

The Sox10 transcription factor is required to maintain as well as specify glial identity, adding new causes for the neuropathies associated with SOX10 mutations.
Abstract
Mutations in the transcription factor SOX10 cause neurocristopathies, including Waardenburg-Hirschsprung syndrome and peripheral neuropathies in humans. This is partly attributed to a requirement for Sox10 in early neural crest for survival, maintenance of pluripotency, and specification to several cell lineages, including peripheral glia. As a consequence, peripheral glia are absent in Sox10-deficient mice. Intriguingly, Sox10 continues to be expressed in these cells after specification. To analyze glial functions after specification, we specifically deleted Sox10 in immature Schwann cells by conditional mutagenesis. Mutant mice died from peripheral neuropathy before the seventh postnatal week. Nerve alterations included a thinned perineurial sheath, increased lipid and collagen deposition, and a dramatically altered cellular composition. Nerve conduction was also grossly aberrant, and neither myelinating nor nonmyelinating Schwann cells formed. Instead, axons of different sizes remained unsorted in large bundles. Schwann cells failed to develop beyond the immature stage and were unable to maintain identity. Thus, our study identifies a novel cause for peripheral neuropathies in patients with SOX10 mutations.
Introduction
Sox10 belongs to the group of HMG (high mobility group) domain–containing transcription factors (Wegner, 1999; Bowles et al., 2000). During vertebrate development, it is highly expressed in the emerging neural crest and later in the developing peripheral nervous system (PNS) and central nervous system, where its occurrence is restricted to glial cells (Kuhlbrodt et al., 1998). Accordingly, loss of Sox10 in the mouse leads to decreased survival of early neural crest cells and additionally to defects in several neural crest–derived lineages (Herbarth et al., 1998; Southard-Smith et al., 1998; Kapur, 1999; Britsch et al., 2001; Paratore et al., 2001; Kim et al., 2003). These also become evident in patients with heterozygous SOX10 mutations as Waardenburg syndrome, Hirschsprung disease, peripheral neuropathies, or combinations thereof (Pingault et al., 1998; Inoue et al., 2004; Bondurand et al., 2007).
In the developing PNS of Sox10-deficient mice, both neurons and glia are affected, but to different extents (Britsch et al., 2001). Whereas at least some neurons are formed, glia are not detected either in vivo or in vitro (Britsch et al., 2001; Paratore et al., 2001). It follows that Sox10 is absolutely required for glial development in the PNS. This includes the development of satellite glia in peripheral ganglia, enteric glia in the gastrointestinal tract, and Schwann cells along nerves.
Schwann cell development has been particularly well studied in the past, and markers are available for all developmental stages (Jessen and Mirsky, 2005). Bfabp (brain fatty acid–binding protein), for instance, is normally induced as Schwann cell precursors are specified from neural crest cells and represents the earliest glial marker (Britsch et al., 2001). From the immature stage onwards, Schwann cells transiently express Sox2, an HMG domain transcription factor distantly related to Sox10 (Le et al., 2005). Sox2 expression again extinguishes as Schwann cells undergo differentiation. For differentiation to myelinating Schwann cells, the transcription factor Oct6, which is indicative of the promyelinating stage, has to be induced first, followed by Krox20 expression and full-blown myelin gene expression (Topilko et al., 1994; Bermingham et al., 1996; Jaegle et al., 1996; Jessen and Mirsky, 2005).
From the fact that neither Bfabp nor ErbB3 as the earliest marker in the Schwann cell lineage are expressed in the developing PNS of Sox10-deficient mice, Sox10 appears already required for glial specification (Britsch et al., 2001). However, even after specification, Sox10 continues to be expressed in PNS glia. As a consequence, Sox10 is not only present in Schwann cell precursors but also in immature and promyelinating Schwann cells and even persists in the myelinating and nonmyelinating Schwann cells of the adult PNS (Kuhlbrodt et al., 1998). This expression pattern argues for additional roles of Sox10 in peripheral glia even after the specification event. Sox10 has indeed been found to activate peripheral myelin genes in tissue culture and to be bound to the responsible regulatory regions both in vitro and in vivo (Peirano et al., 2000; LeBlanc et al., 2007). However, later functions of Sox10 cannot be verified and studied in the available mouse models of Sox10. Therefore, we generated an allele that allows cell type–specific and temporally controlled deletion of Sox10 and used this allele to prove an additional requirement for Sox10 during Schwann cell development at the immature Schwann cell stage.
Results
Generation of mice with a floxed Sox10 allele
The mouse Sox10 gene is encoded by five exons, with the ORF being split among exons 3–5 (Britsch et al., 2001). To generate a Sox10 allele that is sensitive to the presence of Cre recombinase and will undergo Cre-dependent gene inactivation, we replaced the protein-coding information on exons 3–5 by a continuous ORF in such a way that its start codon was placed exactly at the position of the endogenous start codon in exon 3 (Fig. 1 A). Additionally, loxP sequences were introduced immediately preceding and following the continuous Sox10 ORF. The 5′ loxP sequence was placed in the hind part of intron 2 in sufficient distance to branch and splice acceptor site to avoid interference with the splicing of the altered allele. After successful homologous recombination of the targeting construct in embryonic stem (ES) cells, germline transmission, and removal of the neomycin selection cassette (Fig. 1, A and B), both heterozygous and homozygous animals carrying the Sox10fl allele were obtained in Mendelian ratios (Fig. 1 C and not depicted). The additional introduction of an IRES (internal ribosomal entry site)-EGFP cassette furthermore allowed visualization of the unrecombined Sox10fl allele by GFP autofluorescence (Fig. 1 D). Fluorescence was lost upon Cre-mediated recombination, probably because the IRES-EGFP cassette is no longer correctly spliced to exon 2 (unpublished data).
Figure 1.

Generation of a conditional Sox10 allele in mice. (A) Schematic representation, from top to bottom, of the targeting construct, the wild-type Sox10 allele (Sox10+), the conditional Sox10 allele before (Sox10flneo) and after (Sox10fl) removal of the neo selection cassette by Flp recombinase, and the deleted allele (Sox10Δ) after Cre recombination. Sox10 exons (I–V) and the continuous Sox10 ORF used in the conditional allele are shown as boxes, and 4.5- and 1.5-kb-long flanking regions are shown as bars. Regions of homology between wild-type locus and targeting vector are depicted as black bars, introns 3 and 4 are depicted as open bars, and surrounding genomic regions not contained in the targeting construct are depicted as dashed bars. Plasmid backbone sequences of the targeting construct are indicated by a thin line. Restriction sites for NcoI (N), BamHI (B), and ScaI (S) are shown as well as the localization of 5′ and 3′ probes and the start codon of the Sox10 gene (ATG). neo, neomycin resistance cassette; FRT, recognition sites for Flp recombinase (depicted as ellipses); loxP, recognition sites for Cre recombinase (depicted as triangles); Tk, Herpes simplex virus thymidine kinase gene cassette. (B) Southern blot analysis of genomic DNA from Sox10+/+ (wt) and Sox10fl/fl (fl/fl) mice digested with NcoI for use of the 5′ probe and BamHI–ScaI for the 3′ probe. The size of bands corresponding to the wild-type (6.6 kb for the 5′ probe and 4.6 kb for the 3′ probe) and the targeted alleles (4.8 kb for the 5′ probe and 3.7 kb for the 3′ probe) are indicated. (C) PCR genotyping of Sox10+/+ (wt), Sox10+/fl (+/fl), and Sox10fl/fl (fl/fl) mice. Size of DNA fragments in the marker (M) is indicated in kilobases on the left. (D) Whole mount GFP autofluorescence of Sox10fl/fl embryos at 10.5 dpc. (E) Immunohistochemistry with antibodies directed against Sox10 on transverse sections from the thoracic region of Sox10+/+ (wt) and Sox10fl/fl (fl/fl) embryos at 12.5 dpc. (F) Comparison of expression levels for Sox10 in Sox10+/+ (wt) and Sox10fl/fl (fl/fl) embryos at 12.5 dpc with primers recognizing a common sequence in both transcripts using quantitative LightCycler RT-PCR. Transcript levels in each sample were normalized to Rpl8. After normalization, transcript levels in the wild type were arbitrarily set to 1, and transcript levels in Sox10fl/fl are expressed relative to wild-type levels ± SEM. Experiments were repeated twice with material from two independently obtained embryos for each genotype and age. Bars: (D) 1 mm; (E) 100 µm.
