

Abstract
Background:
In Huntington disease (HD), substantial striatal atrophy precedes clinical motor symptoms. Accordingly, neuroprotection should prevent major cell loss before such symptoms arise. To evaluate neuroprotection, biomarkers such as MRI measures are needed. This requires first establishing the best imaging approach.
Methods:
Using a cross-sectional design, we acquired T1-weighted and diffusion-weighted scans in 39 preclinical (pre-HD) individuals and 25 age-matched controls. T1-weighted scans were analyzed with gross whole-brain segmentation and voxel-based morphometry. Analysis of diffusion-weighted scans used skeleton-based tractography. For all imaging measures, we compared pre-HD and control groups and within the pre-HD group we examined correlations with estimated years to clinical onset.
Results:
Pre-HD individuals had lower gross gray matter (GM) and white matter (WM) volume. Voxel-wise analysis demonstrated local GM volume loss, most notably in regions consistent with basal ganglia–thalamocortical pathways. By contrast, pre-HD individuals showed widespread reductions in WM integrity, probably due to a loss of axonal barriers. Both GM and WM imaging measures correlated with estimated years to onset.
Conclusions:
Using automated, observer-independent methods, we found that GM loss in pre-HD was regionally specific, while WM deterioration was much more general and probably the result of demyelination rather then axonal degeneration. These findings provide important information about the nature, relative staging, and topographic specificity of brain changes in pre-HD and suggest that combining GM and WM imaging may be the best biomarker approach. The empirically derived group difference images from this study are provided as regions-of-interest masks for improved sensitivity in future longitudinal studies.
GLOSSARY
- CAG
= cytosine-adenine-guanine;
- EYO
= estimated years to onset;
- FA
= fractional anisotropy;
- FOV
= field of view;
- GM
= gray matter;
- HD
= Huntington disease;
- MRI
= magnetic resonance imaging;
- pre-HD
= preclinical HD;
- TBSS
= tract-based spatial statistics;
- TE
= echo time;
- TI
= inversion time;
- TR
= repetition time;
- UCSD
= University of California at San Diego;
- VBM
= voxel-based morphometry;
- vCSF
= ventricular CSF;
- WM
= white matter.
By the time individuals with the Huntington disease (HD) gene expansion show clinical motor symptoms, striatal volumes are reduced by as much as half.1 Accordingly, efforts are underway to develop neuroprotective strategies to prevent cell loss before overt motor symptoms arise.2 The challenge is to evaluate putative neuroprotection in the absence of these symptoms. This calls for the use of biomarkers, such as neuroimaging measures, which may reveal subtle changes over time associated with the disease process.3,4
Many reports have documented striatal volume reductions in preclinical HD (pre-HD).1,5–17 Recent studies also show loss of white matter (WM) integrity.11,15,16,18,19 However, there have still been comparatively few whole-brain voxel-based studies in pre-HD,11,15,17,19 none of which explored GM and WM changes concurrently. Previous studies mainly employed either manual tracing (laborious and prone to interrater variability) or a region-of-interest approach (requiring a priori hypotheses about the loci of pathology).
We used a multitechnique imaging and analysis strategy for the purpose of evaluating MRI measures as potential biomarkers. We used state-of-the-art tools for analysis of whole-brain MRI data in standard space. All approaches were fully automated, which is important as biomarkers should ideally be observer-independent, longitudinally replicable, cost-effective, and allow for easy multicenter deployment.
We acquired T1-weighted and diffusion-weighted MRI scans in individuals who were positive for the HD gene expansion yet did not fulfill clinical diagnostic criteria and age-matched and sex-matched controls. For the T1-weighted scans, gross whole-brain segmentation and voxel-based morphometry were performed. For diffusion-weighted scans, skeleton-based tractography was carried out.
