing other pathways important for diaphragm development.
References
- 1.Mertins H. Familial abnormality of the diaphragm. Zentralbl Gynakol. 1952;74:951–955. [PubMed] [Google Scholar]
- 2.Rodgers BM, Hawks P. Bilateral congenital eventration of the diaphragms: successful surgical management. J Pediatr Surg. 1986;21:858–864. doi: 10.1016/s0022-3468(86)80008-2. [DOI] [PubMed] [Google Scholar]
- 3.Thomas MP, Stern LM, Morris LL. Bilateral congenital diaphragmatic defects in two siblings. J Pediatr Surg. 1976;11:465–467. doi: 10.1016/s0022-3468(76)80206-0. [DOI] [PubMed] [Google Scholar]
- 4.Varpella E, Lehtovaara R. Familial occurrence of diaphragmatic abnormalities. Ann Chir Gynaecol Fenn. 1962;58:62–64. [PubMed] [Google Scholar]
- 5.Czeizel A, Kovacs M. A family study of congenital diaphragmatic defects. Am J Med Genet. 1985;21:105–117. doi: 10.1002/ajmg.1320210115. [DOI] [PubMed] [Google Scholar]
- 6.Skandalakis JE, Gray SW, Ricketts RR. Embryology for surgeons. Baltimore, MD: Williams & Wilkins; 1994. [Google Scholar]
- 7.Babiuk RP, Greer JJ. Diaphragm defects occur in a CDH hernia model independently of myogenesis and lung formation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1310–L1314. doi: 10.1152/ajplung.00257.2002. [DOI] [PubMed] [Google Scholar]
- 8.Greer JJ, Allan DW, Martin-Caraballo M, et al. An overview of phrenic nerve and diaphragm muscle development in the perinatal rat. J Appl Physiol. 1999;86:779–786. doi: 10.1152/jappl.1999.86.3.779. [DOI] [PubMed] [Google Scholar]
- 9.Dott MM, Wong LY, Rasmussen SA. Population-based study of congenital diaphragmatic hernia: risk factors and survival in Metropolitan Atlanta, 1968-1999. Birth Defects Res A Clin Mol Teratol. 2003;67:261–267. doi: 10.1002/bdra.10039. [DOI] [PubMed] [Google Scholar]
- 10.Torfs CP, Curry CJ, Bateson TF, et al. A population-based study of congenital diaphragmatic hernia. Teratology. 1992;46:555–565. doi: 10.1002/tera.1420460605. [DOI] [PubMed] [Google Scholar]
- 11.Ackerman KG, Vargas SO, Kozakewich HP. Phenotypic Variation of Congenital Diaphragmatic Defects (573). Presented at the annual meeting of The American Society of Human Genetics; 9-13 October; New Orleans, LA. Available at http://wwwashgorg/genetics/ashg05s/2006. [Google Scholar]
- 12.Ackerman KG, Pober BR. Congenital diaphragmatic hernia and pulmonary hypoplasia: new insights from developmental biology and genetics. Am J Med Genet C Semin Med Genet. 2007;145:105–108. doi: 10.1002/ajmg.c.30133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackerman KG, Herron BJ, Vargas SO, et al. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:58–65. doi: 10.1371/journal.pgen.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keijzer R, Liu J, Deimling J, et al. Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol. 2000;156:1299–1306. doi: 10.1016/S0002-9440(10)65000-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rottier R, Tibboel D. Fetal lung and diaphragm development in congenital diaphragmatic hernia. Semin Perinatol. 2005;29:86–93. doi: 10.1053/j.semperi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Colvin J, Bower C, Dickinson JE, et al. Outcomes of congenital diaphragmatic hernia: a population-based study in Western Australia. Pediatrics. 2005;116:e356–e363. doi: 10.1542/peds.2004-2845. [DOI] [PubMed] [Google Scholar]
- 17.Skari H, Bjornland K, Haugen G, et al. Congenital diaphragmatic hernia: a meta-analysis of mortality factors. J Pediatr Surg. 2000;35:1187–1197. doi: 10.1053/jpsu.2000.8725. [DOI] [PubMed] [Google Scholar]
- 18.Lally KP, Lally PA, Lasky RE, et al. Defect size determines survival in infants with congenital diaphragmatic hernia. Pediatrics. 2007;120:e651–e657. doi: 10.1542/peds.2006-3040. [DOI] [PubMed] [Google Scholar]
- 19.Harrison MR, Bjordal RI, Langmark F, et al. Congenital diaphragmatic hernia: the hidden mortality. J Pediatr Surg. 1978;13:227–230. doi: 10.1016/s0022-3468(78)80391-1. [DOI] [PubMed] [Google Scholar]
