

Abstract
In the absence of GABA, neuroactive steroids that enhance GABA-mediated currents modulate binding of [35S]t-butylbicyclophosphorothionate in a biphasic manner, with enhancement of binding at low concentrations (site NS1) and inhibition at higher concentrations (site NS2). In the current study, compound (3α,5β,17β)-3-hydroxy-18-norandrostane-17-carbonitrile (3α5β-18-norACN), an 18-norsteroid, is shown to be a full agonist at site NS1 and a weak partial agonist at site NS2 in both rat brain membranes and heterologously expressed GABAA receptors. 3α5β-18-norACN also inhibits the action of a full neurosteroid agonist, (3α,5α,17β)-3-hydroxy-17-carbonitrile (3α5αACN), at site NS2. Structure-activity studies demonstrate that absence of the C18 methyl group and the 5β-reduced configuration both contribute to the weak agonist effect at the NS2 site. Electrophysiological studies using heterologously expressed GABAA receptors show that 3α5β-18-norACN potently and efficaciously potentiates the GABA currents elicited by low concentrations of GABA but that it has low efficacy as a direct activator of GABAA receptors. 3α5β-18-norACN also inhibits direct activation of GABAA receptors by 3α5αACN. 3α5β-18-norACN also produces loss of righting reflex in tadpoles and mice, indicating that action at NS1 is sufficient to mediate the sedative effects of neurosteroids. These data provide insight into the pharmacophore required for neurosteroid efficacy at the NS2 site and may prove useful in the development of selective agonists and antagonists for neurosteroid sites on the GABAA receptor.
Certain endogenous pregnane steroids and their synthetic analogs modulate the function of GABAA receptors (Lambert et al., 2003). These neuroactive steroids potentiate the actions of GABA at low concentrations and directly open GABAA receptor channels at higher concentrations (Barker et al., 1987; Cottrell et al., 1987). Several lines of evidence suggest that multiple neuroactive steroid recognition sites contribute to GABAA receptor modulation. Radioligand binding studies have shown that pregnanolone [(3α,5β)-3-hydroxypregnan-20-one] modulation of [35S]t-butylbicyclophosphorothionate (TBPS), [3H]flunitrazepam, and [3H]muscimol binding to GABAA receptors has two distinct components (Hawkinson et al., 1994b; Hauser et al., 1995). Likewise, in the absence of added GABA, alphaxalone produces biphasic modulation of [35S]TBPS binding: at 10 to 100 nM alphaxalone, [35S]TBPS binding is stimulated; whereas at 1 to 30 μM, it is inhibited (Srinivasan et al., 1999). The two-component modulation of radioligand binding is observed in heterologously expressed receptor preparations as well as in native tissues in which there are GABAA receptors of various subunit compositions. This indicates that two-site behavior is not merely a reflection of heterogeneous subunit expression but rather an indication that GABAA receptors of defined subunit composition have multiple sites at which neuroactive steroids can modulate function. Recent work using site-directed mutagenesis has identified two putative neurosteroid binding sites, one site that mediates potentiation of the effects of GABA and one site that mediates direct activation of the GABAA receptor (Hosie et al., 2006, 2009). However, the relationship between the two neurosteroid sites observed with radioligand binding and the two sites observed with electrophysiology (coupled with site-directed mutagenesis) has not been rigorously examined.
In this study, we describe the actions of compound (3α,5β,17β)-3-hydroxy-18-norandrostane-17-carbonitrile (3α5β-18-norACN), a neuroactive 18-norsteroid that preferentially affects one of the two neurosteroid binding sites observed in both [35S]TBPS binding assays and electrophysiological assays. The study also examines the structure-activity relationships underlying site selectivity. Finally, we examined the anesthetic efficacy of compound 3α5β-18-norACN to determine whether the anesthetic actions of neurosteroids require agonism at both neurosteroid sites.
Materials and Methods
Prepared Materials.