Similar strategies have previously been used to replace Sox10 by other SoxE genes or mutant versions of Sox10. These genetic manipulations had not led to dramatic alterations of expression levels from the Sox10 locus (Kellerer et al., 2006; Schreiner et al., 2007; Cossais et al., 2010). Expression of the Sox10fl allele was also comparable to the wild type, as evident from similar intensities of Sox10 immunoreactivity in age-matched embryos of both genotypes (Fig. 1 E) and comparable expression levels in quantitative RT-PCR experiments (Fig. 1 F and not depicted). Detailed side by side comparison furthermore showed a very similar, if not identical, temporal and spatial pattern of Schwann cell development in Sox10+/+ and Sox10fl/fl mice so that both genotypes could be used interchangeably as wild-type control throughout this study.
Schwann cell–specific Sox10 deletion
To achieve specific Sox10 deletion in Schwann cells after the specification event, we combined the Sox10fl allele with a Dhh::Cre transgene (Jaegle et al., 2003). This Cre transgene selectively deleted in the Schwann cell lineage and started to be active around 12.5 d postcoitum (dpc) in the Schwann cell precursor stage (Fig. 2 A). The resulting Sox10fl/fl Dhh::Cre mice are referred to as Sox10ΔeSC mice throughout the manuscript. First, we determined the kinetics with which Sox10 disappeared from Schwann cells. Whereas Cre-dependent recombination of a Rosa26stopfloxYFP allele had occurred in ∼70% of cells in the proximal spinal nerve at 13.5 dpc and was more or less complete at 14.5 dpc (Fig. 2, B and C), Sox10 transcripts were still detected in normal amounts at 13.5 dpc (Fig. 2 D). Residual amounts of Sox10 transcripts were still detectable at 14.5 dpc (Fig. 2, E and F), indicating that Cre-dependent recombination may be less efficient for the Sox10fl allele than for the Rosa26stopfloxYFP allele (Fig. 2, E and F). Loss of Sox10 protein as the essential determinant for function took even longer. Sox10 was still expressed in 84% of all DAPI-positive nuclei along the spinal nerve in Sox10ΔeSC mice at 14.5 dpc. This is only slightly lower than the 90% observed in the wild type (Fig. 2, G and J). At this stage, most glia along the spinal nerve have already entered the immature Schwann cell stage (Jessen and Mirsky, 2005). By 16.5 dpc, the percentage of Sox10-positive nuclei along the nerve had dropped to 29% in the mutant compared with 73% in the wild type (Fig. 2, H and J). At 18.5 dpc, only 4% of all nuclei along the mutant nerve were still Sox10 positive, whereas 70% continued to express Sox10 in the wild type (Fig. 2, I and J). We conclude that inefficient recombination at the Sox10 locus and slow turnover of the Sox10 protein lead to a significant delay between the onset of Cre activity and the disappearance of Sox10. As a consequence, Sox10 protein is still present in immature Schwann cells before eventually being deleted during late embryogenesis.
Figure 2.
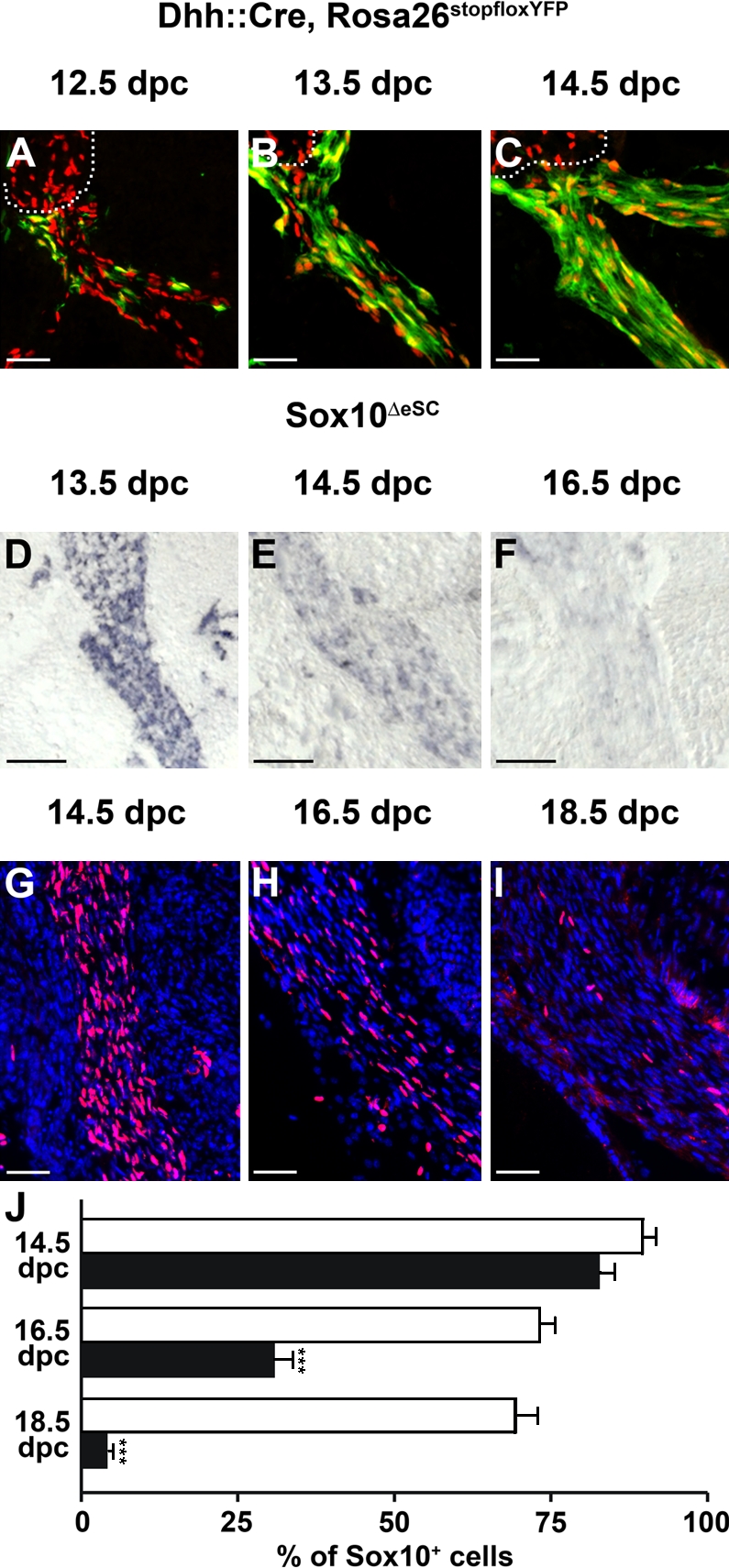
Kinetics of Sox10 deletion in spinal nerves of Sox10ΔeSC embryos. (A–C) Cre activity in spinal nerves was monitored by YFP autofluorescence (green) on transverse sections of Dhh::Cre, Rosa26stopfloxYFP (Rosa26DhhYFP) embryos at 12.5 (A), 13.5 (B), and 14.5 dpc (C). Anti-Sox10 immunofluorescence (red) is shown for comparison. The border between peripheral nerve and dorsal root ganglion is indicated by a dotted line. (D–F) Loss of Sox10 transcripts in spinal nerves of Sox10ΔeSC embryos was determined by in situ hybridization on transverse sections at 13.5 (D), 14.5 (E), and 16.5 dpc (F) using a Sox10-specific antisense riboprobe. (G–I) Disappearance of Sox10 protein in spinal nerves of Sox10ΔeSC embryos was assessed by immunohistochemistry on transverse sections of Sox10ΔeSC embryos at 14.5 (G), 16.5 (H), and 18.5 dpc (I) using a Sox10-specific antibody (red), followed by DAPI staining of nuclei (blue). (A–I) Proximal regions of spinal nerves are shown. (J) The percentage of Sox10-positive nuclei in spinal nerves of wild-type (open bars) and Sox10ΔeSC (closed bars) embryos was quantified at 14.5, 16.5, and 18.5 dpc and is presented as mean ± SEM. At least 15 spinal nerves from two independent embryos were counted for both genotypes. Differences were statistically significant between wild type and Sox10ΔeSC mutant from 16.5 dpc onwards as determined by Student’s t test (***, P ≤ 0.001). Bars, 50 µm.