- 20.Barnewolt CE, Kunisaki SM, Fauza DO, et al. Percent predicted lung volumes as measured on fetal magnetic resonance imaging: a useful biometric parameter for risk stratification in congenital diaphragmatic hernia. J Pediatr Surg. 2007;42:193–197. doi: 10.1016/j.jpedsurg.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 21.Neff KW, Kilian AK, Schaible T, et al. Prediction of mortality and need for neonatal extracorporeal membrane oxygenation in fetuses with congenital diaphragmatic hernia: logistic regression analysis based on MRI fetal lung volume measurements. AJR Am J Roentgenol. 2007;189:1307–1311. doi: 10.2214/AJR.07.2434. [DOI] [PubMed] [Google Scholar]
- 22.Mei-Zahav M, Solomon M, Trachsel D, et al. Bochdalek diaphragmatic hernia: not only a neonatal disease. Arch Dis Child. 2003;88:532–535. doi: 10.1136/adc.88.6.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.You LR, Takamoto N, Yu CT, et al. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci U S A. 2005;102:16351–16356. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clugston RD, Klattig J, Englert C, et al. Teratogeninduced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am J Pathol. 2006;169:1541–1549. doi: 10.2353/ajpath.2006.060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kreidberg JA, Sariola H, Loring JM, et al. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Zhang L, Wang D, et al. Congenital diaphragmatic hernia, kidney agenesis and cardiac defects associated with Slit3-deficiency in mice. Mech Dev. 2003;120:1059–1070. doi: 10.1016/s0925-4773(03)00161-8. [DOI] [PubMed] [Google Scholar]
- 27.Yuan W, Rao Y, Babiuk RP, et al. A genetic model for a central (septum transversum) congenital diaphragmatic hernia in mice lacking Slit3. Proc Natl Acad Sci U S A. 2003;100:5217–5222. doi: 10.1073/pnas.0730709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jay PY, Bielinska M, Erlich JM, et al. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2006;19:1495–1497. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ackerman KG, Greer JJ. Development of the diaphragm and genetic mouse models of diaphragmatic defects. Am J Med Genet C Semin Med Genet. 2007;145:109–116. doi: 10.1002/ajmg.c.30128. [DOI] [PubMed] [Google Scholar]
- 30.Beurskens N, Klaassens M, Rottier R, et al. Linking animal models to human congenital diaphragmatic hernia. Birth Defects Res A Clin Mol Teratol. 2007;79:565–572. doi: 10.1002/bdra.20370. [DOI] [PubMed] [Google Scholar]
- 31.Bielinska M, Jay PY, Erlich JM, et al. Molecular genetics of congenital diaphragmatic defects. Ann Med. 2007;39:261–274. doi: 10.1080/07853890701326883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Philipp EE, Skelton MO. Congenital diaphragmatic hernia in siblings. Br Med J. 1952;1:1283–1284. doi: 10.1136/bmj.1.4771.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burry AF. Congenital diaphragmatic hernia in two siblings. Northwest Med. 1961;60:110–111. [PubMed] [Google Scholar]
- 34.Welch RG, Cooke RT. Congenital diaphragmatic hernia. Lancet. 1962;279:975. [Google Scholar]
- 35.Passarge E, Halsey H, German J. Unilateral agenesis of the diaphragm. Humangenetik. 1968;5:226–230. doi: 10.1007/BF00281959. [DOI] [PubMed] [Google Scholar]
- 36.ten Kate LP, Anders GJ. Unilateral agenesis of the diaphragm. Humangenetik. 1970;8:366–367. doi: 10.1007/BF00280341. [DOI] [PubMed] [Google Scholar]
- 37.Pollack LD, Hall JG. Posterolateral (Bochdalek's) diaphragmatic hernia in sisters. Am J Dis Child. 1979;133:1186–1188. doi: 10.1001/archpedi.1979.02130110094019. [DOI] [PubMed] [Google Scholar]
- 38.Wolff G. Familial congenital diaphragmatic defect: review and conclusions. Hum Genet. 1980;54:1–5. doi: 10.1007/BF00279041. [DOI] [PubMed] [Google Scholar]
- 39.Norio R, Kaariainen H, Rapola J, et al. Familial congenital diaphragmatic defects: aspects of etiology, prenatal diagnosis, and treatment. Am J Med Genet. 1984;17:471–483. doi: 10.1002/ajmg.1320170210. [DOI] [PubMed] [Google Scholar]