The synthesis, spectroscopic and physical properties of 3α5αACN, 3α5βACN, 3α5α-19-norACN, 3α5β-19-norACN, 3α5α-19-norP, and 3α5β-19-norP were reported by us previously (Hu et al., 1993; Han et al., 1996). The 18-nor and 18,19-dinorsteroids were prepared by similar multistep synthetic procedures. In brief, the 18-methyl group was removed from either a 17-ketosteroid or 19-nor-17-ketosteroid precursor to give the corresponding 18-nor or 18,19-dinorsteroids. The seven-step procedure required for removal of the 18-methyl group has been described by us for the preparation of other 18,19-dinor-17-ketosteroids (Han and Covey, 1996). The 17-keto group of the 18-nor or 18,19-dinorsteroids was then converted in two steps into the 17-carbonitrile group using a procedure we described previously (Han et al., 1996). Conversion of the 17-carbonitrile group into the acetyl group of 20-ketopreganes was also described by us previously (Han et al., 1996). The spectroscopic and physical properties of the previously unknown 18-nor and 18, 19-dinorsteroids used in this study are given below.
3α5α-18-norACN.
Colorless crystals (from ethyl acetate/hexanes), m.p. 157–159°C; IR 3413, 2238 cm−1; 1H NMR δ 4.05 (m, 1H, CHOH), 2.31 to 2.21 (m, 1H, CHCN); 0.75 (s, 3H, CH3); 13C NMR δ 122.91 (CN), 66.33 (C-3), 52.89, 52.19, 50.64, 41.75, 38.76, 36.05, 35.68, 32.67, 32.17, 31.94, 29.46, 28.91, 28.06, 27.99, 24.64, 11.05 (CH3). Anal. Calcd for C19H29NO: C, 79.39; H, 10.17, N, 4.87. Found: C, 79.12; H, 10.34, N, 4.68.
3α5β-18-norACN.
Colorless crystals (from ethyl acetate/hexanes), m.p. 179–81°C; IR 3401, 2237 cm−1; 1H NMR δ 3.64 (m, 1H, CHOH)), 2.32–2.22 (m, 1H, CHCN), 0.89 (s, 3H, CH3); 13C NMR δ 122.83 (CN), 71.51 (C-3), 52.05, 50.67, 41.99, 41.73, 39.21, 36.03, 35.16, 34.42, 32.63, 30.35, 29.51, 28.03, 27.92, 26.57, 26.27, 24.66, 23.10 (CH3). Anal. Calcd for C19H29NO: C, 79.39; H, 10.17, N, 4.87. Found: C, 79.16; H, 10.33, N, 4.80.
3α5α-18-norP.
Colorless crystals (from ethyl acetate/hexanes), m.p. 127–128°C; IR 3295, 1708 cm−1; 1H NMR δ 4.04 (m, 1H, CHOH), 2.54 to 2.45 (m, 1H, CHCOCH3), 2.14 (s, 3H, CH3CO), 0.72 (s, 3H, CH3); 13C NMR δ 212.08 (C = O), 66.39 (C-3), 57.36, 53.08, 48.77, 41.87, 38.83, 36.04, 35.76, 32.19, 32.13, 30.41, 29.65, 28.91, 28.53, 28.22, 26.87, 24.93 11.05 (CH3). Anal. Calcd for C20H32O2: C, 78.90; H, 10.59. Found: C, 79.00; H, 10.36.
3α5β-18-norP.
Colorless crystals (from ethyl acetate/hexanes), m.p. 146–148°C; IR 3396, 1705 cm−1; 1H NMR δ 3.65 (m, 1H, CHOH), 2.55 to 2.45 (m, 1H, CHCOCH3), 2.14 (s, 3H, CH3CO), 0.88 (s, 3H, CH3); 13C NMR δ 212.13 (C = O), 71.80 (C-3), 57.42, 53.01, 48.82, 42.20, 41.94, 39.41, 36.22, 35.25, 34.51, 30.56, 30.49, 29.58, 28.54, 26.93, 26.77, 26.49, 25.02, 23.22. Anal. Calcd for C20H32O2: C, 78.90; H, 10.59. Found: C, 79.12; H, 10.63.
3α5α-18,19-dinorACN.