Embryonic Schwann cell development in Sox10ΔeSC mice
As expected from the still widespread occurrence of Sox10 in peripheral nerves of Sox10ΔeSC embryos at 14.5 dpc, Schwann cell markers were similarly expressed in wild type and mutant (unpublished data). From 16.5 dpc onwards, differences between the two genotypes became apparent. Whereas Sox2 was down-regulated in Schwann cells along wild-type nerves, its expression continued at high levels in the mutant (Fig. 3, A–D). Conversely, many Schwann cells had already entered the promyelinating stage at 16.5 dpc in the wild type and expressed Oct6, whereas Oct6 was not expressed in the mutant (Fig. 3, G and H). This is not simply a delay, as Oct6 was still absent in the mutant at 18.5 dpc (Fig. 3, I and J). Up-regulation of Krox20 was also not observed at 18.5 dpc in the mutant nerve but was clearly visible in the wild type concomitant with the onset of myelination (Fig. 3, E and F). Strong expression of myelin genes such as Mbp or Mpz remained restricted to the wild type (Fig. 3, K and L; and not depicted). Thus, the marker gene pattern observed in Sox10ΔeSC embryos is consistent with Schwann cells not progressing beyond the immature stage.
Figure 3.

Expression of Schwann cell markers in peripheral nerves of Sox10ΔeSC embryos. (A–L) Immunohistochemistry (A–J) and in situ hybridizations (K and L) were performed on transverse sections of wild-type (wt; A, C, E, G, I, and K) and Sox10ΔeSC (B, D, F, H, J, and L) sciatic nerves at 16.5 (A, B, G, and H) and 18.5 dpc (C–F and I–L) using antibodies directed against Sox2 (A–D), Krox20 (E and F), Oct6 (G–J), and an antisense riboprobe for Mbp (K and L). (C, F, H, and J) Dotted lines indicate the circumference of the sciatic nerve. Bars, 50 µm.
Postnatal development of Sox10ΔeSC mice
Newborn Sox10ΔeSC mice were indistinguishable from their wild-type littermates but failed to thrive soon after birth. Already at postnatal day (P) 8, body weight was lower in Sox10ΔeSC than in wild-type pups (Fig. 4 A). Weight differences were in the range of 15–25% in the first 2–2.5 wk but became more dramatic after the third week, with Sox10ΔeSC mice being 52% lighter at P24 and 68% lighter at P32 than their wild-type littermates. Sox10ΔeSC mice looked increasingly runted, and locomotion progressively deteriorated. Symptoms included a shiverer-type tremor, an abnormal, straddled position of the hindlimbs, and poor motor coordination, resulting in an unsteady staggering gait. Even when care was taken that Sox10ΔeSC mice had continuous unlimited access to wet chow and drinking water, most succumbed to death during the fifth week. The few remaining survivors invariably died before the end of the seventh week.
Figure 4.

Postnatal development of Sox10ΔeSC mice and their sciatic nerves. (A) Weight of wild-type (open bars) and age-matched Sox10ΔeSC (closed bars) mice was monitored at P8, P16, P24, and P32. Data are shown as means ± SEM (n ≥ 6 for each genotype). Statistically significant differences from wild-type controls were observed from P16 onwards (**, P ≤ 0.01; and ***, P ≤ 0.001 by Student’s t test). (B) Macroscopic appearance of P32 sciatic nerves from wild-type (wt) and Sox10ΔeSC mice. (C) Sciatic nerve thickness was quantified for wild-type and Sox10ΔeSC mice at P8, P16, P24, and P32 by determining the area on proximal nerve sections. At least 20 sections from three mice were used per genotype for quantification. Data are presented as mean ± SEM. Differences were statistically significant between wild type and Sox10ΔeSC mutant from P8 onwards as determined by Student’s t test (**, P ≤ 0.01; ***, P ≤ 0.001). (D–K) Immunohistochemistry was performed on sciatic nerve sections from wild-type (D, F, H, and J) and Sox10ΔeSC (E, G, I, and K) mice at P16 (D, E, H, and I) and P32 (F, G, J, and K) using antibodies directed against Sox2 (D–G) and Krox20 (H–K). (D, F, I, and K) Dotted lines indicate the circumference of the sciatic nerve. (L–S) In situ hybridization was performed on sciatic nerve sections from wild-type (L, N, P, and R) and Sox10ΔeSC (M, O, Q, and S) mice at P16 (L, M, P, and Q) and P32 (N, O, R, and S) using antisense riboprobes for Mpz (L–O) and Mbp (P–S). (T–W) Myelin sheaths were visualized by PPD staining of sciatic nerve sections from wild-type (T and V) and Sox10ΔeSC (U and W) mice at P16 (T and U) and P32 (V and W). (X and Y) Electrophysiology on sciatic nerves of Sox10ΔeSC mice. Compound action potentials were monopolarly recorded from isolated sciatic nerves of wild-type and Sox10ΔeSC mice (n = 2 each). Experiments were performed on both nerves of each animal with identical results within each genotype. Representative superimposed traces are presented for both genotypes showing fast nerve conduction along myelinated fibers (X) and slow conduction along nonmyelinated fibers (Y). The arrows point to components of different conduction velocities (meters/second). Bars: (B) 1 mm; (D–S) 50 µm; (T–W) 3 µm.
Postnatal sciatic nerve development in Sox10ΔeSC mice
To study postnatal peripheral nerve development in more detail, we focused on the sciatic nerve. In contrast to the opaque wild-type sciatic nerve, the sciatic nerve of Sox10ΔeSC mice was translucent, appeared less well defined, and lacked the characteristic banded pattern (Fig. 4 B). From the third week onwards, the mutant sciatic nerve was also significantly thinner, with a mean decrease in thickness by 22% at P24 and by 40% at P32, confirming the degenerative phenotype (Fig. 4 C).
As observed during late embryogenesis, widespread expression of Sox2 continued along the sciatic nerve in Sox10ΔeSC mice throughout the analyzed postnatal period, whereas only few cells along wild-type nerves were Sox2 positive (Fig. 4, D–G; and not depicted). In contrast, Krox20 was only present in wild-type sciatic nerves but was missing from Sox10-deficient sciatic nerves (Fig. 4, H–K; and not depicted). Neither immunohistochemistry nor in situ hybridization detected expression of the myelin genes Mpz and Mbp in sciatic nerves of Sox10ΔeSC mice (Fig. 4, L–S; and not depicted). Paraphenylenediamine (PPD) staining confirmed the complete absence of myelin within the mutant sciatic nerve (Fig. 4, T–W).
Altered nerve conduction in Sox10ΔeSC mice
In agreement with the lack of myelin, compound action potentials of sciatic nerves of 16-d-old Sox10ΔeSC mice lacked the fast component (20 m/s) that originates from conduction along myelinated fibers (Fig. 4 X). Instead, action potentials along mutant sciatic nerves traveled in two compound waves with conduction velocities of 1.3 and 0.65 m/s (Fig. 4 Y). None corresponded to the singular slow component in wild-type sciatic nerves, which exhibited a velocity of 0.56 m/s and is characteristic for nonmyelinated type C fibers, arguing that nonmyelinated fibers are affected as well. Furthermore, amplitudes were grossly reduced in the mutant. Thus, nerve conduction along the sciatic nerve is dramatically altered in Sox10ΔeSC mice.
Cellular turnover in the postnatal sciatic nerve of Sox10ΔeSC mice
In mutant sciatic nerves, the number of cells (quantified as DAPI-stained nuclei) was not decreased (Fig. 5 A). Both in wild-type and mutant sciatic nerve, 220 nuclei were present, on average, per section at 18.5 dpc. Whereas this number increased only gradually to ∼460 in the wild type until P24, it reached already 560 at P8 in the mutant and increased, on average, to a constant level of 730 between P16 and P32. Thus, despite being thinner from P16 onwards, the mutant sciatic nerve contained significantly more cells than the wild type.
Figure 5.

Proliferation and cell death in sciatic nerves of Sox10ΔeSC mice. (A) The total cell number in sciatic nerves was determined for wild-type and Sox10ΔeSC mice at 18.5 dpc, P8, P16, P24, and P32 by quantifying the DAPI-stained nuclei in proximal nerve sections. (B) Proliferative cells were counted in wild-type and Sox10ΔeSC sciatic nerves at P8, P16, P24, and P32 as Ki67-positive cells. (C) Cell death was measured by the number of TUNEL-positive cells in wild-type and Sox10ΔeSC sciatic nerves at P16, P24, and P32. For all quantifications, at least 20 sections from three mice were used per genotype. Data are presented as mean ± SEM. According to the Student’s t test, differences were statistically significant between wild type and Sox10ΔeSC mutant as indicated (**, P ≤ 0.01; ***, P ≤ 0.001).
