

Abstract
The BDNF receptor, TrkB, is critical to limbic epileptogenesis, but the responsible downstream signaling pathways are unknown. We hypothesized that TrkB-dependent activation of phospholipase Cγ1 (PLCγ1) signaling is the key pathway and tested this in trkBPLC/PLC mice carrying a mutation (Y816F) that uncouples TrkB from PLCγ1. Biochemical measures revealed activation of both TrkB and PLCγ1 in hippocampi in the pilocarpine and kindling models in wild-type mice. PLCγ1 activation was decreased in hippocampi isolated from trkBPLC/PLC compared with control mice. Epileptogenesis assessed by development of kindling was inhibited in trkBPLC/PLC compared with control mice. Long-term potentiation of the mossy fiber-CA3 pyramid synapse was impaired in slices of trkBPLC/PLC mice. We conclude that TrkB-dependent activation of PLCγ1 signaling is an important molecular mechanism of limbic epileptogenesis. Elucidating signaling pathways activated by a cell membrane receptor in animal models of CNS disorders promises to reveal novel targets for specific and effective therapeutic intervention.
Introduction
Understanding the mechanisms of limbic epileptogenesis in cellular and molecular terms may lead to novel and specific therapies aimed at preventing onset and/or progression of this disorder. Extensive experimental evidence supports the assertion that the neurotrophin, BDNF, promotes limbic epileptogenesis by activation of its cognate receptor, TrkB. Expression of BDNF is dramatically increased following a seizure in multiple animal models (Ernfors et al., 1991; Isackson et al., 1991; Springer et al., 1994). BDNF mRNA (Murray et al., 2000) and protein content (Takahashi et al., 1999) are also increased in the hippocampus of humans with temporal lobe epilepsy. Enhanced activation of TrkB has been identified in multiple models of limbic epilepsy (Binder et al., 1999; He et al., 2002; Danzer et al., 2004). Administration of BDNF and transgenic overexpression of BDNF enhance limbic epileptogenesis (Croll et al., 1999; Xu et al., 2004). Striking impairments of epileptogenesis in the kindling model were identified in mice carrying only a single BDNF allele (Kokaia et al., 1995), while epileptogenesis was eliminated altogether in mice with a conditional deletion of TrkB in the CNS (He et al., 2004).
Insight into the signaling pathways by which TrkB activation promotes limbic epileptogenesis in vivo may provide clues to the underlying cellular mechanisms as well as novel targets for therapy. BDNF binding to TrkB results in receptor dimerization, enhanced activity of the TrkB tyrosine kinase which results in phosphorylation of Y515 and Y816 in the intracellular domain of TrkB, thereby creating docking sites for adaptor proteins Shc and PLCγ1 respectively. Both Shc and PLCγ1 are phosphorylated by TrkB, thereby initiating Shc/Ras/MAP kinase and PLCγ1 signaling respectively. Because epileptogenesis was similar in controls and trkBSHC/SHC mutant mice (He at al., 2002), we hypothesized that PLCγ1 signaling was activated during epileptogenesis in a TrkB-dependent manner and that this activation promotes limbic epileptogenesis. Substitution of phenylalanine for tyrosine at residue 816 of TrkB (pY816 TrkB) in the trkBPLC/PLC mice selectively eliminates binding and phosphorylation of PLCγ1 by TrkB (Minichiello et al., 2002), thereby permitting study of functional consequences of TrkB-mediated activation of PLCγ1 in vivo.
Materials and Methods
Mice.
Animals were handled according to National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by Duke University Animal Care and Welfare Committee.
TrkBPLC/PLC mutant mice in a C57BL/6-129 background were generated by cDNA knockin approach as described previously (Minichiello et al., 2002). In brief, PCR-based site-directed mutagenesis was used on mouse TrkB cDNA to induce a single point mutation (A to T position 2958) that resulted in substituting phenylalanine for tyrosine 816 (Y816F), thereby disrupting the binding of PLCγ1. The mutant TrkB cDNA (TrkBPLC) and control wild-type (WT) TrkB cDNA (TrkBWT) were knocked into the juxtamembrane exon of the mouse trkB gene. Wild-type (+/+), homozygous mutant trkB (trkBPLC/PLC) and WT knockin trkB (trkBWT/WT) mice were used in this study. In addition, trkBSHC/SHC mutant mice were used in one experiment. trkBSHC/SHC mutant mice were generated as described previously (Minichiello et al., 1998). In brief, PCR-aided mutagenesis was used to introduce a single point mutation (A to T, position 2055) in the trkB receptor that substituted phenylalanine for tyrosine 515 (Y515F). Nonphosphorylatable F515 disrupted the binding of adaptor protein Shc to trkB and abolished Shc site-mediated downstream signaling events.
The genotype of each animal was assessed twice using PCR of genomic DNA isolated from tails (before and after experiments) as previously described (He et al., 2002). In addition to PCR, the genotype of all mice used in the kindling experiments was confirmed by sequencing.
Pilocarpine-induced status epilepticus.
A single intraperitoneal (i.p.) injection of pilocarpine, a muscarinic cholinergic agonist, was administered to induce status epilepticus (SE). To minimize peripheral cholinergic effects, male and female C57BL/6 mice of age 2–3 months were treated with N-methyl scopolamine nitrate (1 mg/kg, i.p.) (Sigma). Fifteen minutes later, either pilocarpine (375 mg/kg) (Sigma) or vehicle (normal saline) was injected i.p. and mice were observed for the appearance of seizure activity and onset of SE for the next 3–4 h. Seizures were classified according to Racine (1972) with slight modifications (Borges et al., 2003). Status epilepticus was defined as occasional or frequent myoclonic jerks, partial-or whole-body clonus, shivering, loss of posture, and /or rearing and falling that was not interrupted by periods of normal behavior. After 3 h of continuous seizure activity, diazepam (10 mg/kg, i.p.) (Hospira) was administered to terminate SE. Pilocarpine-treated animals that failed to exhibit SE or did not survive SE were excluded from the study. Unless specified otherwise, both pilocarpine- and saline-treated mice were decapitated 6 h after the onset of SE for biochemical and immunohistochemical experiments.
To ascertain that pilocarpine-induced status epilepticus assessed by behavioral measures was associated with hippocampal electrographic seizure, a pilot experiment was performed in which a bipolar recording electrode was placed in the right dorsal hippocampus using stereotaxic guidance (2.0 mm posterior and 1.6 mm lateral to bregma and 1.5 mm below dura) under pentobarbital anesthesia. One week thereafter animals were given N-methyl scopolamine and pilocarpine as described in the preceding paragraph; 3 h after onset, status epilepticus was terminated by diazepam. EEG recordings revealed electrographic seizure activity in hippocampus in all animals (3 +/+, 2 trkBWT/WT, and 3 trkBPLC/PLC), the duration of which corresponded to the duration of status epilepticus assessed by behavioral measures (data not shown). Behavioral measures alone were used to assess status epilepticus for the remainder of the experiments with the pilocarpine model.
Surgery and kindling.