- 40.Pober BR, Lin A, Russell M, et al. Infants with Bochdalek diaphragmatic hernia: sibling precurrence and monozygotic twin discordance in a hospital-based malformation surveillance program. Am J Med Genet A. 2005;138:81–88. doi: 10.1002/ajmg.a.30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.David TJ, Illingworth CA. Diaphragmatic hernia in the south-west of England. J Med Genet. 1976;13:253–262. doi: 10.1136/jmg.13.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kantarci S, Al-Gazali L, Hill RS, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957–959. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kling DE, Schnitzer JJ. Vitamin A deficiency (VAD), teratogenic, and surgical models of congenital diaphragmatic hernia (CDH) Am J Med Genet C Semin Med Genet. 2007;145:139–157. doi: 10.1002/ajmg.c.30129. [DOI] [PubMed] [Google Scholar]
- 44.Major D, Cadenas M, Fournier L, et al. Retinol status of newborn infants with congenital diaphragmatic hernia. Pediatr Surg Int. 1998;13:547–549. doi: 10.1007/s003830050399. [DOI] [PubMed] [Google Scholar]
- 45.Yang W, Shaw GM, Carmichael SL, et al. Nutrient intakes in women and congenital diaphragmatic hernia in their offspring. Birth Defects Res A Clin Mol Teratol. 2008;82:131–138. doi: 10.1002/bdra.20436. [DOI] [PubMed] [Google Scholar]
- 46.Bollmann R, Kalache K, Mau H, et al. Associated malformations and chromosomal defects in congenital diaphragmatic hernia. Fetal Diagn Ther. 1995;10:52–59. doi: 10.1159/000264193. [DOI] [PubMed] [Google Scholar]
- 47.Faivre L, Morichon-Delvallez N, Viot G, et al. Prenatal diagnosis of an 8p23.1 deletion in a fetus with a diaphragmatic hernia and review of the literature. Prenat Diagn. 1998;18:1055–1060. doi: 10.1002/(sici)1097-0223(1998100)18:10<1055::aid-pd405>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 48.Garne E, Haeusler M, Barisic I, et al. Congenital diaphragmatic hernia: evaluation of prenatal diagnosis in 20 European regions. Ultrasound Obstet Gynecol. 2002;19:329–333. doi: 10.1046/j.1469-0705.2002.00635.x. [DOI] [PubMed] [Google Scholar]
- 49.Howe DT, Kilby MD, Sirry H, et al. Structural chromosome anomalies in congenital diaphragmatic hernia. Prenat Diagn. 1996;16:1003–1009. doi: 10.1002/(SICI)1097-0223(199611)16:11<1003::AID-PD995>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 50.Philip N, Gambarelli D, Guys JM, et al. Epidemiological study of congenital diaphragmatic defects with special reference to aetiology. Eur J Pediatr. 1991;150:726–729. doi: 10.1007/BF01958765. [DOI] [PubMed] [Google Scholar]
- 51.Thorpe-Beeston JG, Gosden CM, Nicolaides KH. Prenatal diagnosis of congenital diaphragmatic hernia: associated malformations and chromosomal defects. Fetal Ther. 1989;4:21–28. doi: 10.1159/000263386. [DOI] [PubMed] [Google Scholar]
- 52.Tonks A, Wyldes M, Somerset DA, et al. Congenital malformations of the diaphragm: findings of the West Midlands Congenital Anomaly Register 1995 to 2000. Prenat Diagn. 2004;24:596–604. doi: 10.1002/pd.908. [DOI] [PubMed] [Google Scholar]
- 53.Borys D, Taxy JB. Congenital diaphragmatic hernia and chromosomal anomalies: autopsy study. Pediatr Dev Pathol. 2004;7:35–38. doi: 10.1007/s10024-003-2133-7. [DOI] [PubMed] [Google Scholar]
- 54.Lurie IW. Where to look for the genes related to diaphragmatic hernia? Genet Couns. 2003;14:75–93. [PubMed] [Google Scholar]
- 55.Pecile V, Petroni MG, Fertz MC, et al. Deficiency of distal 8p–report of two cases and review of the literature. Clin Genet. 1990;37:271–278. doi: 10.1111/j.1399-0004.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 56.van Dooren MF, Brooks AS, Hoogeboom AJ, et al. Early diagnosis of Wolf-Hirschhorn syndrome triggered by a life-threatening event: congenital diaphragmatic hernia. Am J Med Genet A. 2004;127:194–196. doi: 10.1002/ajmg.a.20613. [DOI] [PubMed] [Google Scholar]
- 57.Holder AM, Klaassens M, Tibboel D, et al. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. 2007;80:825–845. doi: 10.1086/513442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parmar RC, Tullu MS, Bavdekar SB, et al. Morgagni hernia with Down syndrome: a rare association–case report and review of literature. J Postgrad Med. 2001;47:188–190. [PubMed] [Google Scholar]