This went along with significant increases in the number of proliferating cells in the sciatic nerve of Sox10ΔeSC mice, as determined by Ki67 immunoreactivity (Fig. 5 B). In the wild-type sciatic nerve, the number of Ki67-positive cells continuously decreased during the first postnatal weeks from a maximum of 50 cells per section at P8 to <7 from P16 onwards. In contrast, the mutant nerve contained ∼80 Ki67-positive cells at P8 and >50 at P16. A substantial decrease of Ki67-positive cells was not observed before P24 in the mutant sciatic, but even then, numbers remained higher than in the wild type (Fig. 5 B). From P16 onwards, apoptotic cells as determined by TUNEL were also significantly increased in sciatic nerves of Sox10ΔeSC mice relative to the wild type (Fig. 5 C). However, the absolute number of apoptotic cells per section remained fairly low.
Cellular composition in the postnatal sciatic nerve of Sox10ΔeSC mice
To find out whether Schwann cells increased in numbers, we quantified Sox2-positive cells in the sciatic nerves of Sox10ΔeSC mice. Their absolute number increased only slightly from P8 until P32 (Fig. 6 A). As a consequence, the contribution of Sox2-positive cells to the total cell population remained <30% in the mutant sciatic nerve (Fig. 6 B).
Figure 6.

Schwann cell proliferation in sciatic nerves of Sox10ΔeSC mice. (A and B) The absolute number of Sox2-positive cells was determined in sciatic nerves of Sox10ΔeSC mice at P8, P16, P24, and P32 (A) and set in relation to the total number of cells (B). (C) Cell numbers were also determined for YFP-expressing cells in sciatic nerves of Dhh::Cre, Rosa26stopfloxYFP (Rosa26DhhYFP; open bars) and Dhh::Cre, Sox10fl/fl, Rosa26stopfloxYFP mice (Sox10ΔeSC, Rosa26DhhYFP; closed bars) at P8, P16, P24, and P32. (D–G) Coimmunohistochemistry was performed on sciatic nerve sections of Sox10ΔeSC, Rosa26DhhYFP mice at P8 (D), P16 (E), P24 (F), and P32 (G) using antibodies against Sox2 (red) and YFP (green). (H) Proliferation rates of wild-type (wt) and Sox10ΔeSC Schwann cells were determined at P8, P16, P24, and P32 by determining the fraction of Ki67-positive cells among the YFP-labeled cells in Rosa26DhhYFP (open bars) and Sox10ΔeSC, Rosa26DhhYFP mice (closed bars). (I) The proliferation rates of Sox10ΔeSC Schwann cells (see H) were also used to determine the relative contribution of Schwann cells to the overall proliferation in sciatic nerves of Sox10ΔeSC mice. For all quantifications, at least 20 sections from three mice were used per genotype. Data are presented as mean ± SEM. According to the Student’s t test, differences were statistically significant between wild type and Sox10ΔeSC mutant as indicated (**, P ≤ 0.01; ***, P ≤ 0.001). Bars, 10 µm.
Sox2 may not label all cells of the Schwann cell lineage in peripheral nerves of Sox10ΔeSC mice. Therefore, we performed lineage tracing elements by additionally introducing the Rosa26stopfloxYFP allele into the Sox10ΔeSC mice. In these mice, YFP is expressed in all cells along the nerve that at one time expressed Dhh::Cre. The number of YFP-expressing cells along the peripheral nerve of Sox10ΔeSC mice did not significantly increase from P8 to P32 and closely matched the number of Sox2-expressing cells (Fig. 6 C). Coimmunohistochemistry confirmed that the vast majority of YFP-expressing cells were also Sox2 positive and vice versa (Fig. 6, D–G). Direct comparison of the proliferation rate of YFP-expressing cells with similarly YFP-labeled cells in the wild type revealed that it was significantly elevated in peripheral nerves of Sox10ΔeSC mice only at P16 (Fig. 6 H). Furthermore, YFP-expressing cells accounted from P8 to P24 for a smaller fraction of the proliferating cells in the mutant than in the wild type (Fig. 6 I). This argues that the Schwann cell lineage is not predominantly responsible for the increased cell numbers in the mutant sciatic nerves and that their transiently increased proliferation is counteracted by increased apoptosis.
Other major cell types within the peripheral nerve include endothelial cells, pericytes, endoneurial fibroblasts, perineurial cells, and cells of the immune system. Immunohistochemistry with antibodies against Pecam revealed a strong increase in the number of cells (Fig. 7, A–D). In the peripheral nerve, Pecam is expressed in several cell types (Berman et al., 1996). However, 80–90% of the Pecam-positive cells were also labeled in wild-type and mutant nerve with antibodies directed against von Willebrand factor (Fig. 7, C and D), identifying the majority as endothelial cells. This increase in endothelial cells was accompanied by a comparable increase in pericytes, as evident from immunohistochemistry with desmin antibodies (Fig. 7, E–H). Pecam antibodies also stained the perineurial sheath in the wild type (Fig. 7 A). It is unclear whether this staining is specific. However, its absence in the mutant (Fig. 7 B) is indicative of structural alterations in the perineurial sheath of nerves from Sox10ΔeSC mice (see next section). Perineurial cells were present in the mutant sciatic nerve, as transcript levels for the perineurial cell marker Occludin were comparable to the wild type (unpublished data). Unfortunately, we did not manage to visualize these cells directly, as none of the antibodies against the few available markers for this cell type worked in our hands. Similarly, there is no robust specific immunohistochemical marker for endoneurial fibroblasts. Using antibodies against Iba1, we detected a substantial increase in macrophages in the sciatic nerve of Sox10ΔeSC mice (Fig. 7, I and J). T lymphocytes were also significantly increased, as evident from anti-CD3 stainings (Fig. 7, K and L).
Figure 7.

Cellular composition of sciatic nerves in Sox10ΔeSC mice. (A–L) Immunohistochemistry was performed on sciatic nerve sections from wild-type (wt; A, C, E, G, I, and K) and Sox10ΔeSC (B, D, F, H, J, and L) mice at P16 using antibodies directed against Pecam (A–D) and von Willebrand factor (vWF; C and D) as predominantly endothelial markers, desmin (E–H) as a marker for pericytes, Iba1 (I and J) as a marker for macrophages, and CD3 (K and L) as a marker for T lymphocytes and is shown in low (A, B, E, and F) and high (C, D, and G–L) magnification. All markers are in red except Pecam, to which green color was assigned. Nuclei in C, D, and G–L were counterstained by DAPI (blue). (M–O) These and analogous experiments at other time points were used to determine the cellular composition of sciatic nerves in wild-type and Sox10ΔeSC mice at P16 (M), P24 (N), and P32 (O). Quantifications were performed on at least 20 sections from three mice per genotype. Cell numbers are presented as mean ± SEM. Bars: (A, B, E, and F) 50 µm; (C, D, and G–L) 10 µm.
By quantifying the immunohistochemical stainings, we determined the relative contribution of each cell type to the total cell population within wild-type and mutant sciatic nerve from P16 to P32. As evident from Fig. 7 (M–O), not only endothelial cells, pericytes, macrophages, and T lymphocytes increased in number, but also the cells that were not labeled by any of the cell type–specific markers. These include perineurial cells and endoneurial fibroblast as well as some adipocytes in case of the mutant (see next section). Thus, cellular composition is severely altered in postnatal sciatic nerves of Sox10ΔeSC mice.
Nerve ultrastructure in Sox10ΔeSC mice
To define ultrastructural changes in mutant sciatic nerves, we performed electron microscopy at P16 and P32. In wild-type sciatic nerves, large-caliber axons were heavily myelinated at P16 (Fig. 8 A). This was not the case in sciatic nerves of Sox10ΔeSC mice (Fig. 8 B). Remak bundles, in which a nonmyelinating Schwann cell is associated with many small diameter axons and surrounds each and every axon with its membrane, were also present in wild-type nerves (Fig. 8 A, arrows) but missing in Sox10ΔeSC mice (Fig. 8 B). Instead, large- and small-caliber axons were tightly grouped together in mutant nerves and jointly surrounded by a single Schwann cell and its processes (Fig. 8 B). Such a configuration is characteristic of the immature Schwann cell stage in which axonal sorting and establishment of a 1:1 ratio of Schwann cells with large diameter axons has not yet taken place (Jessen and Mirsky, 2005). In mutant sciatic nerves, a dense layer of collagen and elastic fibers was present around each group of axons and associated Schwann cell (Fig. 8 D; for higher magnification of elastic fibers see Fig. 8 F). This was again surrounded in an onion skin style by one or two layers of flat cells with long cytoplasmic processes (Fig. 8 D, black arrows). Furthermore, a considerable number of Schwann cells in the mutant exhibited unusually high amounts of rough endoplasmic reticulum (Fig. 8 D, white arrows) and resembled fibroblasts in their appearance. Their increased protein synthesis likely contributes to the extra collagen and elastic fibers in Sox10ΔeSC mice. Compared with the thick and continuous basal lamina of wild-type Schwann cells (Fig. 8 E), the basal lamina of Schwann cells in Sox10ΔeSC mice was considerably thinner and often incomplete (Fig. 8 F). Axons in mutant sciatic nerves contained microtubules, neurofilaments, and mitochondria (Fig. 8 D) and showed no obvious differences in ultrastructure to wild-type axons (Fig. 8 C). Signs of axonopathy were not observed in the mutant sciatic nerve, and motor neuron numbers in the ventral horn of the spinal cord were unaffected (unpublished data). The presence of adipocytes with lipid inclusions in sciatic nerves was unique to the mutant (Fig. 8 B, asterisks). When compared with the wild type, the perineurial sheath was dramatically thinned (Fig. 8, G and H).