Given prior reports,1,5–17 we expected to find striatal GM loss in pre-HD. However, in this study we were specifically interested in examining if there would be regionally specific cortical GM volume loss as well as WM deterioration that correspond with the well-known pattern of basal ganglia–thalamocortical connections.20 As our whole-brain voxel-based analyses enabled an unbiased search for areas of neuropathologic change, we aimed to create empirically derived regions of interest that may be used in future studies. To examine biomarker potential, we correlated the imaging measures with an estimate of the number of years to clinical (motor) disease onset (EYO).21 A strong correlation would suggest sensitivity to neuropathologic change, while the absence of a correlation points to high interindividual variability unrelated to the disease process and/or relatively unreliable measurements.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Institutional Review Board of the University of California at San Diego (UCSD). All participants provided written consent prior to enrollment in the study.
Participants.
We studied 39 individuals who were positive for the HD gene expansion yet did not fulfill clinical diagnostic criteria for HD (pre-HD) and 25 healthy age-matched and sex-matched controls. Participants were recruited from the UCSD HD Center of Excellence and studied at UCSD facilities over the second half of 2008. Participants were defined as gene expansion positive when they had 36 or more CAG repeats. The control group consisted of spouses and friends of pre-HD individuals. Of the pre-HD individuals, 11 were receiving pharmacologic treatment for depression, anxiety, and/or insomnia.
MRI acquisition and preprocessing.
Imaging was performed on a General Electric (Milwaukee, WI) 1.5 T EXCITE HD scanner with an 8-channel phased-array head coil. High-resolution structural data were acquired using a T1-weighted sequence (echo time [TE] = 2.796 msec, repetition time [TR] = 6.496 msec, inversion time [TI] = 600 msec, flip angle = 12°, bandwidth = 244.141 Hz/pixel, field of view [FOV] = 24 cm, matrix = 256 × 192, slice thickness = 1.2 mm). T1 images were corrected for nonlinear warping.22 Image intensities were corrected for spatial sensitivity inhomogeneities in the head coil by normalizing with the ratio of a body-coil scan to a head-coil scan.
Diffusion-weighted data were acquired using single-shot echoplanar imaging with isotropic 2.5-mm voxels (TE = 80.4 msec, TR = 13,200 msec, matrix = 96 × 96, FOV = 24 cm, 47 axial slices, slice thickness = 2.5 mm, 51 directions, b = 1,000 mm2/s). Preprocessing of diffusion-weighted data were performed according to previously described procedures.23
All postprocessing imaging analysis was performed using the FSL suite of tools (http://www.fmrib.ox.ac.uk/fsl/).24
Gross whole-brain segmentation.
Total brain, GM, WM, cortical (peripheral) GM, and ventricular CSF (vCSF) volume, normalized for subject head size, were estimated by analyzing preprocessed T1-weighted MRI data with SIENAX.25,26 SIENAX starts by extracting brain and skull images from the single whole-head input data. The brain image is then affine-registered to Montreal Neurological Institute–152 space, using the skull image to determine the registration scaling. Next, tissue-type segmentation with partial volume estimation is carried out in order to calculate total volume of brain tissue (including estimates of volumes of GM, WM, cortical GM, and vCSF).
Voxel-based GM analysis.
Local changes in GM volume were estimated by analyzing preprocessed T1-weighted MRI data with a VBM-style analysis.27 First, structural images were brain-extracted. Next, tissue-type segmentation was carried out. The resulting GM partial volume images were then aligned to Montreal Neurological Institute–152 space using affine registration, followed by nonlinear registration. From the resulting images, those of all 25 controls as well as of 25 age-matched and sex-matched pre-HD individuals were averaged to create a study-specific template (an equal number of participants from each group was used to avoid registration bias). All 63 of the native GM images were nonlinearly reregistered to this study-specific template. The registered partial volume images were then modulated by dividing by the Jacobian of the warp field and smoothed with an isotropic Gaussian kernel with a sigma of 3 mm. The resulting data were fed into voxel-wise cross-subject statistics.
Voxel-based WM analysis.