Twelve +/+, 12 trkBWT/WT and 10 trkBPLC/PLC mice were included in the kindling experiment. Procedures for surgery and kindling were performed as described previously (He et al., 2002, 2004) by an individual blinded to genotype of the animals. Briefly, under pentobarbital (60 mg/kg) (Ovation) anesthesia, a bipolar electrode used for stimulation and recording was stereotactically implanted in the right amygdala. Following a postoperative recovery period of 2 weeks, the electrographic seizure threshold (EST) in the amygdala was determined and stimulations at the intensity of the EST were subsequently administered twice daily, 5 d per week as described previously (He et al., 2002, 2004). The behavioral manifestations of seizures were classified according to a modification of the description of Racine (1972) as described previously. Mice were stimulated until fully kindled as defined by the occurrence of 3 consecutive seizures of class 4 or greater. Unstimulated control animals of each genotype underwent surgical implantation of an electrode in amygdala and were handled identically but were not stimulated. Six hours after the last stimulation, the stimulated and unstimulated mice were decapitated for further study. Accuracy of electrode placements were verified by histological analysis and only animals with correct electrode placement in the amygdala were included in the statistical analysis for kindling experiment. All kindling data are presented as mean ± SEM and analyzed by one-way ANOVA with post hoc Bonferroni's test.
Biochemistry.
Following decapitation, the mouse head was quickly dipped into liquid nitrogen for 4 s to rapidly cool the brain. The hippocampi were rapidly dissected on ice and homogenized in lysis buffer [20 mm Tris, pH 8.0, 137 mm NaCl, 1% NP40, 10% glycerol, 1 mm sodium orthovanadate (NaOV), 1 mm phenylmethylsulfonylfuoride (PMSF), and 1 Complete Mini protease inhibitor tablet (Mini, Roche)/10 ml]. The supernatant was saved following centrifugation at 16,000 × g for 10 min, aliquoted and stored at −80°C for further biochemical analysis.
In experiments studying a synaptosomal membrane fraction, hippocampi were homogenized in an isotonic sucrose buffer (0.32 m sucrose, 4 mm HEPES, 1 mm NaOV, 1 mm PMSF, and 1 Mini tablet/10 ml, pH 7.4), centrifuged at 325 × g for 10 min at 4°C, and the supernatant was collected and centrifuged at 16,000 × g for 15 min to provide a crude synaptosomal pellet. Crude synaptosomes underwent osmotic shock by addition of ice-cold deionized H2O and rapidly returned to osmotic balance with 1 m HEPES pH 7.4; following centrifugation at 16,000 × g for 30 min, the pellet consisting of an enriched synaptosomal membrane fraction was collected. BCA kit (Thermo Scientific) was used to determine the protein concentration.
Western blotting was performed to analyze phosphorylated and nonphosphorylated TrkB and PLCγ1 using procedures as described previously (He et al., 2004; Huang et al., 2008). The following antibodies were used in these experiments: p-Trk (Y816) (a gift from Dr. Moses Chao, New York University, New York, NY); p-PLCγ1 (Y783) (Biosource); TrkB (BD Biosciences); PLCγ1 (Cell Signaling Technology); β-actin (Sigma). The results from Western blotting were quantified by a method described previously (Huang et al., 2008). Briefly, the immunoreactivity of individual band on Western blots was measured by ImageQuant software and normalized to TrkB or β-actin content; similar results were obtained with the two methods. Student's t test and one-way ANOVA were used for statistical analyses. Results are presented as mean ± SEM for the designated number of experiments.
P-Trk immunohistochemistry.
P-Trk immunohistochemistry was performed using the protocol described previously (Danzer and McNamara, 2004; Danzer et al., 2010). Briefly, under pentobarbital anesthesia (200 mg/kg), mice were perfused with 4% paraformaldehyde in PBS and the brains were removed, postfixed and cryoprotected. Forty micrometer coronal sections were cut and used for immunofluorescent staining. After 1 h incubation with blocking solution (5% NGS, 0.5% NP40 in PBS buffer with 1 mm NaOV), pY816 antibody was applied to floating sections overnight at 4°C. Alexa Fluor 594 goat anti-rabbit secondary antibody (Invitrogen) was used to visualize the immunofluorescent staining. The sections from experimental and control animals of different genotypes were processed simultaneously in the same incubation plates using the identical solutions and protocol so that valid comparisons could be made. Images were captured and quantified using a Leica TCS SL confocal system. Immunoreactivity over the corpus callosum was sampled in each section as internal control because of its low immunoreactivity. In addition values were collected from a square of fixed size over CA1 stratum oriens, CA1 stratum lacunosum-moleculare, and CA3a stratum lucidum (supplemental Fig. 1b, available at www.jneurosci.org as supplemental material) and presented as percentage of value of corpus callosum. The specificity of pY816 antibody for TrkB pY816 was verified by the reductions of immunoreactivity in stratum lucidum of trkBPLC/PLC compared with control mice (supplemental Fig. 1a, available at www.jneurosci.org as supplemental material). All results from experimental mice and their controls were analyzed by Student's t test.
Hippocampal slice preparation and electrophysiology.
Mice (postnatal day 28–42) were anesthetized with pentobarbital and decapitated. The brain was quickly removed and placed in ice-cold buffer containing the following (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7.0 MgCl2, and 5 dextrose, saturated with 95% O2 plus 5% CO2, pH 7.4. Following dissection of hippocampi, transverse slices (400 μm in thickness) were cut with a vibratome and incubated in oxygenated artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 1.75 KCl, 1.25 KH2PO4, 26 NaHCO3, 2.4 CaCl2, 1.3 MgCl2, and 10 dextrose for at least 1 h at 32–34° before recording. The slices were then transferred to a recording chamber mounted on Zeiss Axioskop upright microscope.
The following criteria were applied to be considered a mossy fiber excitatory postsynaptic field potentials (fEPSP): (1) the ratio for paired pulse facilitation (PPF) at 60 ms interval was 1.75 or greater; (2) frequency facilitation at 20 Hz was 2.0 or greater as determined by the ratio of the amplitude of the response to the third pulse compared with the first pulse (Toth et al., 2000); and (3) application of the Group II metabotropic glutamate receptor (mGluR) II agonist 2-(2, 3-dicarboxycyclopropy) glycine (DCG-IV) 1 μm at the end of the experiment reduced the amplitude of the evoked fEPSP by at least 70%. Addition of picrotoxin, which blocks feedforward inhibition of CA3 pyramids evoked by mossy fiber activation of interneurons in stratum lucidum, did not modify the latency, amplitude, or waveform of the mossy fiber (mf)-CA3 pyramid fEPSP. The mossy fiber-CA3 pyramid fEPSPs were induced by a bipolar tungsten stimulating electrode placed at the junction of the granule cell layer and hilus near the midpoint of the suprapyramidal blade of the dentate. Extracellular recordings were obtained with a glass micropipette filled with 2 m NaCl, 2–6 MΩ resistance placed in stratum lucidum near the junction of CA3a and CA3b. An input-output curve was obtained by hilar stimulation (0.2 ms square pulses delivered at 0.03 Hz) with a Digitmer constant current stimulator (DS3, Digitimer Ltd.). A stimulus intensity sufficient to induce a fEPSP amplitude approximating 30% of the maximum amplitude was used for these experiments. D, L-APV (100 μm) was included in perfusion solution to eliminate contamination of associational-commissural afferents (Zalutsky and Nicoll, 1990). LTP was induced by applying a total of 4 trains of high-frequency stimulation (HFS) (each train consisting of 0.2 ms pulses at 100 Hz for 1 s and intensity sufficient to induce maximum fEPSP amplitude and intertrain interval of 10 s). To assure objectivity, the individual performing all experiments with wild-type and mutant mice was blinded as to genotype.