Figure 8.

Sciatic nerve ultrastructure in Sox10ΔeSC mice at P16. (A–H) Myelin sheaths and Remak bundles (white arrows in A) were only present in wild-type nerves (A, C, E, and G), whereas large- and small-caliber axons were jointly surrounded by a single Schwann cell in nerves of Sox10ΔeSC mice (B, D, F, and H). Collagen and elastic fibers were densely layered around each group of axons and associated Schwann cell in mutant sciatic nerves, again surrounded in an onion skin style by flat cells with long cytoplasmic processes (black arrows in D). Adipocytes with lipid inclusions (asterisks in B) were present in mutant sciatic nerves and Schwann cells frequently had an enlarged rough endoplasmic reticulum (white arrows in D). Axons in mutant and wild-type sciatic nerves looked similar. Myelin sheaths in wild-type nerves were surrounded by a complete basal lamina (black arrows in E). In contrast, in nerves of Sox10ΔeSC mice the basal lamina of Schwann cells was considerably thinner and often incomplete (black arrows in F). In addition, elastic fibers (white arrow in F) were frequently observed in close contact with Schwann cells of mutant nerves but were essentially absent in wild-type nerves. The perineurial sheath was dramatically thicker in wild-type nerves (bracket in G) than in mutant nerves (bracket in H). Bars: (A) 2 µm; (B) 5 µm; (C and D) 1 µm; (E–H) 500 nm.
Compared with P16, myelinated axons and Remak bundles in wild-type nerves were less tightly grouped together at P32 and separated by loose endoneural extracellular matrix (Fig. 9 A). Both structures were still absent at P32 in mutant sciatic nerves, and the dense fibrillar extracellular matrix had expanded even further (Fig. 9 B). Many axon bundles were no longer completely surrounded by a Schwann cell and its processes (Fig. 9 C, white arrows) but were in direct contact with collagen fibrils and elastic fibers in the surrounding extracellular matrix (Fig. 9 C, asterisk). Several Schwann cells were observed in mutant sciatic nerves that showed condensed and fragmented chromatin indicative of apoptotic cell death (Fig. 9 D, asterisk).
Figure 9.

Sciatic nerve ultrastructure in Sox10ΔeSC mice at P32. (A) Myelinated axons and Remak bundles in wild-type sciatic nerves were separated by loose endoneural extracellular matrix. (B) In mutant nerves, axon bundles and their associated Schwann cells were separated from other units by a dense fibrillar extracellular matrix. (C) Axon bundles in mutant nerves were frequently not completely covered by a Schwann cell and its processes (white arrows) but in direct contact to the surrounding extracellular matrix which contains collagen fibrils (black arrow) and elastic fibers (asterisk). (D) Several Schwann cells in mutant sciatic nerves show condensed and fragmented chromatin (asterisk), strongly indicating apoptotic cell death. Bars: (A and B) 2.5 µm; (C) 250 nm; (D) 1 µm.
Gene expression of Schwann cells in the postnatal sciatic nerve of Sox10ΔeSC mice
The fact that Schwann cells possessed only a poorly developed basal lamina, exhibited a distended endoplasmic reticulum, and, in some instances, had loosened the contact to their associated axon bundle by P32 is evidence that these cells had lost some of their typical features. To characterize the mutant Schwann cells in greater detail, we also analyzed their expression pattern by comparing transcript levels for Schwann cell markers in wild-type and mutant sciatic nerves by quantitative RT-PCR at P16. Myelin gene expression was used as a control and was indeed dramatically reduced in the mutant relative to the wild type (Fig. 10, A and B). In contrast, c-Jun expression was strongly up-regulated in the mutant (Fig. 10 C). This was expected, as c-Jun and Sox2 are both characteristic of the immature Schwann cell stage (Jessen and Mirsky, 2005). However, surprisingly, there was also a dramatic down-regulation of Dhh, S100-β, and ErbB3, which should all be expressed in the immature Schwann cell stage (Fig. 10, D–F). This indicates that Schwann cells had not maintained their normal expression pattern in the absence of Sox10.
Figure 10.

Gene expression experiments on sciatic nerves of Sox10ΔeSC mice. (A–F) Quantitative RT-PCR was performed on cDNA prepared from P16 sciatic nerves of wild-type (open bars) and Sox10ΔeSC (closed bars) mice using primers directed against Mbp (A), Mpz (B), c-Jun (C), Dhh (D), S100-β (E), and ErbB3 (F). Transcript levels in each sample were normalized to Rpl8. After normalization, transcript levels in the wild type were arbitrarily set to 1, and transcript levels in Sox10ΔeSC mice are expressed relative to wild-type levels ± SEM. Experiments were repeated twice with material from two independent sciatic nerve preparations for each genotype.
Discussion
In this study, we show that Sox10 is not only required for Schwann cell specification but also at later stages for maintaining Schwann cell identity and proper lineage progression. To obtain these results, we generated a conditional Sox10 allele and deleted it with a Dhh::Cre transgene. This transgene has previously been reported to become active already in Schwann cell precursors (Jaegle et al., 2003). Nevertheless, all available morphological data and marker gene studies indicate that it is not before the immature Schwann cell stage that most Schwann cells lose Sox10 protein.
Sox10 is expressed at high levels during Schwann cell development. Therefore, if stable, it may take a while before cells become Sox10 negative even if the gene is already deleted in the Schwann cell precursor. Alternatively, the loxP sites in the Sox10 allele may not be easily accessible to Cre recombinase in Schwann cells or recombine with fairly low efficiency for other reasons. In fact, slow protein turnover and inefficient recombination both seem to contribute, as Sox10 protein is lost at a significantly slower pace than Sox10 transcripts, which in turn persist longer than it takes for the Rosa26stopfloxYFP allele to recombine.
Because Schwann cells do not lose Sox10 protein before the immature stage, we cannot answer whether Sox10 is required in Schwann cell precursors to allow progression into immature Schwann cells. What we can conclude is that immature Schwann cells depend on the presence of Sox10 to develop into myelinating as well as nonmyelinating Schwann cells. As a consequence, large- and small-caliber axons remain jointly bundled in nerves from Sox10ΔeSC mice. In the absence of myelination and axonal sorting, fast nerve conduction is missing, and conduction in nonmyelinated fibers is grossly altered. The absence of myelinating as well as nonmyelinating Schwann cells may be one reason for the severity of the resulting phenotype. In contrast, defects are largely restricted to myelinating Schwann cells in the Oct6- and Krox20-null mutants (Topilko et al., 1994; Bermingham et al., 1996; Jaegle et al., 1996) as well as the various myelin gene mutants (for review see Martini, 1997). In these mutants, axonal sorting occurs, and Schwann cells progress at least into the promyelinating stage, where they establish a 1:1 ratio with large-caliber axons and wrap one turn and a half of their plasma membrane around the axon.
However, there is also a set of mutants in which additional sorting defects occur. These include the Nfat1c, Rac1, and β1 integrin mouse mutants (Feltri et al., 2002; Benninger et al., 2007; Nodari et al., 2007; Kao et al., 2009). However, in Nfat1c mutants, the effect on axonal sorting is much less pronounced (Kao et al., 2009). In contrast, Schwann cell–specific Rac1 mutants exhibit major but not complete myelination and axonal sorting defects. In contrast to our mouse model, quite a bit of normal myelin appears in Rac1 mutants at older ages (Benninger et al., 2007). Complete disruption of axonal sorting was observed in Schwann cell–specific β1 integrin mutants, and there is evidence that Rac1 functions downstream of β1 integrin (Feltri et al., 2002; Nodari et al., 2007). In this respect, Schwann cell–specific β1 integrin mutants resemble our Sox10ΔeSC mice phenotypically. However, the underlying defect is different, as β1 integrin exerts its main influence directly on the interaction of Schwann cells with axons, whereas Sox10 rather influences cell identity and lineage progression.