- 59.Marin J, Lopoo J. An infant with trisomy 21 and tachypnea. Pediatr Emerg Care. 2006;22:170–172. doi: 10.1097/01.pec.0000202457.64978.3d. [DOI] [PubMed] [Google Scholar]
- 60.Schaefer GB, Jochar A, Muneer R, et al. Clinical variability of tetrasomy 12p. Clin Genet. 1997;51:102–108. doi: 10.1111/j.1399-0004.1997.tb02429.x. [DOI] [PubMed] [Google Scholar]
- 61.Mathieu M, Piussan C, Thepot F, et al. Collaborative study of mosaic tetrasomy 12p or Pallister-Killian syndrome (nineteen fetuses or children) Ann Genet. 1997;40:45–54. [PubMed] [Google Scholar]
- 62.Doray B, Girard-Lemaire F, Gasser B, et al. Pallister-Killian syndrome: difficulties of prenatal diagnosis. Prenat Diagn. 2002;22:470–477. doi: 10.1002/pd.342. [DOI] [PubMed] [Google Scholar]
- 63.Struthers JL, Cuthbert CD, Khalifa MM. Parental origin of the isochromosome 12p in Pallister-Killian syndrome: molecular analysis of one patient and review of the reported cases. Am J Med Genet. 1999;84:111–115. [PubMed] [Google Scholar]
- 64.Klaassens M, Galjaard RJ, Scott DA, et al. Prenatal detection and outcome of congenital diaphragmatic hernia (CDH) associated with deletion of chromosome 15q26: two patients and review of the literature. Am J Med Genet A. 2007;143:2204–2212. doi: 10.1002/ajmg.a.31892. [DOI] [PubMed] [Google Scholar]
- 65.Castiglia L, Fichera M, Romano C, et al. Narrowing the candidate region for congenital diaphragmatic hernia in chromosome 15q26: contradictory results. Am J Hum Genet. 2005;77:892–894. doi: 10.1086/497082. author reply 894-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klaassens M, van Dooren M, Eussen HJ, et al. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pereira FA, Tsai MJ, Tsai SY. COUP-TF orphan nuclear receptors in development and differentiation. Cell Mol Life Sci. 2000;57:1388–1398. doi: 10.1007/PL00000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kantarci S, Donahoe PK. Congenital diaphragmatic hernia (CDH) etiology as revealed by pathway genetics. Am J Med Genet C Semin Med Genet. 2007;145:217–226. doi: 10.1002/ajmg.c.30132. [DOI] [PubMed] [Google Scholar]
- 69.Scott DA, Klaassens M, Holder AM, et al. Genome-wide oligonucleotide-based array comparative genome hybridization analysis of non-isolated congenital diaphragmatic hernia. Hum Mol Genet. 2007;16:424–430. doi: 10.1093/hmg/ddl475. [DOI] [PubMed] [Google Scholar]
- 70.Slavotinek AM, Moshrefi A, Davis R, et al. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1-15q26. 2 Eur J Hum Genet. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- 71.Hutchinson R, Wilson M, Voullaire L. Distal 8p deletion (8p23.1——8pter): a common deletion? J Med Genet. 1992;29:407–411. doi: 10.1136/jmg.29.6.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lopez I, Bafalliu JA, Bernabe MC, et al. Prenatal diagnosis of de novo deletions of 8p23.1 or 15q26.1 in two fetuses with diaphragmatic hernia and congenital heart defects. Prenat Diagn. 2006;26:577–580. doi: 10.1002/pd.1468. [DOI] [PubMed] [Google Scholar]
- 73.Shimokawa O, Miyake N, Yoshimura T, et al. Molecular characterization of del(8)(p23.1p23.1) in a case of congenital diaphragmatic hernia. Am J Med Genet A. 2005;136:49–51. doi: 10.1002/ajmg.a.30778. [DOI] [PubMed] [Google Scholar]
- 74.Slavotinek A, Lee SS, Davis R, et al. Fryns syndrome phenotype caused by chromosome microdeletions at 15q26.2 and 8p23.1. J Med Genet. 2005;42:730–736. doi: 10.1136/jmg.2004.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ackerman KG, Wang J, Luo L, et al. Gata4 is necessary for normal pulmonary lobar development. Am J Respir Cell Mol Biol. 2006;36:391–397. doi: 10.1165/rcmb.2006-0211RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Capellini A, Sala E, Colombo D, et al. Monosomy 8q and features of Fryns syndrome. Eur J Hum Genet. 1996;4(Suppl 1):29. [Google Scholar]