For the LTP experiment, the amplitude of fEPSPs was measured and LTP was plotted as mean percentage change in the fEPSP amplitude 50–60 min after HFS relative to the 10 min of fEPSP amplitude immediately preceding the HFS. The numbers listed in the figure legends and text refer to the number of animals. Results are typically obtained and averaged from at least two slices from each animal and the average value is presented as a single value for each animal. Data were collected from slices at room temperature using a Multi 700A amplifier and pClamp 9.2 software (Molecular Devices). The synaptic responses were filtered at 2 kHz and digitized at 5 kHz. All data were presented as mean ± SEM and analyzed by Student's t test with Excel (Microsoft) and Prism (GraphPad Software) software.
Results
Biochemical study of TrkB and PLCγ signaling during limbic epileptogenesis
Induction of continuous seizure activity for a couple h by systemically administered pilocarpine is followed by emergence of spontaneous recurrent seizures arising weeks thereafter, thereby recapitulating some features of temporal lobe epilepsy (TLE) in humans (Lemos and Cavalheiro, 1995; Klitgaard et al., 2002). To test whether TrkB and PLCγ1 underwent activation in the pilocarpine model, Western blots were prepared from hippocampal homogenates isolated from wild-type (+/+) mice 6 h following the onset of status epilepticus induced by injection of pilocarpine. Status epilepticus was associated with increased tyrosine phosphorylation of Trk as evidenced by increased immunoreactivity of a 145 kDa band detected by an antibody specific to pY816 Trk (Fig. 1 a, top). Note that the increased size of the pY816 Trk band in the status epilepticus treatment (Fig. 1 a, top) compared with vehicle is similar to that observed by Iwakura et al. (2008), (see Fig. 4) upon BDNF treatment of heterologous cells expressing TrkB using the same antibody; the increased size of the band likely reflects TrkB molecules phosphorylated to different extents resulting in small differences of migration within the SDS gel. No significant increase of TrkB content was detected (Fig. 1 a, top). Quantitative analysis of Western blot data 6 h after onset of status epilepticus revealed a 2.3-fold increase of pY816 relative to TrkB in the pilocarpine-treated group (n = 7) compared with normal saline (NS) controls (n = 6) (p < 0.05), Student's t test. The increased pY816 immunoreactivity was time dependent as revealed by modest increases evident at 30 min and 3 h, more marked increases at 6–24 h, and a return to baseline values 1 week later (Fig. 1 a, bottom). The 3.5-fold increase of pY816 Trk relative to TrkB in 6 h group (a separate group from that with 2.3-fold increase described above) is significantly higher than in NS controls (p < 0.001, one-way ANOVA).
Figure 1.

TrkB-PLCγ1 signaling is increased in the pilocarpine (pilo) model. a, Top, Representative Western blot of pY816 TrkB and TrkB in hippocampal homogenate isolated 6 h after onset of status epilepticus. Bottom, Quantitative analysis of Western blot of pY816 TrkB at multiple times (30 min, 3 h, 6 h, 12 h, 24 h and 1 week) after onset of pilo-induced status epilepticus. The-fold increase of pY816 Trk relative to TrkB in 6 h group is significantly higher than in NS controls (p < 0.001). Western blots were quantified and presented as mean ± SEM of fold increase of pY816 relative to TrkB in pilo mice (n = 4 for each time point) compared with NS controls (n = 4). Note that different groups of animals were studied at 6 h after pilo in bottom panel compared with top panel. b, Top, Representative Western blot of pY783 PLCγ1 and PLCγ1 in hippocampal homogenate isolated 6 h after onset of status epilepticus. Bottom, Quantitative analysis of Western blot of pY783 relative to PLCγ1 immunoreactivity at multiple times after onset of pilo-induced status epilepticus. The fold increases of pY783 PLCγ1 relative to PLCγ1 in 6 h (p < 0.01) and 12 h (p < 0.001) groups are significantly higher than in NS controls. Data are presented as mean ± SEM of fold increase of pY783 relative to PLCγ1 in pilo mice (n = 4 for each time point) compared with NS controls (n = 4). Note that different groups of animals were studied at 6 h after pilo in the bottom panel compared with the top panel.
Figure 4.
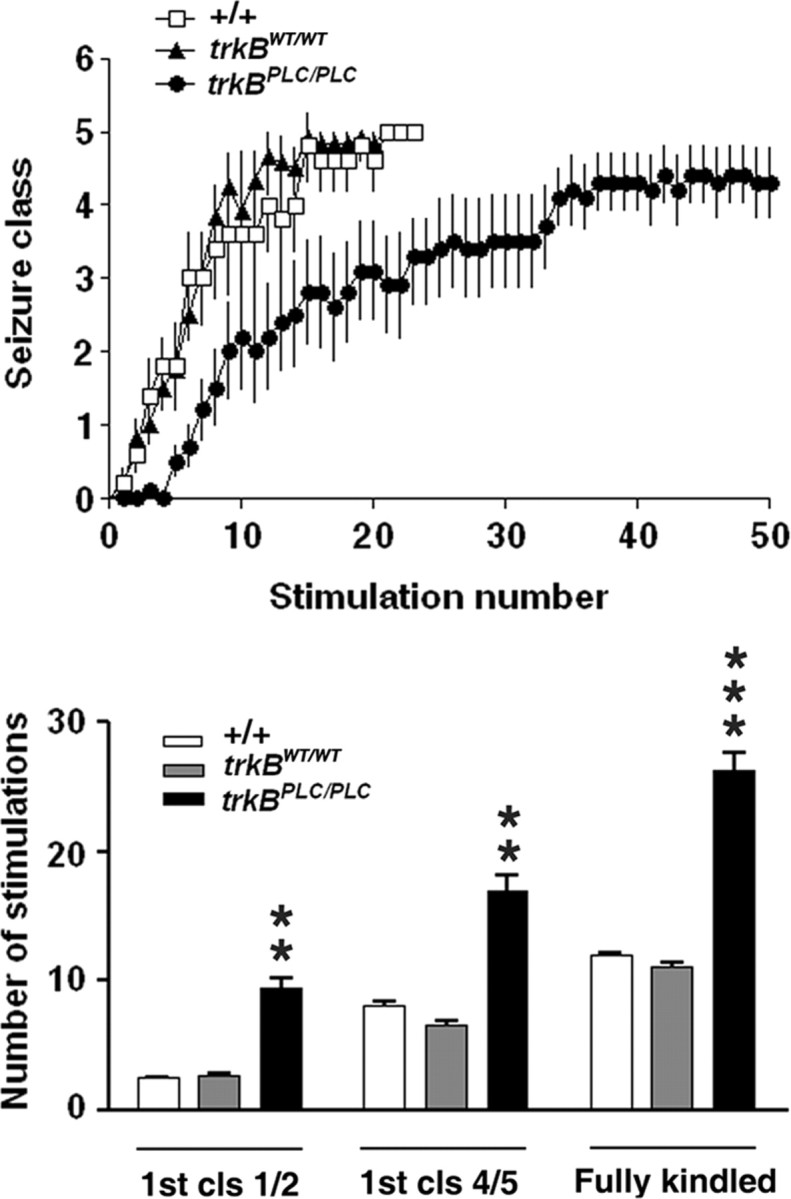
Top, Kindling development is inhibited in trkBPLC/PLC mutants. Kindling development is presented as behavioral seizure class (y-axis). Stimulation number (x-axis) refers to the number of stimulations that evoked an electrographic seizure with duration of at least 5 s. Bottom, number of stimulations required to reach different seizure classes in wild-type (+/+) (n = 12), trkBWT/WT (n = 12), and trkBPLC/PLC (n = 10). Fully kindled stage is defined by the occurrence of three consecutive seizures of class 4 or 5. For the number reaching first class1 or 2, +/+ versus trkBPLC/PLC, p < 0.01; trkBWT/WT versus trkBPLC/PLC, p < 0.01. For the number reaching first class 4 or 5, +/+ versus trkBPLC/PLC, p < 0.05; trkBWT/WT versus trkBPLC/PLC, p < 0.01. For the number reaching fully kindled stage, +/+ versus trkBPLC/PLC, p < 0.01; trkBWT/WT versus trkBPLC/PLC, p = 0.001. All data are presented as mean ± SEM; one-way ANOVA with post hoc Bonferroni's test.