Loss of Sox10 in immature Schwann cells did not negatively affect their proliferation. We even observed a slight transient increase in Schwann cell proliferation rates during the first postnatal weeks. However, this was counteracted by simultaneous mild increases in apoptosis. We conclude from these findings that Sox10 does not primarily function in immature Schwann cells as a proliferation or cell survival factor. Thus, Sox10ΔeSC mice are different from Schwann cell–specific Cdc42 mutants in which Schwann cell proliferation is significantly decreased (Benninger et al., 2007).
Sox10 rather appears to be important in immature Schwann cells for lineage progression as neither Oct6 nor Krox20 was induced in these cells in the absence of Sox10. This agrees with previous reports of a direct dependence of Krox20 expression on Sox10 (Ghislain and Charnay, 2006; Kao et al., 2009; Reiprich et al., 2010) and predicts that Oct6 expression is also dependent on Sox10, either directly or indirectly.
In addition to its requirement for lineage progression, Sox10 is also needed to maintain the identity of immature Schwann cells. In the absence of Sox10, immature Schwann cells lose expression of some of their typical markers, including S100-β and Dhh. Schwann cell–derived Dhh has previously been reported to be essential for perineurial sheath formation (Parmantier et al., 1999). Thus, its loss may explain the dramatically thinned perineurial sheath in peripheral nerves of Sox10ΔeSC mice. An additional reduction in ErbB3 expression may contribute to the inability of mutant immature Schwann cells to further mature. Dependency of ErbB3 expression on Sox10 has already been reported for earlier periods of Schwann cell development (Britsch et al., 2001).
By expression profiling, we also found up-regulation of the proangiogenic factors angiopoietin 1 and vascular endothelial growth factor C, the monocyte chemoattractant Ccl8, and the macrophage chemoattractants Ccl6 and Cxcl14 in peripheral nerves of Sox10ΔeSC mice (unpublished data). In nerves, Cxcl14 is predominantly expressed in Schwann cells, and expression levels appear to correlate with less differentiated or pathological states (Barbaria et al., 2009). Angiopoietin 1 and vascular endothelial growth factor C have also been detected in Schwann cells, and higher expression in neurofibromin-deficient Schwann cells was associated with increased vasculogenesis (Thomas and De Vries, 2007). In Sox10ΔeSC mice, increased Schwann cell expression of proangiogenic factors may thus promote vasculogenesis, whereas heightened chemokine production may attract macrophages, which in turn help to recruit T lymphocytes to the mutant nerve (Martini et al., 2008). This offers a plausible explanation for the origin of the altered cellular composition in mutant sciatic nerves.
Altered gene expression in the immature Sox10-deficient Schwann cells can also be inferred from the poorly developed basal lamina and their strong overall increase of protein synthesis in Sox10ΔeSC mice. At least some of this synthesis can be attributed to the production of collagen fibrils and elastin-containing fibers. Furthermore, mutant Schwann cells loosened their contact with axons and became more fibroblast-like in appearance. Our study supports the notion that Schwann cells are unable to maintain their full identity and acquire some characteristics that are normally associated with fibroblasts. However, we do not suggest that mutant Schwann cells fully transform into fibroblasts. Previous experiments have indicated that a small percentage of neural crest cells in peripheral nerves give rise to endoneurial fibroblasts, whereas the majority develops into Schwann cells (Joseph et al., 2004). No single marker exists that identifies these endoneurial fibroblasts unequivocally. Thus, the relationship between our mutant Schwann cells and these neural crest–derived endoneurial fibroblasts remains unclear at present.
Our findings also have important implications for human pathology. Several reported SOX10 mutations cause peripheral neuropathies in the heterozygous state (Inoue et al., 2004). Such neuropathies are not detected in mice with a single functional Sox10 allele and are likely caused by the dominant action of the mutant human SOX10 protein. Our results now show that this dominant action may not only affect the Schwann cell specification process but may also interfere with the normal function of Sox10 during further lineage progression.
Materials and methods
Construction of targeting vectors, gene targeting, generation, and genotyping of mouse mutants
To generate a conditional Sox10 allele, the ORF for rat Sox10 was placed between 5′ and 3′ Sox10 genomic regions as homology arms (4.3 kb and 1.5 kb, respectively) in the context of a pPNT vector backbone in such a way that the start codon of the ORF was placed exactly at the position of the endogenous ATG in exon 3 (Britsch et al., 2001; Ludwig et al., 2004; Kellerer et al., 2006). The targeting vector thereby replaced the Sox10 coding exons by a continuous Sox10 reading frame. Additionally, a neomycin resistance cassette with flanking loxP and Flp recognition target (FRT) sites and a linked IRES-EGFP cassette were placed immediately behind the Sox10 ORF using a SalI restriction site. Finally, another loxP site was introduced into a StuI site 394 bp in front of the splice acceptor site in intron 2 in the same orientation as the ones flanking the neomycin resistance cassette (Fig. 1 A). The construct was linearized with SspI and electroporated into embryonic day 14.1 ES cells, which were then selected with 400 µg/ml G418 and 2 µM ganciclovir. Selected ES cell clones were screened by Southern blotting as described previously (Schreiner et al., 2007). Two targeted ES cell lines carrying the Sox10flneo allele (Fig. 1 A) were injected into C57BL/6J blastocysts to generate chimeras. Chimeric males from two independent clones were crossed to FLPe deleter mice (Rodríguez et al., 2000) to achieve germline transmission and simultaneous FRT-mediated deletion of the neomycin resistance cassette. As a result, heterozygous mice with a Sox10fl allele were obtained (Fig. 1 A). Structural integrity of the Sox10fl allele was confirmed by Southern blot experiments with a 0.6-kb 5′ probe, which recognized a 6.6-kb fragment for the wild-type allele and a 4.8-kb fragment for the Sox10fl allele in NcoI-digested genomic DNA, and a 0.6-kb 3′ probe, which recognized a 4.6-kb fragment for the wild-type allele and a 3.7-kb fragment for the Sox10fl allele in BamHI–ScaI-digested genomic DNA (Fig. 1 B). Direct sequencing of the modified Sox10 locus additionally verified the accuracy of the Sox10fl allele on the nucleotide level. Heterozygous mice were intercrossed to generate Sox10fl/fl mice or bred to Dhh::Cre transgenic (Jaegle et al., 2003) and Rosa26stopfloxYFP mice (gift from F. Costantini, Columbia University, New York, NY; Srinivas et al., 2001) to produce Sox10+/fl, Dhh::Cre double transgenic and Sox10+/fl, Rosa26stopfloxYFP, Dhh::Cre triple transgenic mice. Crossing these double and triple transgenic mice with Sox10fl/fl mice yielded the genotypes analyzed in this study. Genotyping was routinely performed on DNA from tail tips or, in case of embryos, from yolk sacs by PCR analysis. Primers for the Dhh::Cre transgene and the Rosa26stopfloxYFP allele were as described previously (Srinivas et al., 2001; Jaegle et al., 2003). For Sox10 genotyping, we used an upper primer located 81 bp upstream of the start codon (5′-CAGGTGGGCGTTGGGCTCTT-3′) and two lower primers located 487 bp downstream of the start codon in intron 3 of the Sox10 gene (5′-TCCCAGGCTAGCCCTAGTG-3′) or 617 bp downstream of the start codon in the Sox10 reading frame (5′-GTGAGCCTGGATAGCAGCAG-3′). Genotyping revealed a 568-bp fragment for the wild-type allele and a 698-bp fragment in the case of the Sox10fl allele (Fig. 1 C).
Electrophysiology
Animals were sacrificed in a pure CO2 atmosphere, and the sciatic nerves of both legs were excised from the trifurcation up to the lumbar plexus and stored in carbogen-gased physiological buffer. Compound action potentials were recorded monopolarly in a Perspex chamber filled with fluorocarbon oil (FC-43; 3M Company) at 38°C temperature. The distal end of the isolated nerve was electrically stimulated through gold wire electrodes using a voltage-constant stimulus isolator and supramaximal intensities: ∼1 V at 0.1 ms for myelinated nerve fibers and ∼12 V at 1 ms for nonmyelinated ones. Gold wire electrodes and a custom-made differential amplifier were used for recording and filtering (1–5,000 Hz), and the output was digitized (DAP 1200; Microstar) and stored in a PC using the Spike/Spidi software package (Forster and Handwerker, 1990).
RNA preparation from embryos or adult sciatic nerves for quantitative RT-PCR
Embryos were obtained from staged pregnancies at 12.5 dpc, and sciatic nerves were prepared from 16-d-old mice. Both were processed for RNA isolation using TRIZOL reagent (Invitrogen).