- 77.Harnsberger J, Carey JC, Morgan M, et al. Interstitial deletion of the long arm of the number 8 chromosome and the Langer-Giedion syndrome (LG) Proceedings of the 1982 Birth Defects Conference. 1982;19:175–176. [Google Scholar]
- 78.Temple IK, Barber JC, James RS, et al. Diaphragmatic herniae and translocations involving 8q22 in two patients. J Med Genet. 1994;31:735–737. doi: 10.1136/jmg.31.9.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Casaccia G, Mobili L, Braguglia A, et al. Distal 4p microdeletion in a case of Wolf-Hirschhorn syndrome with congenital diaphragmatic hernia. Birth Defects Res A Clin Mol Teratol. 2006;76:210–213. doi: 10.1002/bdra.20235. [DOI] [PubMed] [Google Scholar]
- 80.Sergi C, Schulze BR, Hager HD, et al. Wolf-Hirschhorn syndrome: case report and review of the chromosomal aberrations associated with diaphragmatic defects. Pathologica. 1998;90:285–293. [PubMed] [Google Scholar]
- 81.Tapper JK, Zhang S, Harirah HM, et al. Prenatal diagnosis of a fetus with unbalanced translocation (4;13)(p16;q32) with overlapping features of Patau and Wolf-Hirschhorn syndromes. Fetal Diagn Ther. 2002;17:347–351. doi: 10.1159/000065383. [DOI] [PubMed] [Google Scholar]
- 82.Van Buggenhout G, Melotte C, Dutta B, et al. Mild Wolf-Hirschhorn syndrome: micro-array CGH analysis of atypical 4p16.3 deletions enables refinement of the genotype-phenotype map. J Med Genet. 2004;41:691–698. doi: 10.1136/jmg.2003.016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim EH, Cohen RS, Ramachandran P, et al. Trisomy 22 with congenital diaphragmatic hernia and absence of corpus callosum in a liveborn premature infant. Am J Med Genet. 1992;44:437–438. doi: 10.1002/ajmg.1320440410. [DOI] [PubMed] [Google Scholar]
- 84.Ladonne JM, Gaillard D, Carre-Pigeon F, et al. Fryns syndrome phenotype and trisomy 22. Am J Med Genet. 1996;61:68–70. doi: 10.1002/(SICI)1096-8628(19960102)61:1<68::AID-AJMG13>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 85.Kadir RA, Hastings R, Economides DL. Prenatal diagnosis of supernumerary chromosome derivative (22) due to maternal balanced translocation in association with diaphragmatic hernia: a case report. Prenat Diagn. 1997;17:761–764. [PubMed] [Google Scholar]
- 86.Lin AE, Bernar J, Chin AJ, et al. Congenital heart disease in supernumerary der(22), t(11;22) syndrome. Clin Genet. 1986;29:269–275. doi: 10.1111/j.1399-0004.1986.tb01254.x. [DOI] [PubMed] [Google Scholar]
- 87.Schinzel A, Schmid W, Auf der Maur P, et al. Incomplete trisomy 22. I. Familial 11/22 translocation with 3:1 meiotic disjunction. Delineation of a common clinical picture and report of nine new cases from six families. Hum Genet. 1981;56:249–262. doi: 10.1007/BF00274675. [DOI] [PubMed] [Google Scholar]
- 88.Kantarci S, Casavant D, Prada C, et al. Findings from aCGH in patients with congenital diaphragmatic hernia (CDH): a possible locus for Fryns syndrome. Am J Med Genet A. 2006;140:17–23. doi: 10.1002/ajmg.a.31025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rogers JC, Harris DJ, Pasztor LM. Interstitial deletion of the long arm of chromosome 1: del(1)(pter→42.11::q42.3→ qter) Am J Hum Genet. 1995;57 [Google Scholar]
- 90.Smith SA, Martin KE, Dodd KL, et al. Severe microphthalmia, diaphragmatic hernia and Fallot's tetralogy associated with a chromosome 1;15 translocation. Clin Dysmorphol. 1994;3:287–291. [PubMed] [Google Scholar]
- 91.Youssoufian H, Chance P, Tuck-Muller CM, et al. Association of a new chromosomal deletion [del(1)(q32q42)] with diaphragmatic hernia: assignment of a human ferritin gene. Hum Genet. 1988;78:267–270. doi: 10.1007/BF00291674. [DOI] [PubMed] [Google Scholar]
- 92.Shaffer LG, Theisen A, Bejjani BA, et al. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- 93.Van Hove JL, Spiridigliozzi GA, Heinz R, et al. Fryns syndrome survivors and neurologic outcome. Am J Med Genet. 1995;59:334–340. doi: 10.1002/ajmg.1320590311. [DOI] [PubMed] [Google Scholar]