Eddy-current and movement correction of preprocessed diffusion-weighted MR data were performed using in-house software. Voxel-wise analysis of the diffusion data was carried out using TBSS.28 First, fractional anisotropy (FA), mean diffusivity, longitudinal diffusivity (λ∥), and transverse diffusivity (λ⊥) images were created by fitting a tensor model to the raw diffusion data. Brain extraction was then run. All subjects' diffusion data were then aligned into a common space using nonlinear registration. Next, the mean FA image was created and thinned to create a mean FA skeleton that represents the centers of all tracts common to the whole group. Each subject's aligned FA, mean diffusivity, λ∥, and λ⊥ data were then projected onto this skeleton and the resulting data were fed into voxel-wise cross-subject statistics.
Statistical analysis.
Between-group differences in sex distribution were analyzed with χ2 tests; differences in age and SIENAX volumes were analyzed with t tests.
Voxel-wise cross-subject statistics were carried out using t tests and permutation-based nonparametric inference,29 utilizing threshold-free cluster enhancement.30 For analyses of GM distribution, the overall mean GM segmented image was used as a mask. For all analyses of WM tract integrity (FA, mean diffusivity, λ∥, and λ⊥), the overall mean FA skeleton was used as a mask (thresholded at a mean FA value of 0.2). The number of permutations was set to 50,000 in all voxel-wise analyses.
The statistical analyses were carried out at a significance level of 5% (2-tailed) using either PASW Statistics 17.0.2 (SPSS Inc., Chicago, IL) or FSL's randomize tool, version 4.1.3.24 Clusters found in voxel-based analyses were localized using standard brain atlases.
RESULTS
Participant characteristics and confounders.
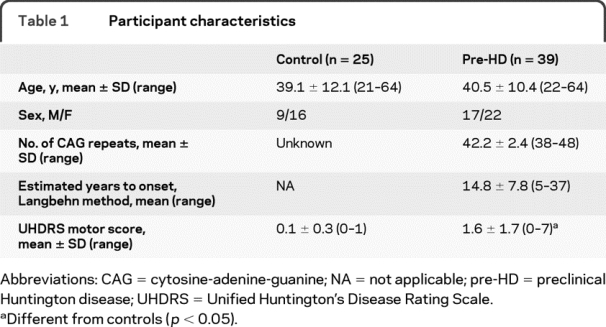
All pre-HD individuals scored below two on the Unified Huntington's Disease Rating Scale31 confidence rating, confirming their preclinical status. One individual had 38 repeats and two had 39 repeats (reduced penetrance). There were no significant differences in sex distribution or age between groups. The pre-HD group had higher Unified Huntington's Disease Rating Scale motor scores (t[62] = 4.44, p < 0.001) compared to controls. One participant was excluded from the SIENAX and FSL-VBM analyses and two from the TBSS analysis due to technical problems with image acquisition or image quality. Participant characteristics are listed in table 1.
Table 1 Participant characteristics
Gross whole-brain segmentation.
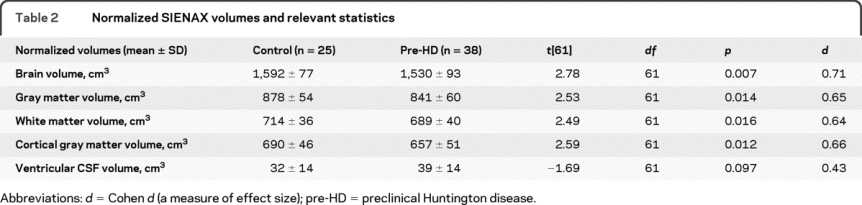
SIENAX analysis showed significantly lower normalized total brain, GM, WM, and cortical GM, and a trend toward higher vCSF volume, in pre-HD relative to controls (table 2).
Table 2 Normalized SIENAX volumes and relevant statistics
Correlation analyses showed a significant positive relation between EYO and volumes of overall brain, GM, WM, and cortical GM volumes in pre-HD individuals (figure 1, A–D).

Figure 1 Gross whole-brain segmentation
Scatterplots are shown for estimated years to onset in the preclinical Huntington's disease group (n = 38) against normalized brain (A), gray matter (B), white matter (C), and cortical gray matter (D) volumes from the SIENAX analysis. R2 values are shown in the bottom right corner; solid horizontal lines indicate the mean volume in controls (n = 25), dashed horizontal lines indicate the 95% confidence interval around this mean.