Reverse transcription was performed in a 25-µl reaction using 2 µg RNA, oligo (dT) primers, and Moloney murine leukemia virus reverse transcription (New England Biolabs, Inc.). Quantitative polymerase chain reaction with 0.2 µl of the obtained cDNA was performed on a LightCycler (Roche) according to the manufacturer’s instructions using the ABsolute QPCR SYBR green mix (Thermo Fisher Scientific) with an annealing temperature of 60°C. Rpl8 transcripts were used for normalization (Kellerer et al., 2006). The following primers were used: 5′-ACCCCCACTCAGTTCTTGTG-3′ and 5′-AGTTGCTGAGGTTGGCGTAG-3′ for c-Jun, 5′-CATGTGGCCCGGAGTACGCC-3′ and 5′-CGCTGCATCAGCGGCCAGTA-3′ for Dhh, 5′-GAGCGGGGTGACGGGAGTAA-3′ and 5′-GGGTCGCGAACAGTTCTCCC-3′ for ErbB3, 5′-CCAAGTTCACCCCTACTCCA-3′ and 5′-TAAGTCCCCGTTTCCTGTTG-3′ for Mbp, 5′-CTGGTCCAGTGAATGGGTCT-3′ and 5′-CATGTGAAAGTGCCGTTGTC-3′ for Mpz, 5′-GACTGGGTCAGGGAATATCCACC-3′ and 5′-AGCAGCAGCCATGTACTCTTCAC-3′ for Occludin, 5′-GACTCCAGCAGCAAAGGTGAC-3′ and 5′-CATCTTCGTCCAGCGTCTCCA-3′ for S100-β, and 5′-CAGGTGTGGCTCTGCCCACG-3′ and 5′-GTGTAGAGGGGCCGCTGGGA-3′ for Sox10.
In situ hybridization and immunohistochemistry
Embryos were obtained from staged pregnancies at 12.5, 14.5, 16.5, and 18.5 dpc, and sciatic nerves were obtained from postnatal mice at various ages. After fixation in 4% paraformaldehyde, specimens were cryoprotected by overnight incubation at 4°C in 30% sucrose and embedded in tissue-freezing medium (Leica) compound at −80°C. Frozen sections were prepared at 14-µm thickness and used for in situ hybridizations or at 10-µm thickness and used for immunohistochemistry.
In situ hybridizations were performed using digoxigenin-labeled antisense riboprobes for Sox10, Mbp, and Mpz as described previously (Britsch et al., 2001; Stolt et al., 2002). Micrographs were taken from a stereomicroscope (MZ FLIII; Leica) equipped with a 10× Plan-Apochromat objective at room temperature in air as imaging medium using a camera (AxioCam HRc; Carl Zeiss, Inc.) and Axio Vision (Carl Zeiss, Inc.) as acquisition software.
For immunohistochemistry, the following primary antibodies were used in various combinations: anti-Sox10 guinea pig antiserum (1:1,000; Maka et al., 2005), anti-Oct6 rabbit antiserum (1:2,000; Friedrich et al., 2005), anti-Krox20 rabbit antiserum (1:200; Covance), anti-Sox2 rabbit antiserum (1:500), anti-Ki67 rabbit antiserum (1:500; Thermo Fisher Scientific), anti-CD3 rabbit antiserum (1:500; Abcam), anti-GFP rabbit antiserum (1:500; Invitrogen), anti-Iba1 rabbit antiserum (1:250; Wako Chemicals USA, Inc.) antidesmin rabbit antiserum (1:1,000; Abcam), anti–von Willebrand factor rabbit antiserum (1:800; Abcam), and anti-PECAM rat antiserum (1:200; BD). Secondary antibodies conjugated to Cy2, Cy3, or Alexa Fluor 488 immunofluorescent dyes (Dianova) were used for detection. Micrographs were taken from a microscope (DM-IRB; Leica) equipped with 10× NA 0.3 or 20× NA 0.5 HC PL Fluotar objectives at room temperature in air as imaging medium using a charge-coupled device camera (SPOT model 1.3.0; Diagnostic Instruments, Inc.) and IP Laboratory (Scanalytics) as acquisition software. All images were processed using Photoshop CS2 (Adobe).
Histology and electron microscopic analysis
Mice were anaesthetized and perfused transcardially using 0.9% NaCl solution followed by 1% glutaraldehyde and 2.5% paraformaldehyde in 0.1 M Na cacodylate buffer, pH 7.5. Sciatic nerves were dissected and underwent overnight fixation at 4°C in cacodylate-buffered fixative containing 2.5% paraformaldehyde and 2.5% glutaraldehyde. After extensive washing in 0.1 M Na cacodylate, pH 7.3, tissues were postfixed in cacodylate-buffered 1% osmium ferrocyanide for 2 h, dehydrated in an ascending ethanol series, and embedded in Epon resin. For light microscopy, semithin sections (1-µm thickness) were stained with PPD (Estable-Puig et al., 1965). Micrographs were taken from a microscope (DMR; Leica) equipped with a 40× NA 0.75 HCX PL Fluotar objective at room temperature in air as imaging medium using a camera (AxioCam MRc; Carl Zeiss, Inc.) and Axio Vision as acquisition software. For electron microscopy, ultrathin sections (50-nm thickness) were stained with uranyl acetate and lead citrate and examined with an electron microscope (Libra; Carl Zeiss, Inc.; Friedrich et al., 2005).
Acknowledgments
We thank F. Costantini for the Rosa26stopfloxYFP mice and M. Schimmel for excellent technical help with electron microscopy.
This work was supported by the Interdisziplinäres Zentrum für Klinische Forschung Erlangen and grants from the Deutsche Forschungsgemeinschaft to M. Wegner (We1326/8-1) and to E.R. Tamm (FOR 1075, TP5).
Footnotes
Abbreviations used in this paper:
- dpc
- day postcoitum
- ES
- embryonic stem
- FRT
- Flp recognition target
- IRES
- internal ribosomal entry site
- PNS
- peripheral nervous system
- PPD
- paraphenylenediamine
References
- Barbaria E.M., Kohl B., Buhren B.A., Hasenpusch-Theil K., Kruse F., Küry P., Martini R., Müller H.W. 2009. The alpha-chemokine CXCL14 is up-regulated in the sciatic nerve of a mouse model of Charcot-Marie-Tooth disease type 1A and alters myelin gene expression in cultured Schwann cells. Neurobiol. Dis. 33:448–458 10.1016/j.nbd.2008.11.014 [DOI] [PubMed] [Google Scholar]
- Benninger Y., Thurnherr T., Pereira J.A., Krause S., Wu X., Chrostek-Grashoff A., Herzog D., Nave K.A., Franklin R.J., Meijer D., et al. 2007. Essential and distinct roles for cdc42 and rac1 in the regulation of Schwann cell biology during peripheral nervous system development. J. Cell Biol. 177:1051–1061 10.1083/jcb.200610108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman M.E., Xie Y., Muller W.A. 1996. Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and beta 2 integrin activation. J. Immunol. 156:1515–1524 [PubMed] [Google Scholar]
- Bermingham J.R., Jr, Scherer S.S., O’Connell S., Arroyo E., Kalla K.A., Powell F.L., Rosenfeld M.G. 1996. Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 10:1751–1762 10.1101/gad.10.14.1751 [DOI] [PubMed] [Google Scholar]
- Bondurand N., Dastot-Le Moal F., Stanchina L., Collot N., Baral V., Marlin S., Attie-Bitach T., Giurgea I., Skopinski L., Reardon W., et al. 2007. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 81:1169–1185 10.1086/522090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles J., Schepers G., Koopman P. 2000. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227:239–255 10.1006/dbio.2000.9883 [DOI] [PubMed] [Google Scholar]
- Britsch S., Goerich D.E., Riethmacher D., Peirano R.I., Rossner M., Nave K.A., Birchmeier C., Wegner M. 2001. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15:66–78 10.1101/gad.186601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossais F., Sock E., Hornig J., Schreiner S., Kellerer S., Bösl M.R., Russell S., Wegner M. 2010. Replacement of mouse Sox10 by the Drosophila ortholog Sox100B provides evidence for co-option of SoxE proteins into vertebrate-specific gene-regulatory networks through altered expression. Dev. Biol. 341:267–281 10.1016/j.ydbio.2010.01.038 [DOI] [PubMed] [Google Scholar]
- Estable-Puig J.F., Bauer W.C., Blumberg J.M. 1965. Technical note: paraphenylendiamine staining of osmium-fixed plastic embedded tissue for light and phase microscopy. J. Neuropathol. Exp. Neurol. 24:531–536 10.1097/00005072-196507000-00012 [DOI] [Google Scholar]
- Feltri M.L., Graus Porta D., Previtali S.C., Nodari A., Migliavacca B., Cassetti A., Littlewood-Evans A., Reichardt L.F., Messing A., Quattrini A., et al. 2002. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J. Cell Biol. 156:199–209 10.1083/jcb.200109021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster C., Handwerker H.O. 1990. Automatic classification and analysis of microneurographic spike data using a PC/AT. J. Neurosci. Methods. 31:109–118 10.1016/0165-0270(90)90155-9 [DOI] [PubMed] [Google Scholar]