Because phosphorylation of Y816 of TrkB activates PLCγ1 signaling in vitro in cultured neurons and recombinant systems, the increased pY816 immunoreactivity predicted enhanced activation of PLCγ1 itself. Consistent with this prediction, increased immunoreactivity of a 150 kDa band detected by an antibody specific to pY783 PLCγ1 was evident in hippocampal homogenates isolated 6 h after onset of pilocarpine-induced status epilepticus (Fig. 1 b, top). No change in content of PLCγ1 itself was found (Fig. 1 b). Quantitative analysis of Western blot data 6 h after onset of status epilepticus revealed a 1.8-fold increase of pY783 relative to PLCγ1 in pilo (n = 7) compared with NS controls (n = 6) (p < 0.01), Student's t test. The increased pY783 immunoreactivity was also time dependent as revealed by modest increases evident at 30 min and 3 h, more marked increases at 6–12 h, and a return to baseline values 1 week later (Fig. 1 b, bottom). The 2.7- and 3.4-fold increases of pY783 PLCγ1 relative to PLCγ1 at 6 and 12 h respectively are significantly higher in the pilo-treated group compared with NS controls (6 h vs NS, p < 0.01; 12 h vs NS, p < 0.001, one-way ANOVA).
To test whether TrkB and PLCγ signaling were activated in a distinct model of limbic epileptogenesis, Western blots were prepared from hippocampal homogenates isolated from wild-type mice 6 h following a class 4/5 kindled seizure evoked by amygdala stimulation. The kindled seizure also resulted in increased pY816 Trk immunoreactivity (Fig. 2 a). No significant increase of TrkB content was detected (Fig. 2 a). Quantitative analyses of Western blots revealed a 1.8-fold increase of pY816 relative to TrkB in mice killed 6 h after a class 4/5 kindled seizure (K) (n = 4) compared with unstimulated controls (C) (n = 3) (p < 0.05), Student's t test. Consistent with this increase of pY816 Trk immunoreactivity, a kindled seizure also induced increased tyrosine phosphorylation of PLCγ1 itself 6 h afterward as evidenced by increased pY783 PLCγ1 immunoreactivity (Fig. 2 b). No change in content of PLCγ1 itself was detected (Fig. 2 b). The 1.9-fold increase of pY783 relative to PLCγ1 in K (n = 4) compared with C (n = 3) was significant (p < 0.05), Student's t test.
Figure 2.

TrkB-PLCγ1 signaling is increased in the kindling model. a, Representative Western blot of pY816 TrkB and TrkB in hippocampal homogenate isolated 6 h after last stimulation-induced class 4/5 kindled seizure. b, Representative Western blot of pY783 PLCγ1 and PLCγ1 in hippocampal homogenate isolated 6 h after last Class 4/5 kindled seizure.
The correlation of increased pY816 Trk and pY783 PLCγ1 immunoreactivity at 6 h after seizures in two distinct models of limbic epilepsy together with similarity of time course in the pilocarpine model provided circumstantial evidence that the enhanced PLCγ1 activation induced by status epilepticus was a consequence of TrkB activation. The availability of trkBPLC/PLC mice in which substitution of phenylalanine for tyrosine at residue 816 of TrkB selectively eliminates binding and phosphorylation of PLCγ1 by TrkB enabled us to test directly in vivo whether activation of PLCγ1 during status epilepticus was a consequence of TrkB activation. We first examined pY816 Trk immunoreactivity in synaptic membranes isolated from trkBWT/WT and trkBPLC/PLC mice isolated 6 h following status epilepticus. Consistent with findings in Figure 1, status epilepticus was associated with increased pY816 Trk immunoreactivity in hippocampal synaptic membranes isolated from trkBWT/WT mice (Fig. 3, top). Quantification of pY816 immunoreactivity revealed a 1.6-fold increase in trkBWT/WT animals killed 6 h after status epilepticus (Fig. 3 a, bottom, n = 3, p < 0.001). Analysis of pY816 immunoreactivity in trkBPLC/PLC following treatment with normal saline revealed a 40% reduction compared with trkBWT/WT animals (Fig. 3 a, n = 3, p < 0.05), demonstrating that phosphorylation of pY816 of TrkB itself contributes to pY816 immunoreactivity measured under basal conditions. Likewise following status epilepticus, the pY816 immunoreactivity in trkBWT/WT exceeded that in trkBPLC/PLC mice by 1.7-fold (Fig. 3 a, n = 3, p < 0.001), demonstrating that the increased pY816 immunoreactivity following status epilepticus is due mainly to phosphorylation of TrkB. A small increase of pY816 immunoreactivity of 145 kDa band was evident following status epilepticus in trkBPLC/PLC mice (Fig. 3 a, n = 3, p < 0.05), suggesting the possibility that status epilepticus may also result in increased pY816 immunoreactivity of TrkC.
Figure 3.

Effect of trkBPLC/PLC mutation on TrkB-PLCγ signaling. a, Top, Representative Western blot of pY816 TrkB and TrkB in hippocampal synaptosomal membranes isolated 6 h after onset of pilo-induced status epilepticus from trkBPLC/PLC or trkBWT/WT mice. Bottom, Quantitative analysis of Western blot. The fold increases of pY816 to TrkB from 3 experiments in trkBPLC/PLC were compared with that from trkBWT/WT mice. One-way ANOVA (p < 0.001). b, Top, Representative Western blot of pY783 PLCγ1 and PLCγ1 in hippocampal synaptosomal membranes isolated 6 h after onset of pilo-induced status epilepticus from trkBPLC/PLC or trkBWT/WT mice. Bottom, Quantitative analysis of Western blot. The fold increases of p-PLCγ1 relative to PLCγ1 from 3 experiments in trkBPLC/PLC were compared with that from trkBWT/WT mice. Data are presented as means ± SEM, one-way ANOVA (p < 0.01).