Colorless crystals (from ethyl acetate), m.p. 174–175°C; IR 3317, 2238 cm−1; 1H NMR δ 4.08 (m, 1H, CHOH), 2.32 to 2.23 (m, 1H, CHCN); 13C NMR δ 122.89 (CN), 66.10 (C-3), 51.10, 50.72, 47.48, 46.74, 46.56, 40.34, 35.55, 33.20, 32.75, 32.63, 30.87, 29.20, 29.06, 27.95, 27.74, 23.57. Anal. Calcd for C18H27NO: C, 79.07; H, 9.95; N, 5.12. Found: 78.87; H, 9.89; N, 5.09.
3α5β-18,19-dinorACN.
Colorless crystals (from ethyl acetate/hexanes), m.p. 158–160°C; IR 3299, 2236 cm−1; 1H NMR δ 3.69 to 3.59 (m, 1H, CHOH), 2.33 to 2.23 (m, 1H, CHCN); 13C NMR δ 122.80 (CN), 71.46 (C-3), 51.01, 50.80, 48.01, 39.74, 37.11, 36.10, 35.33, 32.78, 31.18, 29.59, 29.38, 29.24, 28.09, 27.93, 26.23, 25.93. Anal. Calcd for C18H27NO: C, 79.07; H, 9.95; N, 5.12. Found: C, 79.03; H, 9.76; N, 5.07.
Vector Construction.
cDNA constructs for GABAA receptor subunits were provided by A. Tobin [University of California, Los Angeles, CA (rat α1)] and D. Weiss [University of Texas, San Antonio, TX (rat β2)]. The expression constructs for the rat α1F pcDNA3, rat β2 pcDNA3, and rat α1myc were described previously (Ueno et al., 1996; Darbandi-Tonkabon et al., 2003). The rat β2F construct was made by polymerase chain reaction mutagenesis inserting the FLAG between amino acids 4 and 5 using the following oligonucleotides: rat β2F forward, 5′-gattacaaggacgatgacgacaaggaccctagtaatatgtcgctgg-3′ and rat β2F reverse, 5′-cttgacgacatcgtccttgtaatcattgacactctgagcacagacagc-3′. All inserts were sequenced through the entire coding region.
Tissue Culture.
Quail fibroblast (QT-6) cells were maintained in culture using standard methods and passaged at subconfluent densities. A stably transfected cell line with rat α1myc rat β2Flag was produced in QT-6 cells by standard methods. In brief, QT-6 cells were transfected with the cDNA using the calcium phosphate precipitation method or using Effectene (QIAGEN, Valencia, CA). Cells resistant to G418 were selected. A population of cells expressing high levels of surface FLAG was selected by immunoselection using the anti-FLAG antibody (M2; Sigma-Aldrich, St. Louis, MO) (Chen et al., 1995).
Membrane Preparation.
Rat brains were purchased from Pel-Freez Biologicals (Rogers, AK) and stored until use at −80°C. Cerebella and brain stem were trimmed from the frozen brains, and the cerebral hemispheres were used. Membranes for structure-activity relationship experiments (Tables 1 and 2) were prepared using minor modifications of the method of Hawkinson et al. (1994a), as described previously (Covey et al., 2000). For all other studies, GABA-depleted membranes were prepared using minor modifications of the method described by Srinivasan et al. (1999). In brief, brains were immersed in ice-cold 0.32 M sucrose (10 ml/g) and homogenized using a Teflon pestle in a motor-driven homogenizer. The homogenate was centrifuged for 10 min at 1000g at 4°C and the pellet was discarded. The supernatant was then centrifuged for 45 min at 100,000g. The resultant pellet was then resuspended in distilled water (12 ml/brain) and stirred for 30 min at 4°C. Membranes were then collected by centrifugation for 45 min at 100,000g at 4°C. The pellet was washed twice with buffer (20 mM potassium phosphate and 50 mM KCl, pH 7.5). The membranes were pelleted after each wash by centrifugation for 45 min at 100,000g at 4°C. After the final centrifugation, membranes were resuspended in assay buffer (10 mM potassium phosphate and 100 mM KCl, pH 7.5) at approximately 5 mg/ml membrane protein and stored at −80°C.