- Friedrich R.P., Schlierf B., Tamm E.R., Bösl M.R., Wegner M. 2005. The class III POU domain protein Brn-1 can fully replace the related Oct-6 during schwann cell development and myelination. Mol. Cell. Biol. 25:1821–1829 10.1128/MCB.25.5.1821-1829.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain J., Charnay P. 2006. Control of myelination in Schwann cells: a Krox20 cis-regulatory element integrates Oct6, Brn2 and Sox10 activities. EMBO Rep. 7:52–58 10.1038/sj.embor.7400573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbarth B., Pingault V., Bondurand N., Kuhlbrodt K., Hermans-Borgmeyer I., Puliti A., Lemort N., Goossens M., Wegner M. 1998. Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc. Natl. Acad. Sci. USA. 95:5161–5165 10.1073/pnas.95.9.5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K., Khajavi M., Ohyama T., Hirabayashi S.-i., Wilson J., Reggin J.D., Mancias P., Butler I.J., Wilkinson M.F., Wegner M., Lupski J.R. 2004. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36:361–369 10.1038/ng1322 [DOI] [PubMed] [Google Scholar]
- Jaegle M., Mandemakers W., Broos L., Zwart R., Karis A., Visser P., Grosveld F., Meijer D. 1996. The POU factor Oct-6 and Schwann cell differentiation. Science. 273:507–510 10.1126/science.273.5274.507 [DOI] [PubMed] [Google Scholar]
- Jaegle M., Ghazvini M., Mandemakers W., Piirsoo M., Driegen S., Levavasseur F., Raghoenath S., Grosveld F., Meijer D. 2003. The POU proteins Brn-2 and Oct-6 share important functions in Schwann cell development. Genes Dev. 17:1380–1391 10.1101/gad.258203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen K.R., Mirsky R. 2005. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6:671–682 10.1038/nrn1746 [DOI] [PubMed] [Google Scholar]
- Joseph N.M., Mukouyama Y.S., Mosher J.T., Jaegle M., Crone S.A., Dormand E.L., Lee K.F., Meijer D., Anderson D.J., Morrison S.J. 2004. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. 131:5599–5612 10.1242/dev.01429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S.C., Wu H., Xie J., Chang C.P., Ranish J.A., Graef I.A., Crabtree G.R. 2009. Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science. 323:651–654 10.1126/science.1166562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur R.P. 1999. Early death of neural crest cells is responsible for total enteric aganglionosis in Sox10(Dom)/Sox10(Dom) mouse embryos. Pediatr. Dev. Pathol. 2:559–569 10.1007/s100249900162 [DOI] [PubMed] [Google Scholar]
- Kellerer S., Schreiner S., Stolt C.C., Scholz S., Bösl M.R., Wegner M. 2006. Replacement of the Sox10 transcription factor by Sox8 reveals incomplete functional equivalence. Development. 133:2875–2886 10.1242/dev.02477 [DOI] [PubMed] [Google Scholar]
- Kim J., Lo L., Dormand E., Anderson D.J. 2003. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 38:17–31 10.1016/S0896-6273(03)00163-6 [DOI] [PubMed] [Google Scholar]
- Kuhlbrodt K., Herbarth B., Sock E., Hermans-Borgmeyer I., Wegner M. 1998. Sox10, a novel transcriptional modulator in glial cells. J. Neurosci. 18:237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N., Nagarajan R., Wang J.Y., Araki T., Schmidt R.E., Milbrandt J. 2005. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc. Natl. Acad. Sci. USA. 102:2596–2601 10.1073/pnas.0407836102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc S.E., Ward R.M., Svaren J. 2007. Neuropathy-associated Egr2 mutants disrupt cooperative activation of myelin protein zero by Egr2 and Sox10. Mol. Cell. Biol. 27:3521–3529 10.1128/MCB.01689-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A., Schlierf B., Schardt A., Nave K.A., Wegner M. 2004. Sox10-rtTA mouse line for tetracycline-inducible expression of transgenes in neural crest cells and oligodendrocytes. Genesis. 40:171–175 10.1002/gene.20083 [DOI] [PubMed] [Google Scholar]
- Maka M., Stolt C.C., Wegner M. 2005. Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Dev. Biol. 277:155–169 10.1016/j.ydbio.2004.09.014 [DOI] [PubMed] [Google Scholar]
- Martini R. 1997. Animal models for inherited peripheral neuropathies. J. Anat. 191:321–336 10.1046/j.1469-7580.1997.19130321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R., Fischer S., López-Vales R., David S. 2008. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia. 56:1566–1577 10.1002/glia.20766 [DOI] [PubMed] [Google Scholar]
- Nodari A., Zambroni D., Quattrini A., Court F.A., D’Urso A., Recchia A., Tybulewicz V.L., Wrabetz L., Feltri M.L. 2007. β1 integrin activates Rac1 in Schwann cells to generate radial lamellae during axonal sorting and myelination. J. Cell Biol. 177:1063–1075 10.1083/jcb.200610014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratore C., Goerich D.E., Suter U., Wegner M., Sommer L. 2001. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development. 128:3949–3961 [DOI] [PubMed] [Google Scholar]
- Parmantier E., Lynn B., Lawson D., Turmaine M., Namini S.S., Chakrabarti L., McMahon A.P., Jessen K.R., Mirsky R. 1999. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron. 23:713–724 10.1016/S0896-6273(01)80030-1 [DOI] [PubMed] [Google Scholar]
- Peirano R.I., Goerich D.E., Riethmacher D., Wegner M. 2000. Protein zero gene expression is regulated by the glial transcription factor Sox10. Mol. Cell. Biol. 20:3198–3209 10.1128/MCB.20.9.3198-3209.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingault V., Bondurand N., Kuhlbrodt K., Goerich D.E., Préhu M.-O., Puliti A., Herbarth B., Hermans-Borgmeyer I., Legius E., Matthijs G., et al. 1998. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18:171–173 10.1038/ng0298-171 [DOI] [PubMed] [Google Scholar]
- Reiprich S., Kriesch J., Schreiner S., Wegner M. 2010. Activation of Krox20 gene expression by Sox10 in myelinating Schwann cells. J. Neurochem. 112:744–754 10.1111/j.1471-4159.2009.06498.x [DOI] [PubMed] [Google Scholar]
- Rodríguez C.I., Buchholz F., Galloway J., Sequerra R., Kasper J., Ayala R., Stewart A.F., Dymecki S.M. 2000. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25:139–140 10.1038/75973 [DOI] [PubMed] [Google Scholar]
- Schreiner S., Cossais F., Fischer K., Scholz S., Bösl M.R., Holtmann B., Sendtner M., Wegner M. 2007. Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development. 134:3271–3281 10.1242/dev.003350 [DOI] [PubMed] [Google Scholar]
- Southard-Smith E.M., Kos L., Pavan W.J. 1998. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 18:60–64 10.1038/ng0198-60 [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.S., William C.M., Tanabe Y., Jessell T.M., Costantini F. 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1:4 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt C.C., Rehberg S., Ader M., Lommes P., Riethmacher D., Schachner M., Bartsch U., Wegner M. 2002. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 16:165–170 10.1101/gad.215802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S.L., De Vries G.H. 2007. Angiogenic expression profile of normal and neurofibromin-deficient human Schwann cells. Neurochem. Res. 32:1129–1141 10.1007/s11064-007-9279-z [DOI] [PubMed] [Google Scholar]
- Topilko P., Schneider-Maunoury S., Levi G., Baron-Van Evercooren A., Chennoufi A.B., Seitanidou T., Babinet C., Charnay P. 1994. Krox-20 controls myelination in the peripheral nervous system. Nature. 371:796–799 10.1038/371796a0 [DOI] [PubMed] [Google Scholar]
- Wegner M. 1999. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 27:1409–1420 10.1093/nar/27.6.1409 [DOI] [PMC free article] [PubMed] [Google Scholar]