Next we asked whether the status epilepticus-induced activation of PLCγ1 was dependent upon TrkB activation, again probing Western blots of hippocampal synaptic membranes isolated from trkBWT/WT and trkBPLC/PLC with an antibody specific to pY783 PLCγ1. Increased pY783 PLCγ1 immunoreactivity was evident following status epilepticus in trkBWT/WT mice (Fig. 3 b, top). Quantification of the pY783 immunoreactivity revealed a 2.0-fold increase in trkBWT/WT animals killed 6 h after status epilepticus (Fig. 3 b, bottom, n = 3, p = 0.051). Analysis of pY783 PLCγ1 immunoreactivity in trkBPLC/PLC following treatment with normal saline revealed a 38% reduction compared with trkBWT/WT animals which was not statistically significant (Fig. 3 b, n = 3, p > 0.05). Following status epilepticus, pY783 PLCγ1 immunoreactivity in trkBWT/WT exceeded that in trkBPLC/PLC mice by 1.9-fold (Fig. 3 b, n = 3, p < 0.05), demonstrating that the status epilepticus-induced increase of pY783 PLCγ1 is due predominantly to TrkB activation. The small absolute increase of pY783 PLCγ1 immunoreactivity in trkBPLC/PLC mice following status epilepticus (Fig. 3 b, n = 3, p > 0.05) was not statistically significant.
Effect of limiting TrkB-dependent PLCγ1 signaling on limbic epileptogenesis in vivo
The evidence of enhanced TrkB-dependent activation of PLCγ1 signaling during status epilepticus together with previous evidence of a requirement for TrkB for induction of epileptogenesis in the kindling model (He et al., 2004) raised the question as to whether TrkB activation of PLCγ1 signaling is critical to epileptogenesis. To address this question, we examined epileptogenesis in the kindling model in trkB PLC/PLC mice that selectively prevents activation of the PLCγ1 signaling pathway by TrkB. trkBPLC/PLC mice exhibited a marked inhibition of the rate of kindling development as evident in the increased number of stimulations required to elicit behavioral seizures compared with both +/+ and trkBWT/WT mice (Fig. 4, top). The number of stimulations required to evoke a limbic seizure termed class 1 or 2 (Fig. 4, bottom) was increased by >3-fold in trkBPLC/PLC mice (9.5 ± 2.5, n = 10) compared with either of two controls (+/+ 2.5 ± 0.5, n = 12, p < 0.01) (trkBWT/WT, 2.8 ± 0.4, n = 12, p < 0.01). Likewise the number of stimulations required to evoke the third consecutive clonic tonic seizure (class 4 or greater) (Fig. 4, bottom) was increased by >2-fold in trkBPLC/PLC (26.2 ± 4.6) compared with either of two controls (+/+ 12.0 ± 0.9, p < 0.01) or trkBWT/WT (11.1 ± 1.0, p = 0.001). By contrast, no significant difference was evident in the electrographic seizure duration during kindling development among 3 genotypes. Likewise no significant differences were detected in the current required to evoke an initial electrographic seizure duration in the three groups (+/+ 150.0 ± 27.3 μA; trkBWT/WT 172.7 ± 24.5 μA; trkBPLC/PLC 128 ± 14.1 μA; p > 0.05). Together, these results demonstrate that selectively limiting activation of PLCγ signaling by TrkB markedly inhibits epileptogenesis in the kindling model.
Immunohistochemical localization of pY816 Trk Immunoreactivity in limbic epileptogenesis
The pivotal role of TrkB-dependent PLCγ1 signaling in epileptogenesis in the kindling model raised the question as to potential cellular consequences of the enhanced activation of TrkB and PLCγ1 that might contribute to epileptogenesis. Insight into the anatomic locale of the enhanced TrkB activation would provide a valuable clue as to the nature and locale of potential cellular mechanisms. Our previous results provided immunohistochemical evidence that TrkB receptors undergo increased phosphorylation during epileptogenesis in a spatially specific pattern in the hippocampus, that is, increased p-Trk (pY515) was evident in the mossy fiber pathway in multiple models (Binder et al., 1999; He et al., 2002). That said, the anatomic locale of enhanced pY816 Trk immunoreactivity detected by Western blotting in the pilocarpine and kindling models is unknown. To address this question, we performed pY816 immunohistochemistry in these models.
The immunohistochemical pattern in sections prepared from WT mice killed 6 h after onset of status epilepticus revealed increased pY816 Trk immunoreactivity in the stratum lucidum of CA3a bilaterally (only one hippocampus shown) in all brain sections examined (Fig. 5 a, top); no overt changes of p-Trk immunoreactivity were noted elsewhere in the hippocampus. Quantification revealed a 1.7-fold increase of pY816 immunoreactivity in CA3a stratum lucidum in pilocarpine (n = 6) compared with normal saline (n = 5)-treated animals (p < 0.05) (Fig. 5 a, bottom). By contrast, no significant changes were detected in stratum oriens or lacunosum-moleculare of CA1. Like the pilocarpine model, increased pY816 Trk immunoreactivity was detected in the mossy fiber pathway of hippocampus bilaterally of animals killed 6 h after the last class 4/5 seizure evoked by amygdala stimulation in the kindling model compared with sham-stimulated controls (Fig. 5 b, top). Quantification revealed 2.6-fold increase of pY816 immunoreactivity in CA3a stratum lucidum in kindled (n = 4) compared with control group (n = 3) (p < 0.05) (Fig. 5 b, bottom). By contrast, no significant changes were detected in stratum oriens or lacunosum-moleculare of CA1.
Figure 5.

Immunohistochemical localization of pY816 TrkB Immunoreactivity in limbic epileptogenesis. a, pY816 immunoreactivity is increased in pilo model. Top, representative images in low magnification (Low mag) and high magnification (High mag) from stratum lucidum of CA3a in hippocampus of pY816 immunoreactivity in sections prepared 6 h after onset of status epilepticus. Note that the increased pY816 immunoreactivity was found mainly in the mossy fiber pathway as denoted by arrowheads. Bottom, Quantitative analysis of pY816 immunoreactivity in hippocampal subregions of mice treated with NS or after 6 h of pilo-induced status epilepticus (pilo). The pY816 immunoreactivity in CA3a stratum lucidum was increased 1.7 fold in pilo (n = 6) compared with NS (n = 5)-treated mice (p < 0.05, Student's t test). b, pY816 immunoreactivity is increased in the kindling model. Top, Representative images in low magnification (Low mag) and high magnification (High mag) of pY816 TrkB immunoreactivity in hippocampal sections prepared 6 h after last stimulation-induced class 4/5 kindled seizure. Note the increased pY816 immunoreactivity in the mossy fiber pathway as denoted by arrowheads. Bottom, quantitative analysis of pY816 immunoreactivity in hippocampal subregions of kindled and control mice. The pY816 immunoreactivity in CA3a stratum lucidum was increased 2.6 fold in kindled (n = 4) compared with control group (n = 3) (p < 0.05). Data are presented as means ± SEM, Student's t test. Scale bar, 300 μm in low magnification; 50 μm in high magnification.