Voxel-based GM analysis.
VBM-style comparison of GM distribution revealed significant local GM volume loss in pre-HD (figure 2A). The most conspicuous subcortical clusters were localized bilaterally in the caudate and putamen. Within the cortex, volume loss was found in the subcallosal cortex and bilaterally in the insular cortex, Heschl's gyrus, precentral and postcentral gyrus, middle and inferior frontal gyrus, and supplementary motor area.

Figure 2 Voxel-based gray matter analysis
(A) Regions of significant gray matter (GM) volume reduction in the preclinical Huntington's disease (pre-HD) group (n = 38) relative to controls (n = 25), whole-brain corrected. (B) Regions where local GM volume decreases with decreasing estimated years to onset in the pre-HD group, whole-brain corrected. (C) Anatomic masks based on the Harvard-Oxford probabilistic labels atlas (25% probability threshold) for reference. Clusters and masks are overlaid on axial slices from the Montreal Neurological Institute–152 template, displayed according to neurologic convention (left = left). Z coordinates in Montreal Neurological Institute space of the individual slices, color labels for the various anatomic masks, and a color bar indicating significance level are displayed at the bottom.
Voxel-level correlations with EYO showed that volume loss was most severe in those pre-HD individuals who were closest to estimated onset (figure 2B). This included regions in which between-group differences had previously been found, but additionally involved the frontal pole, frontal orbital cortex, superior frontal gyrus, central opercular cortex, inferior and superior temporal gyrus, temporal pole, and cerebellum. No correlations were found between volume loss and number of CAG repeats.
Skeleton-based WM analysis.
TBSS mapping of FA differences demonstrated significantly reduced FA in many parts of the WM skeleton in pre-HD relative to controls (figure 3A). The most conspicuous changes were localized in the corpus callosum (from the splenium to the genu), and bilaterally in the thalamic radiations, cingulum, longitudinal fasciculi, corona radiata, internal capsule, and cerebral peduncles. Even at much higher significance thresholds, there were substantial differences all over the skeleton. Adding total normalized WM volume as a nuisance variable did not change the results.

Figure 3 Skeleton-based tractography of major white matter (WM) tracts
(A) Regions of significantly lower fractional anisotropy (FA) in the preclinical Huntington's disease (pre-HD) group (n = 38) relative controls (n = 24), whole-brain corrected. (B) Regions of significantly higher transverse diffusivity in pre-HD relative to controls, whole-brain corrected. (C) Regions of significantly higher longitudinal diffusivity in pre-HD relative to controls, whole-brain corrected. (D) Regions where FA values decrease with decreasing estimated years to onset in the pre-HD group, whole-brain corrected. (E) Anatomic masks based on the Johns Hopkins University ICBM-DTI-81 WM labels atlas for reference. Clusters and masks are overlaid on coronal slices from the mean FA image, displayed according to neurologic convention (left = left). The mean FA skeleton, in which voxel-wise permutation testing was performed for all the above results, is shown in green. For better visibility, significant clusters (p values <0.05) have been thickened to fill out local tracts in the FA image. Clusters are shown in yellow with red boundaries. Y coordinates in Montreal Neurological Institute space of the individual slices and color labels for the various anatomic masks are displayed at the bottom.
TBSS mapping of differences in diffusion coefficients demonstrated increased λ∥ in the thalamic radiations, internal capsule, and external capsule (figure 3B) and increased λ⊥ (3C) as well as mean diffusivity (not depicted) in all parts of the WM skeleton in pre-HD relative to controls.
Voxel-level correlations of EYO with FA showed that FA was lowest in those pre-HD individuals who were closest to estimated onset (figure 3D). Significant correlations with FA were found in all regions in which between-group differences had previously been found, but were more widespread and additionally involved the external capsule. No correlations were found between FA and number of CAG repeats. Voxel-level correlations of EYO with λ∥ and λ⊥ showed that pre-HD individuals with the highest λ⊥ were those who were closest to estimated onset (not depicted), while no correlations were found with λ∥ (not depicted). Significant correlations with λ⊥ were found in all regions in which between-group differences were found.