- 94.Kantarci S, O'Neill F, Russell MK, et al. The first report of a de novo heterozygous missense DISP1 mutation in a patient with congenital diaphragmatic hernia (CDH) and additional malformations. Presented at the annual meeting of The American Society of Human Genetics; 26 October 2007; San Diego, California. 2007. Available at http://wwwashgorg/genetics/ashg07s/indexshtml. [Google Scholar]
- 95.Pober BR, Russell MR, Ackerman KG. GeneReviews at genetests: medical genetics information resource. Seattle, WA: University of Washington; 2006. Congenital diaphragmatic hernia overview. (database online). Copyright 1997-2007. Available at http://wwwgenetestsorg. [Google Scholar]
- 96.Gillis LA, McCallum J, Kaur M, et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet. 2004;75:610–623. doi: 10.1086/424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martinez-Frias ML, Bermejo E, Felix V, et al. Brachmannde-Lange syndrome in our population: clinical and epidemiological characteristics. An Esp Pediatr. 1998;48:293–298. [PubMed] [Google Scholar]
- 98.Marino T, Wheeler PG, Simpson LL, et al. Fetal diaphragmatic hernia and upper limb anomalies suggest Brachmann-de Lange syndrome. Prenat Diagn. 2002;22:144–147. doi: 10.1002/pd.281. [DOI] [PubMed] [Google Scholar]
- 99.Saavedra D, Richieri-Costa A, Guion-Almeida ML, et al. Craniofrontonasal syndrome: study of 41 patients. Am J Med Genet. 1996;61:147–151. doi: 10.1002/(SICI)1096-8628(19960111)61:2<147::AID-AJMG8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 100.Vasudevan PC, Twigg SR, Mulliken JB, et al. Expanding the phenotype of craniofrontonasal syndrome: two unrelated boys with EFNB1 mutations and congenital diaphragmatic hernia. Eur J Hum Genet. 2006;14:884–887. doi: 10.1038/sj.ejhg.5201633. [DOI] [PubMed] [Google Scholar]
- 101.Wieland I, Makarov R, Reardon W, et al. Dissecting the molecular mechanisms in craniofrontonasal syndrome: differential mRNA expression of mutant EFNB1 and the cellular mosaic. Eur J Hum Genet. 2008;16:184–191. doi: 10.1038/sj.ejhg.5201968. [DOI] [PubMed] [Google Scholar]
- 102.Compagni A, Logan M, Klein R, et al. Control of skeletal patterning by ephrinB1-EphB interactions. Dev Cell. 2003;5:217–230. doi: 10.1016/s1534-5807(03)00198-9. [DOI] [PubMed] [Google Scholar]
- 103.Twigg SR, Matsumoto K, Kidd AM, et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: frequent somatic mosaicism and explanation of the paucity of carrier males. Am J Hum Genet. 2006;78:999–1010. doi: 10.1086/504440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wieacker P, Wieland I. Clinical and genetic aspects of craniofrontonasal syndrome: towards resolving a genetic paradox. Mol Genet Metab. 2005;86:110–116. doi: 10.1016/j.ymgme.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 105.Donnai D, Barrow M. Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: a newly recognized autosomal recessive disorder? Am J Med Genet. 1993;47:679–682. doi: 10.1002/ajmg.1320470518. [DOI] [PubMed] [Google Scholar]
- 106.Chassaing N, Lacombe D, Carles D, et al. Donnai-Barrow syndrome: four additional patients. Am J Med Genet A. 2003;121:258–262. doi: 10.1002/ajmg.a.20266. [DOI] [PubMed] [Google Scholar]
- 107.Devriendt K, Standaert L, Van Hole C, et al. Proteinuria in a patient with the diaphragmatic hernia-hypertelorismmyopia-deafness syndrome: further evidence that the facio-oculo-acoustico-renal syndrome represents the same entity. J Med Genet. 1998;35:70–71. doi: 10.1136/jmg.35.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holmes LB, Schepens CL. Syndrome of ocular and facial anomalies, telecanthus and deafness. J Pediatr. 1972;81:552–555. doi: 10.1016/s0022-3476(72)80189-6. [DOI] [PubMed] [Google Scholar]