Inhibition of LTP of mossy fiber-CA3 pyramid synapse in trkBPLC/PLC mice
The anatomic localization of the increased pY816 Trk immunoreactivity to the mossy fiber pathway directed study of potential cellular consequences of TrkB activation to this locale. One consequence of TrkB activation in this locale that might promote limbic epileptogenesis is development of LTP of the excitatory synapse of mf axons of dentate granule cells with CA3 pyramidal cells. Our previous work demonstrated that inhibiting TrkB kinase activity eliminated LTP of this synapse induced by HFS of the dentate granule cells (Huang et al., 2008). To determine whether TrkB signaling through PLCγ in particular is required for LTP of this synapse, the effects of HFS of the mf on the efficacy of this synapse were compared in trkBPLC/PLC and control mice. Significant (p < 0.01) impairments of HFS-induced LTP of the mf-CA3 pyramid synapse were detected in slices isolated from trkBPLC/PLC (115 ± 3%, n = 7) compared with WT (155 ± 9%, n = 8) or trkBWT/WT (148 ± 4%, n = 7) control mice (Fig. 6). Importantly, no differences in basal synaptic transmission were detected between trkBPLC/PLC and control mice as evident in part by similar ratios of paired pulse facilitation of the fEPSP in the three groups (PPF: +/+, 2.56 ± 0.5, n = 5; trkBPLC/PLC, 1.83 ± 0.3, n = 5, p > 0.05, t test and trkBWT/WT 1.95 ± 0.3, n = 5, p > 0.05, t test). Moreover, the impairment of mf-LTP was specific to the PLCγ1 signaling pathway because no differences in LTP of the mf-CA3 pyramid synapse were detected in trkBSHC/SHC compared with WT control mice (+/+, 144 ± 7%, n = 6; trkBSHC/SHC, 145 ± 7%, n = 5, p > 0.05, t test). Together, these data demonstrate that TrkB-dependent signaling through the PLCγ1 but not the Shc pathway is required for LTP of the mf-CA3 pyramid synapse.
Figure 6.
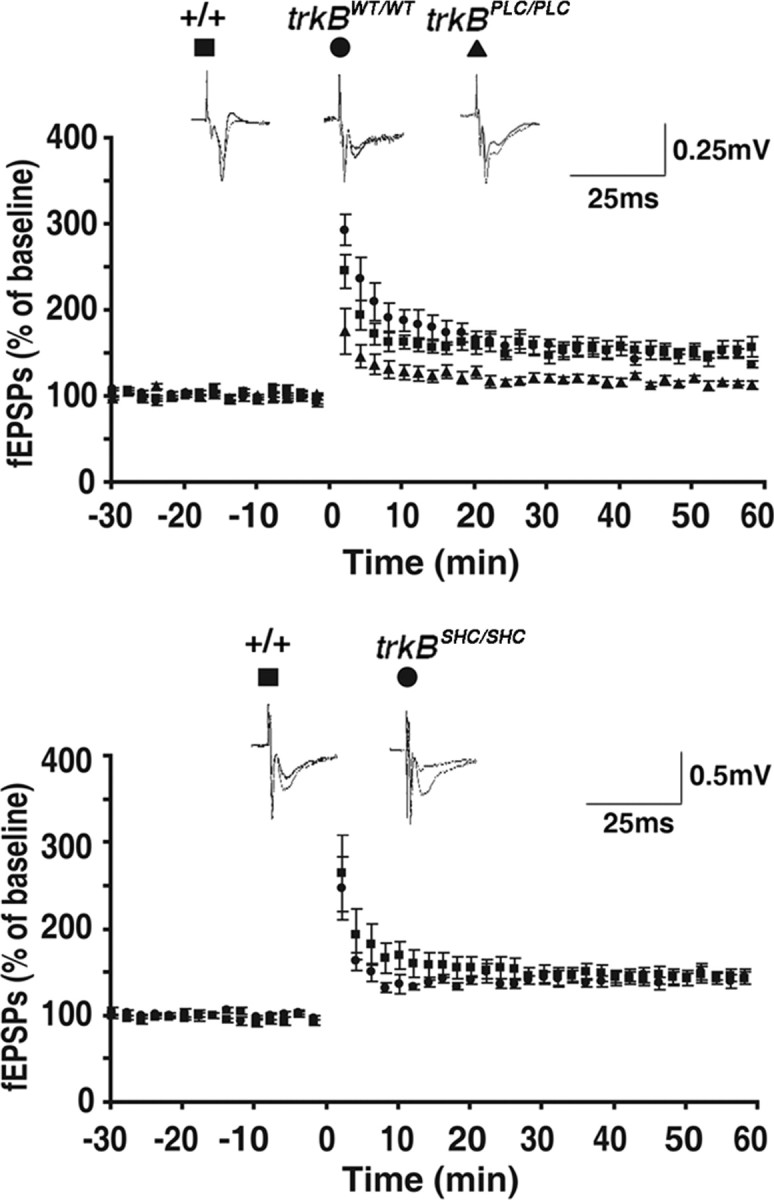
Mf-CA3 LTP is impaired in trkBPLC/PLC mutants. Hippocampal slices were isolated from wild-type or mutant mice and mf-evoked fEPSPs were recorded. Graphs represent mean ± SEM of the responses evoked compared with baseline. Traces of representative experiments are shown above each graph. Top, HFS-induced mf LTP is impaired in trkBPLC/PLC mutant mice. Significant (p < 0.01) impairments of HFS-induced LTP of the mf-CA3 pyramid synapse were detected in slices isolated from trkBPLC/PLC (115 ± 3%, n = 7) compared with WT (155 ± 9%, n = 8) or trkBWT/WT (148 ± 4%, n = 7) control mice. Slices isolated from trkBWT/WT mice exhibited increases of fEPSP (148 ± 4%, n = 7) similar to wild-type animals (+/+) (155 ± 9%, n = 8). Scale bar, 0.25 mV, 25 ms. Bottom, By contrast, no differences in HFS-induced LTP of the mf-CA3 pyramid synapse were detected in trkBSHC/SHC compared with WT control mice (+/+, 144 ± 7%, n = 6; trkBSHC/SHC, 145 ± 7%, n = 5, p > 0.05, Student's t test). Scale bar, 0.5 mV, 25 ms.