DISCUSSION
We performed a fully automated whole-brain analysis of GM and WM changes in pre-HD individuals vs controls using state-of-the-art analysis tools. Gross whole-brain segmentation analysis confirmed loss of GM (whole brain as well as cortical) and WM volume in pre-HD. Voxel-based analysis showed that GM loss was largely confined to the striatum and frontal cortical regions known to be connected with the striatum. In contrast, there was a very widespread loss of WM integrity, most likely as a result of deterioration of axonal barriers in pre-HD. Both GM loss and WM deterioration were more severe in subjects closer to estimated onset. Taken together, these findings provide important information about the staging and relative specificity of neuropathology in pre-HD.
Gross whole-brain segmentation analysis showed significant decreases of GM volume, both overall and at the cortical level, and WM volume in pre-HD relative to controls. The advantage of using a fully automated, gross segmentation technique is that it yields a few easily interpretable values. For use as a biomarker, this technique may seem less appealing, as better sensitivity may arise by focusing on specific regions of known pathologic change. However, all segmentation techniques are characterized by a trade-off between regional specificity and signal-to-noise ratio. When tracking longitudinal change, the latter may prove more important than high spatial resolution. Brain volume accuracy of SIENA(X) in cross-sectional analyses has been reported to be between 0.5% and 1%,26 while longitudinal analyses have estimated the error in brain volume change to be as low as 0.15%26 and shown higher sensitivity in the detection of subtle differences in atrophy rates when compared to semi-automated methods.32 In conclusion, although topographic information is lost, the use of gross whole-brain measures to monitor longitudinal change may prove more reliable than methods that retain a high spatial resolution.
The topography of GM changes we observed in pre-HD individuals was striking in that it was fairly specific to regions which are part of the basal ganglia–thalamocortical loops.20 We observed volume loss in the supplementary motor area, primary motor cortex, and putamen, which together with the pallidum and thalamus make up the motor fronto-basal-ganglia-pallidal-thalamic loop. There were also significant changes in the middle frontal gyrus and caudate, consistent with the so-called associative fronto-basal-ganglia-pallidal-thalamic loop.
Although VBM was initially controversial,33,34 it has since become an established method for evaluating GM. VBM's strength is that it does not require a priori information about the loci of possible differences (in contrast to region-of-interest approaches). However, its validity strongly depends on accurate image registration and there is loss of spatial resolution due to spatial smoothing. This could lead to inability to detect potentially interesting morphologic abnormalities. Also, we cannot definitively exclude the possibility that observed cortical differences are due to dissimilar gyrification patterns between groups rather than reduced cortical thickness. Nevertheless, our use of nonlinear registration techniques, moderate spatial smoothing, the large participant sample, and the observed regionally specific pattern of GM loss increase confidence that observed differences reflect veridical atrophy. Moreover, observed changes are highly consistent with our results in the same sample when using FreeSurfer's subject-specific subcortical segmentation35 and cortical thickness mapping (unpublished observations). In conclusion, VBM could be a useful biomarker that is unbiased, reproducible, and sensitive to neuropathology in the preclinical disease state.
Using the recent innovation of TBSS, we observed widespread decreases in WM integrity in pre-HD. This technique overcomes many of the difficulties of aligning multisubject diffusion data, allowing for an objective whole-brain voxel-wise analysis of regional WM changes. Our findings clearly suggest that changes in WM microstructure are an early and widespread phenomenon in the pre-HD brain. A disadvantage of using the skeleton-based approach is that smaller tracts are often not included in the skeleton due to alignment issues. Future analyses using probabilistic tractography will be needed to closely investigate individual tracts projecting from atrophied basal ganglia nuclei in pre-HD.