- 109.Schowalter DB, Pagon RA, Kalina RE, et al. Facio-oculo-acoustico-renal syndrome: case report and review. Am J Med Genet. 1997;69:45–49. doi: 10.1002/(sici)1096-8628(19970303)69:1<45::aid-ajmg9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 110.Willnow TE, Hilpert J, Armstrong SA, et al. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. 1996;93:8460–8464. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kantarci S, Thomas S, Robinson D, et al. Complete paternal uniparental isodisomy for chromosome 2 resulting in a homozygous LRP2 mutation in a child with Donnai-Barrow syndrome. Am J Med Genet A. doi: 10.1002/ajmg.a.32381. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lin AE, Pober BR, Mullen MP, et al. Cardiovascular malformations in Fryns syndrome: is there a pathogenic role for neural crest cells? Am J Med Genet A. 2005;139:186–193. doi: 10.1002/ajmg.a.31023. [DOI] [PubMed] [Google Scholar]
- 113.Fryns JP. Fryns syndrome: a variable MCA syndrome with diaphragmatic defects, coarse face, and distal limb hypoplasia. J Med Genet. 1987;24:271–274. doi: 10.1136/jmg.24.5.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wilgenbus KK, Engers R, Crombach G, et al. Two fetuses with Fryns syndrome without diaphragmatic defects. J Med Genet. 1994;31:962–964. doi: 10.1136/jmg.31.12.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chitayat D, Sroka H, Keating S, et al. The PDAC syndrome (pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia, and cardiac defect) (Spear syndrome, Matthew-Wood syndrome): report of eight cases including a living child and further evidence for autosomal recessive inheritance. Am J Med Genet A. 2007;143:1268–1281. doi: 10.1002/ajmg.a.31788. [DOI] [PubMed] [Google Scholar]
- 116.Pasutto F, Sticht H, Hammersen G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007;80:550–560. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Park Y, Gong G, Choe G, et al. Jarcho-Levin syndrome– a report of an autopsy case with cytogenetic analysis. J Korean Med Sci. 1993;8:471–475. doi: 10.3346/jkms.1993.8.6.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lam YH, Eik-Nes SH, Tang MH, et al. Prenatal sonographic features of spondylocostal dysostosis and diaphragmatic hernia in the first trimester. Ultrasound Obstet Gynecol. 1999;13:213–215. doi: 10.1046/j.1469-0705.1999.13030213.x. [DOI] [PubMed] [Google Scholar]
- 119.Day R, Fryer A. Diaphragmatic hernia and preaxial polydactyly in spondylothoracic dysplasia. Clin Dysmorphol. 2003;12:277–278. doi: 10.1097/00019605-200310000-00014. [DOI] [PubMed] [Google Scholar]
- 120.Rodriguez LM, Garcia-Garcia I, Correa-Rivas MS, et al. Pulmonary hypoplasia in Jarcho-Levin syndrome. P R Health Sci J. 2004;23:65–67. [PubMed] [Google Scholar]
- 121.Slavotinek AM. Single gene disorders associated with congenital diaphragmatic hernia. Am J Med Genet C Semin Med Genet. 2007;145:172–183. doi: 10.1002/ajmg.c.30125. [DOI] [PubMed] [Google Scholar]
- 122.Cornier AS, Ramirez N, Arroyo S, et al. Phenotype characterization and natural history of spondylothoracic dysplasia syndrome: a series of 27 new cases. Am J Med Genet A. 2004;128:120–126. doi: 10.1002/ajmg.a.30011. [DOI] [PubMed] [Google Scholar]
- 123.Turnpenny PD, Alman B, Cornier AS, et al. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev Dyn. 2007;236:1456–1474. doi: 10.1002/dvdy.21182. [DOI] [PubMed] [Google Scholar]
- 124.Neri G, Gurrieri F, Zanni G, et al. Clinical and molecular aspects of the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1998;79:279–283. doi: 10.1002/(sici)1096-8628(19981002)79:4<279::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 125.Sakazume S, Okamoto N, Yamamoto T, et al. GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am J Med Genet A. 2007;143:1703–1707. doi: 10.1002/ajmg.a.31822. [DOI] [PubMed] [Google Scholar]
- 126.Li M, Shuman C, Fei YL, et al. GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson-Golabi-Behmel syndrome. Am J Med Genet. 2001;102:161–168. doi: 10.1002/1096-8628(20010801)102:2<161::aid-ajmg1453>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 127.Scott DA, Cooper ML, Stankiewicz P, et al. Congenital diaphragmatic hernia in WAGR syndrome. Am J Med Genet A. 2005;134:430–433. doi: 10.1002/ajmg.a.30654. [DOI] [PubMed] [Google Scholar]