Discussion
We hypothesized that the neurotrophin receptor, TrkB, promotes limbic epileptogenesis by activation of the PLCγ1 signaling pathway. We used biochemical, immunohistochemical, and electrophysiological studies of trkBWT/WT and trkBPLC/PLC mice to test this hypothesis. Four principal findings emerged. (1) Time-dependent increases of both pY816 Trk and pY783 PLCγ1 immunoreactivity were detected in hippocampi of WT mice in the pilocarpine and kindling models. The enhanced pY783 PLCγ1 immunoreactivity in the pilocarpine model was decreased in hippocampi isolated from trkBPLC/PLC mice. (2) Limbic epileptogenesis as measured by development of kindling was markedly inhibited in trkBPLC/PLC mice. (3) The enhanced pY816 Trk immunoreactivity in WT mice was selectively localized to the mossy fiber pathway within hippocampus in these models. (4) LTP of the mossy fiber-CA3 pyramid synapse was impaired in slices of trkBPLC/PLC mice. We conclude that activation of pY783 PLCγ1 is due mainly to TrkB activation in these models and that TrkB-induced PLCγ1 signaling promotes limbic epileptogenesis.
The spatial and temporal patterns of TrkB activation are notable. While the precise identity of the endogenous ligand(s) promoting TrkB activation in these models is uncertain, the prototypic agonist of TrkB, BDNF, is a leading candidate. Yet persistence of increased pY515 TrkB following seizures in BDNF conditional knock-out mice (He et al., 2004) led to the discovery that the divalent cation, zinc, can transactivate TrkB by a BDNF independent mechanism in vitro (Huang et al., 2008). The localization of increased pY816 TrkB immunoreactivity exclusively to stratum lucidum is puzzling because both BDNF and zinc are thought to reside in synaptic vesicles of axons of CA3 and CA1 pyramids and to be released during hippocampal seizures; this should result in increased pY816 TrkB in strata oriens and radiatum of CA3 and CA1 yet no increase of pY816 was found in these regions (Fig. 5). The localization of increased pY816 TrkB immunoreactivity to stratum lucidum correlates with the highest concentrations of BDNF protein and vesicular zinc within hippocampus and forebrain (Yan et al., 1997; Cole TB et al., 1999; Frederickson et al., 2005). Thus low concentrations of BDNF and zinc together with limited sensitivity of the immunohistochemical method likely contribute to our inability to find increases in hippocampal regions apart from stratum lucidum. We suspect that similar factors contribute to an additional unexpected result, namely the absence of increased pY816 TrkB immunoreactivity in Western blots 30 min or 3 h following onset of status epilepticus (Fig. 1). The lack of increase at 30 min and 3 h is unexpected for several reasons: (1) both endogenous BDNF and zinc are released in an activity-dependent fashion (Balkowiec and Katz, 2002; Qian and Noebels, 2005; Matsumoto et al., 2008); (2) the synchronous, high-frequency firing of populations of hippocampal neurons (Labiner et al., 1993; Alexander et al., 2009) almost certainly triggers synaptic release of both BDNF and zinc during the seizures; (3) application of either BDNF or zinc to cultured neurons triggers striking activation of TrkB within 5–15 min (Huang et al., 2008); (4) impairments of LTP in slices from BDNF knock-out mice or with zinc chelators provide functional evidence of TrkB activation as early as 15 min following high-frequency stimulation (Korte et al., 1995; Patterson et al., 1996; Huang et al., 2008; Matsumoto et al., 2008). Collectively, this suggests that TrkB is activated at 30 min and 3 h following onset of status epilepticus yet escapes detection. Perhaps higher concentrations of BDNF mediated by seizure-evoked increases of transcription and translation results in a greater and more readily detectable activation of TrkB at later time points, a suggestion consistent with increased BDNF mRNA and protein 3–7 h after onset of hippocampal seizures (Ernfors et al., 1991; Isackson et al., 1991; Nawa et al., 1995; Yan et al., 1997). If BDNF activates TrkB at these later time points in WT animals, then compensatory increases of other neurotrophins (e.g., NT-3) and/or zinc may mediate the late increases of TrkB activation detected in conditional BDNF knock-out mice (He et al., 2004). That said, the latency of several hours between seizure onset and detectable increases of TrkB activation may provide a therapeutic window within which to intervene with an inhibitor to limit progressive severity of epilepsy.
The marked inhibition of development of kindling of trkBPLC/PLC mice establishes a causal role for TrkB-dependent PLCγ1 signaling in limbic epileptogenesis in vivo. Given the enormous diversity of cell surface receptors presumably undergoing activation during an event as complex as a seizure (McNamara et al., 2006), the activation of PLCγ1 almost exclusively by TrkB (Fig. 3) is remarkable. Also remarkable is the striking specificity of signaling pathways downstream of TrkB with respect to the phenotype of epileptogenesis. That is, increases of both pY515 and pY816 immunoreactivity in diverse models of limbic epileptogenesis (Binder et al., 1999; He et al., 2004) suggest that TrkB activates both Shc and PLCγ1 signaling. Yet in contrast to the marked inhibition of development of kindling in trkBPLC/PLC mice, no differences in development of kindling were detected between WT and trkBSHC/SHC mice (He et al., 2002). Although inhibition of kindling is marked in trkBPLC/PLC mice, the magnitude of inhibition was less than reported previously with conditional trkB-nulls in which trkB was recombined from CNS neurons by crossing synapsin-cre with floxed trkB mice (He et al., 2004). Notably, the mutation of the trkBPLC/PLC is in the germline whereas the onset of trkB recombination is delayed until late in embryonic development in the synapsin-cre trkBFLOX/FLOX; perhaps perturbing TrkB signaling earlier in the life of the trkBPLC/PLC mice compared with the conditional null mutants facilitates emergence of a compensatory mechanism that promotes epileptogenesis. The residual immunoreactivity detected by the pY816 Trk antibody migrating at ∼145 kDa in SDS-PAGE (Fig. 3) of hippocampi of trkBPLC/PLC mice likely represents p-TrkC; if so, this might be a compensatory mechanism promoting epileptogenesis. Alternatively, perhaps TrkB-mediated activation of the Shc pathway promotes epileptogenesis in the absence but not presence of TrkB-mediated activation of PLCγ1 signaling.
The inhibition of epileptogenesis in the trkBPLC/PLC mice provides clues to cellular mechanisms by which enhanced activation of TrkB promotes limbic epileptogenesis. Both ex vivo and in vivo studies of animal models suggest that LTP of excitatory synapses between principal cells contributes to limbic epileptogenesis (Sutula and Steward, 1987); potentiation of these synapses may facilitate propagation of seizure activity through synaptically coupled neuronal populations in the limbic system and beyond. Evidence that the mf-CA3 pyramid synapse undergoes LTP in vivo emerged in the kainic acid model of limbic epilepsy (Goussakov et al., 2000). The requirement for TrkB-dependent PLCγ1 signaling for LTP of this synapse together with evidence of increased pY816 immunoreactivity in the mf pathway in sections ex vivo from these models suggests that TrkB-mediated activation of PLCγ1 signaling in vivo may contribute to LTP of this synapse during epileptogenesis. The fact that LTP of these synapses remains intact in the trkBSHC/SHC mice is consistent with findings at the Schaffer collateral-CA1 synapse (Minichiello et al., 2002; Minichiello, 2009) and correlates with similar rates of kindling development in trkBSHC/SHC and control mice (He et al., 2002).