We know of 5 imaging studies that have investigated diffusion in pre-HD. The first study to demonstrate altered diffusion in pre-HD used region-of-interest–based techniques; gene carriers showed increased mean diffusion in the striatum, periventricular WM, and whole brain.18 A voxel-based analysis showed decreased FA in superior frontal, middle frontal, postcentral, and precentral WM in pre-HD.15 Using several manually placed regions of interest, decreased FA in the corticospinal tracts and corpus callosum was recently demonstrated in pre-HD.16 A study using machine learning was able to classify a high proportion of diffusion-weighted scans as pre-HD vs controls.11 A very recent study of small sample size employing the TBSS technique was the first to suggest longitudinal WM degeneration in HD.19
While GM change in pre-HD was fairly specific to regions consistent with basal ganglia–thalamocortical pathways, WM deterioration was much more general. In correspondence with the GM abnormalities found bilaterally in cortical motor areas, TBSS analysis of FA showed reduced anisotropy in the major motor output pathway, the corticospinal tract, in pre-HD individuals. However, there were changes in many other parts of the WM skeleton, corresponding to a diverse range of tracts, including, for example, the superior and inferior longitudinal fasciculus, which do not relate strongly to the loci of GM change. This suggests that loss of WM tract integrity is not merely a consequence of GM atrophy.
To further probe WM in pre-HD, we examined λ⊥, which reflects the integrity of nonrandom longitudinal barriers to diffusion (i.e., the myelin sheath), and λ∥, which is hypothesized to relate to axonal genesis/injury/degeneration.36–38 We found modest, highly localized increases in λ∥, while λ⊥ showed widespread increases in pre-HD. These findings suggest that loss of WM integrity is not due to axonal degeneration, but relates to demyelination, which further supports the notion that WM degeneration is not merely a consequence of GM loss. Moreover, demyelination is consistent with neuropathologic findings in pre-HD, which show increased density of oligodendrocytes (which are responsible for axonal myelination production/repair), which was suggested to be a developmental effect of the HD gene expansion.39 Provocatively, demyelination might therefore predate GM loss. Additionally, a recent imaging study specifically pointed to myelin breakdown in HD.40 By contrast the increases in longitudinal diffusivity are puzzling: they could indicate increases in axonal thickness related to a compensatory increase in the number of fibers in pre-HD. Future studies will be required to confirm our findings and shed more light on the pathophysiologic basis of WM changes in pre-HD.
In pre-HD individuals, we found strong correlations between EYO and multiple imaging measures used in the current study, both at the gross segmented and voxel-based level, which suggests that these measures are very sensitive to neuropathologic change. In contrast, no correlations with CAG repeat length were observed. This is likely due to the large age variance in our pre-HD sample; CAG length in itself is a predictor of age at onset, not years to onset and (progression of) disease pathology.
Although one cannot easily compare the results from our voxel-based analyses of GM and WM with those obtained using gross whole-brain segmentation, our results clearly suggest that all 3 techniques may provide surrogate markers of disease progression before clinical onset. Analyses using multivariate pattern classification techniques are needed to show whether a combination of imaging techniques is indeed superior to an approach aimed at a single region of interest or neuropathologic substrate, and whether adding patterns of voxel-level information improves sensitivity to the disease process. To facilitate biomarker development, standard space masks of the between-group differences in voxel-based analyses of GM and WM have been provided (www.aronlab.org/hdmasks110109.zip). These may be used to validate our results in different samples and to serve as regions of interest to improve sensitivity in future longitudinal neuroprotective trials.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. D. Stoffers.
ACKNOWLEDGMENT
The authors thank Rebecca Theilman, Cecilia Kemper, and Melissa Generoso for logistical assistance, Anders Dale for technical input, and Tim Behrens for comments. Funding support from CHDI is gratefully acknowledged.