- 128.Suri M, Kelehan P, O’Neill D, et al. WT1 mutations in Meacham syndrome suggest a coelomic mesothelial origin of the cardiac and diaphragmatic malformations. Am J Med Genet A. 2007;143:2312–2320. doi: 10.1002/ajmg.a.31924. [DOI] [PubMed] [Google Scholar]
- 129.Cho HY, Lee BS, Kang CH, et al. Hydrothorax in a patient with Denys-Drash syndrome associated with a diaphragmatic defect. Pediatr Nephrol. 2006;21:1909–1912. doi: 10.1007/s00467-006-0273-5. [DOI] [PubMed] [Google Scholar]
- 130.Devriendt K, Deloof E, Moerman P, et al. Diaphragmatic hernia in Denys-Drash syndrome. Am J Med Genet. 1995;57:97–101. doi: 10.1002/ajmg.1320570120. [DOI] [PubMed] [Google Scholar]
- 131.Denamur E, Bocquet N, Baudouin V, et al. WT1 splice-site mutations are rarely associated with primary steroidresistant focal and segmental glomerulosclerosis. Kidney Int. 2000;57:1868–1872. doi: 10.1046/j.1523-1755.2000.00036.x. [DOI] [PubMed] [Google Scholar]
- 132.Killeen OG, Kelehan P, Reardon W. Double vagina with sex reversal, congenital diaphragmatic hernia, pulmonary and cardiac malformations–another case of Meacham syndrome. Clin Dysmorphol. 2002;11:25–28. doi: 10.1097/00019605-200201000-00005. [DOI] [PubMed] [Google Scholar]
- 133.Moore AW, Schedl A, McInnes L, et al. YAC transgenic analysis reveals Wilms' tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech Dev. 1998;79:169–184. doi: 10.1016/s0925-4773(98)00188-9. [DOI] [PubMed] [Google Scholar]
- 134.Lin AE, Pober BR, Adatia I. Congenital diaphragmatic hernia and associated cardiovascular malformations: type, frequency, and impact on management. Am J Med Genet C Semin Med Genet. 2007;145:201–216. doi: 10.1002/ajmg.c.30131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dillon E, Renwick M. Antenatal detection of congenital diaphragmatic hernias: the northern region experience. Clin Radiol. 1993;48:264–267. doi: 10.1016/s0009-9260(05)81015-3. [DOI] [PubMed] [Google Scholar]
- 136.Migliazza L, Otten C, Xia H, et al. Cardiovascular malformations in congenital diaphragmatic hernia: human and experimental studies. J Pediatr Surg. 1999;34:1352–1358. doi: 10.1016/s0022-3468(99)90010-6. [DOI] [PubMed] [Google Scholar]
- 137.Migliazza L, Xia H, Alvarez JI, et al. Heart hypoplasia in experimental congenital diaphragmatic hernia. J Pediatr Surg. 1999;34:706–710. doi: 10.1016/s0022-3468(99)90360-3. [DOI] [PubMed] [Google Scholar]
- 138.Robert E, Kallen B, Harris J. The epidemiology of diaphragmatic hernia. Eur J Epidemiol. 1997;13:665–673. doi: 10.1023/a:1007395727819. [DOI] [PubMed] [Google Scholar]
- 139.Dillon E, Renwick M, Wright C. Congenital diaphragmatic herniation: antenatal detection and outcome. Br J Radiol. 2000;73:360–365. doi: 10.1259/bjr.73.868.10844860. [DOI] [PubMed] [Google Scholar]
- 140.Czeizel A, Vitez M. Birth prevalence of five congenital abnormalities of medium frequency in Budapest. Acta Paediatr Acad Sci Hung. 1981;22:299–308. [PubMed] [Google Scholar]
- 141.van Dooren MF, Brooks AS, Tibboel D, et al. Association of congenital diaphragmatic hernia with limb-reduction defects. Birth Defects Res A Clin Mol Teratol. 2003;67:578–584. doi: 10.1002/bdra.10079. [DOI] [PubMed] [Google Scholar]
- 142.Gallot D, Marceau G, Coste K, et al. Congenital diaphragmatic hernia: a retinoid-signaling pathway disruption during lung development? Birth Defects Res A Clin Mol Teratol. 2005;73:523–531. doi: 10.1002/bdra.20151. [DOI] [PubMed] [Google Scholar]
- 143.Andersen DH. Effect of diet during pregnancy upon the incidence of congenital hereditary diaphragmatic hernia in the rat. Am J Pathol. 1949;25:163–185. [PMC free article] [PubMed] [Google Scholar]
- 144.Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation Am J Anat. 1953;92:189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 145.Mendelsohn C, Lohnes D, Decimo D, et al. Function of the retinoic acid receptors (RARs) during development (II) Multiple abnormalities at various stages of organogenesis in RAR double mutants Development. 1994;120:2749–2771. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]