Notably, enhanced excitability in models of epilepsy is often accompanied and likely caused by both enhanced function of excitatory synapses and impaired function of inhibitory synapses. Might enhanced activation of PLCγ1 signaling by TrkB somehow compromise inhibitory function and thereby contribute to the increased excitability of limbic epilepsy? One interesting possibility is that enhanced TrkB-dependent activation of PLCγ1 signaling reduces expression of the K-Cl cotransporter, KCC2, resulting in accumulation of [Cl−]i and a shift of EGABA in a depolarizing direction (Rivera et al., 2004). Collectively, study of human epileptic tissue (Cohen et al., 2002; Huberfeld et al., 2007) buttressed by study of diverse in vivo and in vitro models (Rivera et al., 2002, 2004; Woo et al., 2002; Pathak et al., 2007; Li et al., 2008; Blaesse et al., 2009) advance reduced expression of KCC2 and resulting accumulation of [Cl−]i as an important molecular and cellular mechanism contributing to limbic epilepsy. Interestingly, in vitro studies reveal that TrkB-mediated activation of PLCγ1 signaling can suppress KCC2 expression (Rivera et al., 2002, 2004). Whether TrkB-mediated activation of PLCγ1 signaling promotes reductions of KCC2 expression described in the kindling and pilocarpine models (Rivera et al., 2002; Li et al., 2008) in vivo is unclear.
Our work elucidates a single signaling pathway activated by a single receptor contributing to limbic epileptogenesis in vivo, namely TrkB-mediated activation of PLCγ1. Whereas a pharmacological approach would be expected to inhibit PLCγ1 activated by diverse membrane receptors, only PLCγ1 activated by TrkB is inhibited in the trkBPLC/PLC mutants. That epileptogenesis is inhibited in trkBPLC/PLC but not trkBSHC/SHC mice (He et al., 2002) implies that anti-epileptogenic therapies need not necessarily target TrkB itself, thereby circumventing potential unwanted consequences of global inhibition of TrkB. Novel downstream targets suggested by the present findings include PLCγ1 itself or uncoupling TrkB from PLCγ1. Dissecting signaling pathways directly coupled to a single cell membrane receptor in vivo in models of CNS disorders may elucidate novel targets for specific and effective therapeutic intervention.
Footnotes
This work was supported by National Institutes of Health Grant 5 R01 NS056217 (J.O.M.), and in part by a grant from the European Union (EU FP6 StemStroke, 037526) to L.M. We thank Dr. Moses Chao for his kind gift of antibody and Drs. Ram Puranam and Yangzhong Huang for useful discussions of this work.
References
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci. 1999;19:4616–4626. doi: 10.1523/JNEUROSCI.19-11-04616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A. 1999;96:1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll SD, Suri C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, Wiegand SJ, Rudge JS, Scharfman HE. Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience. 1999;93:1491–1506. doi: 10.1016/s0306-4522(99)00296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC, McNamara JO. Localization of brain-derived neurotrophic factor to distinct terminals of mossy fiber axons implies regulation of both excitation and feedforward inhibition of CA3 pyramidal cells. J Neurosci. 2004;24:11346–11355. doi: 10.1523/JNEUROSCI.3846-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC, He XP, McNamara JO. Ontogeny of seizure-induced increases in BDNF immunoreactivity and TrkB receptor activation in rat hippocampus. Hippocampus. 2004;14:345–355. doi: 10.1002/hipo.10190. [DOI] [PubMed] [Google Scholar]
- Danzer SC, He XP, Loepke AW, McNamara JO. Structural plasticity of dentate granule cell mossy fibers during the development of limbic epilepsy. Hippocampus. 2010;20:113–124. doi: 10.1002/hipo.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Goussakov IV, Fink K, Elger CE, Beck H. Metaplasticity of mossy fiber synaptic transmission involves altered release probability. J Neurosci. 2000;20:3434–3441. doi: 10.1523/JNEUROSCI.20-09-03434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XP, Minichiello L, Klein R, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trkB receptors in the mossy fiber pathway of adult mouse hippocampus. J Neurosci. 2002;22:7502–7508. doi: 10.1523/JNEUROSCI.22-17-07502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57:546–558. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, Nawa H, Sora I, Chao MV. Dopamine D1 receptor-induced signaling through TrkB receptors in striatal neurons. J Biol Chem. 2008;283:15799–15806. doi: 10.1074/jbc.M801553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klitgaard H, Matagne A, Vanneste-Goemaere J, Margineanu DG. Pilocarpine-induced epileptogenesis in the rat: Impact of initial duration of status epilepticus on electrophysiological and neuropathological alterations. Epilepsy Res. 2002;51:93–107. doi: 10.1016/s0920-1211(02)00099-2. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmér E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labiner DM, Butler LS, Cao Z, Hosford DA, Shin C, McNamara JO. Induction of c-fos mRNA by kindled seizures: complex relationship with neuronal burst firing. J Neurosci. 1993;13:744–751. doi: 10.1523/JNEUROSCI.13-02-00744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos T, Cavalheiro EA. Suppression of pilocarpine-induced status epilepticus and the late development of epilepsy in rats. Exp Brain Res. 1995;102:423–428. doi: 10.1007/BF00230647. [DOI] [PubMed] [Google Scholar]
- Li X, Zhou J, Chen Z, Chen S, Zhu F, Zhou L. Long-term expressional changes of Na+-K+-Cl− co-transporter 1 (NKCC1) and K+-Cl− co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE) Brain Res. 2008;1221:141–146. doi: 10.1016/j.brainres.2008.04.047. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;356:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Murray KD, Isackson PJ, Eskin TA, King MA, Montesinos SP, Abraham LA, Roper SN. Altered mRNA expression for brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase in the hippocampus of patients with intractable temporal lobe epilepsy. J Comp Neurol. 2000;418:411–422. doi: 10.1002/(sici)1096-9861(20000320)418:4<411::aid-cne4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fiber synapse. J Physiol. 2005;566:747–758. doi: 10.1113/jphysiol.2005.089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Gwag BJ, Sessler FM. Neurotrophic factor mRNA expression in dentate gyrus is increased following in vivo stimulation of the angular bundle. Brain Res Mol Brain Res. 1994;23:135–143. doi: 10.1016/0169-328x(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Sutula T, Steward O. Facilitation of kindling by prior induction of long-term potentiation in the perforant path. Brain Res. 1987;420:109–117. doi: 10.1016/0006-8993(87)90245-9. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Hayashi S, Kakita A, Wakabayashi K, Fukuda M, Kameyama S, Tanaka R, Takahashi H, Nawa H. Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res. 1999;818:579–582. doi: 10.1016/s0006-8993(98)01355-9. [DOI] [PubMed] [Google Scholar]
- Toth K, Suares G, Lawrence JJ, Philips-Tansey E, McBain CJ. Differential mechanisms of transmission at three types of mossy fiber synapse. J Neurosci. 2000;20:8279–8289. doi: 10.1523/JNEUROSCI.20-22-08279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Xu B, Michalski B, Racine RJ, Fahnestock M. The effects of brain-derived neurotrophic factor (BDNF) administration on kindling induction, Trk expression and seizure-related morphological changes. Neuroscience. 2004;126:521–531. doi: 10.1016/j.neuroscience.2004.03.044. [DOI] [PubMed] [Google Scholar]
- Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]