DISCLOSURE
Dr. Stoffers and S. Sheldon report no disclosures. Dr. Kuperman was funded as Postdoctoral Scholar on NIH grants U54 NS056883-01 and 1P50MH081755-01. J. Goldstein reports no disclosures. Dr. Corey-Bloom has received travel expenses and/or honoraria for lectures or educational activities not funded by industry and receives research support from Medivation, Inc., NeuroSearch, Elan Corporation, the University of Rochester, the Alzheimer Disease Cooperative Study, and the Huntington Study Group. Dr. Aron serves on the editorial board of Neuropsychologia and receives research support from the NIH (DA026452 [PI]), the National Science Foundation (0921168 [PI]), CHDI and NARSAD.
Address correspondence and reprint requests to Dr. A.R. Aron, Department of Psychology, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0109 adamaron@ucsd.edu
Study funding: Supported by the Cure Huntington's Disease Initiative.
Disclosure: Author disclosures are provided at the end of the article.
Received September 16, 2009. Accepted in final form February 1, 2010.
REFERENCES
- 1.Aylward EH, Sparks BF, Field KM, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004;63:66–72. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF, Ferrante RJ. Experimental therapeutics in transgenic mouse models of Huntington's disease. Nat Rev Neurosci 2004;5:373–384. [DOI] [PubMed] [Google Scholar]
- 3.Hersch SM, Rosas HD. Neuroprotection for Huntington's disease: ready, set, slow. Neurotherapeutics 2008;5:226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohanna I, Georgiou-Karistianis N, Hannan AJ, Egan GF. Magnetic resonance imaging as an approach towards identifying neuropathological biomarkers for Huntington's disease. Brain Res Rev 2008;58:209–225. [DOI] [PubMed] [Google Scholar]
- 5.Aylward EH, Brandt J, Codori AM, et al. Reduced basal ganglia volume associated with the gene for Huntington's disease in asymptomatic at-risk persons. Neurology 1994;44:823–828. [DOI] [PubMed] [Google Scholar]
- 6.Aylward EH, Codori AM, Barta PE, et al. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Arch Neurol 1996;53:1293–1296. [DOI] [PubMed] [Google Scholar]
- 7.Aylward EH, Codori AM, Rosenblatt A, et al. Rate of caudate atrophy in presymptomatic and symptomatic stages of Huntington's disease. Mov Disord 2000;15:552–560. [DOI] [PubMed] [Google Scholar]
- 8.Campodonico JR, Aylward E, Codori AM, et al. When does Huntington's disease begin? J Int Neuropsychol Soc 1998;4:467–473. [DOI] [PubMed] [Google Scholar]
- 9.Harris GJ, Codori AM, Lewis RF, et al. Reduced basal ganglia blood flow and volume in pre-symptomatic, gene-tested persons at-risk for Huntington's disease. Brain 1999;122:1667–1678. [DOI] [PubMed] [Google Scholar]
- 10.Kipps CM, Duggins AJ, Mahant N, et al. Progression of structural neuropathology in preclinical Huntington's disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatry 2005;76:650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kloppel S, Draganski B, Golding CV, et al. White matter connections reflect changes in voluntary-guided saccades in pre-symptomatic Huntington's disease. Brain 2008;131:196–204. [DOI] [PubMed] [Google Scholar]
- 12.Nopoulos P, Magnotta VA, Mikos A, et al. Morphology of the cerebral cortex in preclinical Huntington's disease. Am J Psychiatry 2007;164:1428–1434. [DOI] [PubMed] [Google Scholar]
- 13.Paulsen JS, Hayden M, Stout JC, et al. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol 2006;63:883–890. [DOI] [PubMed] [Google Scholar]
- 14.Paulsen JS, Magnotta VA, Mikos AE, et al. Brain structure in preclinical Huntington's disease. Biol Psychiatry 2006;59:57–63. [DOI] [PubMed] [Google Scholar]
- 15.Reading SA, Yassa MA, Bakker A, et al. Regional white matter change in pre-symptomatic Huntington's disease: a diffusion tensor imaging study. Psychiatry Res 2005;140:55–62. [DOI] [PubMed] [Google Scholar]
- 16.Rosas HD, Tuch DS, Hevelone ND, et al. Diffusion tensor imaging in presymptomatic and early Huntington's disease: selective white matter pathology and its relationship to clinical measures. Mov Disord 2006;21:1317–1325. [DOI] [PubMed] [Google Scholar]
- 17.Thieben MJ, Duggins AJ, Good CD, et al. The distribution of structural neuropathology in pre-clinical Huntington's disease. Brain 2002;125:1815–1828. [DOI] [PubMed] [Google Scholar]
- 18.Mascalchi M, Lolli F, Della Nave R, et al. Huntington disease: volumetric, diffusion-weighted, and magnetization transfer MR imaging of brain. Radiology 2004;232:867–873. [DOI] [PubMed] [Google Scholar]
- 19.Weaver KE, Richards TL, Liang O, et al. Longitudinal diffusion tensor imaging in Huntington's disease. Exp Neurol 2009;216:525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986;9:357–381. [DOI] [PubMed] [Google Scholar]
- 21.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet 2004;65:267–277. [DOI] [PubMed] [Google Scholar]
- 22.Jovicich J, Czanner S, Greve D, et al. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. Neuroimage 2006;30:436–443. [DOI] [PubMed] [Google Scholar]
- 23.Hagler DJ, Jr., Ahmadi ME, Kuperman J, et al. Automated white-matter tractography using a probabilistic diffusion tensor atlas: application to temporal lobe epilepsy. Hum Brain Mapp 2008;30:1535–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004;23:S208–S219. [DOI] [PubMed] [Google Scholar]
- 25.Smith SM, De Stefano N, Jenkinson M, Matthews PM. Normalized accurate measurement of longitudinal brain change. J Comput Assist Tomogr 2001;25:466–475. [DOI] [PubMed] [Google Scholar]
- 26.Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross-sectional brain change analysis. Neuroimage 2002;17:479–489. [DOI] [PubMed] [Google Scholar]
- 27.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. Neuroimage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 28.Smith SM, Jenkinson M, Johansen-Berg H, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage 2006;31:1487–1505. [DOI] [PubMed] [Google Scholar]
- 29.Nichols TE, Holmes AP. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 2002;15:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith SM, Nichols TE. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 2009;44:83–98. [DOI] [PubMed] [Google Scholar]
- 31.Huntington Study Group. Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 32.Sormani MP, Rovaris M, Valsasina P, et al. Measurement error of two different techniques for brain atrophy assessment in multiple sclerosis. Neurology 2004;62:1432–1434. [DOI] [PubMed] [Google Scholar]
- 33.Ashburner J, Friston KJ. Why voxel-based morphometry should be used. Neuroimage 2001;14:1238–1243. [DOI] [PubMed] [Google Scholar]
- 34.Bookstein FL. “Voxel-based morphometry” should not be used with imperfectly registered images. Neuroimage 2001;14:1454–1462. [DOI] [PubMed] [Google Scholar]
- 35.Stoffers D, Kuperman J, Sheldon S, et al. Automated segmentation shows atrophy of striatum and pallidum in preclinical HD, which strongly predict estimated years to onset. Movement Disorders Society 13th International Congress; Paris; 2009.
- 36.Beaulieu C. The basis of anisotropic water diffusion in the nervous system: a technical review. NMR Biomed 2002;15:435–455. [DOI] [PubMed] [Google Scholar]
- 37.Song SK, Sun SW, Ramsbottom MJ, et al. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage 2002;17:1429–1436. [DOI] [PubMed] [Google Scholar]
- 38.Concha L, Gross DW, Wheatley BM, Beaulieu C. Diffusion tensor imaging of time-dependent axonal and myelin degradation after corpus callosotomy in epilepsy patients. Neuroimage 2006;32:1090–1099. [DOI] [PubMed] [Google Scholar]
- 39.Gomez-Tortosa E, MacDonald ME, Friend JC, et al. Quantitative neuropathological changes in presymptomatic Huntington's disease. Ann Neurol 2001;49:29–34. [PubMed] [Google Scholar]
- 40.Bartzokis G, Lu PH, Tishler TA, et al. Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications. Neurochem Res 2007;32:1655–1664. [DOI] [PubMed] [Google Scholar]