TABLE 1.
C17-carbonitrile neuroactive steroids: modulation of [35S]TBPS binding in rat brain membranes
Concentration-response curves were generated for the inhibition of specific [35S]TBPS binding in the presence of 5 μM GABA. Curves were fit to a single-component Hill equation; Hill coefficients, IC50 values (mean ± S.D. of triplicate determinations), and minimal binding are reported for all compounds.
| Compound | Hill Slope | IC50 | Minimal Binding |
|---|---|---|---|
| nM | % | ||
| 3α5α-ACN | 0.96 | 46 ± 4 | 2 ± 2 |
| 3α5α-18-norACN | 0.75 | 59 ± 7 | 15 ± 2 |
| 3α5α-19-norACN | 0.88 | 76 ± 4 | 0 ± 1 |
| 3α5α-18,19-dinorACN | 0.87 | 133 ± 24 | 19 ± 3 |
| 3α5β-ACN | 0.90 | 63 ± 7 | 4 ± 2 |
| 3α5β-18-norACN | 1.27 | 49 ± 4 | 41 ± 1 |
| 3α5β-19-norACN | 0.82 | 22 ± 3 | 3 ± 2 |
| 3α5β-18,19-dinorACN | 1.05 | 73 ± 16 | 31 ± 3 |
TABLE 2.
C17-acetyl neuroactive steroids: modulation of [35S]TBPS binding in rat brain membranes
Concentration-response curves were generated for the inhibition of specific [35S]TBPS binding in the presence of 5 μM GABA. Curves were fit to a single-component Hill equation; Hill coefficients, IC50 values (mean ± S.D. of triplicate determinations), and minimal binding are reported for all compounds.
| Compound | Hill Slope | IC50 | Minimal Binding |
|---|---|---|---|
| nM | % | ||
| 3α5αP | 0.94 | 69 ± 8 | 2 ± 2 |
| 3α5α-18-norP | 0.90 | 157 ± 16 | 11 ± 2 |
| 3α5α-19-norP | 0.99 | 86 ± 7 | 2 ± 2 |
| 3α5βP | 0.65 | 51 ± 16 | 7 ± 5 |
| 3α5β-18-norP | 0.85 | 35 ± 11 | 22 ± 4 |
| 3α5β-19-norP | 1.01 | 12 ± 1 | 9 ± 1 |
QT-6 cell membranes were prepared as follows. Cells were grown in monolayer culture to 70 to 80% confluence on 150-cm plates. The plates were washed twice with 5 ml of ice-cold phosphate-buffered saline containing 0.1% protease inhibitor cocktail (Sigma-Aldrich). Five milliliters of TEN (10 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, pH 7.5, and 0.1% protease inhibitor cocktail) was added to each plate, and cells were scraped from the plate with a rubber cell scraper. The plates were washed with 5 ml of TEN, and the harvested cells were collected by centrifugation for 10 min at 5000g at 4°C. Cells were resuspended in TEN and homogenized using an Ultra-Turrax high-performance disperser (10 5-s bursts at 4°C; Tekmar-Dohrmann, Mason, OH). Membranes were then collected by centrifugation for 30 min at 30,000g at 4°C, resuspended in TEN (2–3 mg protein/ml), and stored in aliquots at −80°C.
[35S]TBPS Binding.
[35S]TBPS binding assays were performed using previously described methods (Hawkinson et al., 1994b; Covey et al., 2000), with modification. In brief, aliquots of membrane suspension (0.5 mg/ml final protein concentration in assay) were incubated with 1 to 2 nM [35S]TBPS (60–100 Ci/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA) and 5-μl aliquots of steroid in Me2SO solution (final steroid concentrations ranged from 1 nM to 10 μM), in a total volume of 1 ml of assay buffer. For rat brain membranes, the assay buffer was 100 mM KCl and 10 mM potassium phosphate buffer, pH 7.5; for QT-6 cell membranes, assay buffer was 20 mM Tris-HCl and 1 M NaCl, pH 7.5; for the structure-activity screens shown in tables, the assay buffer was 50 mM potassium phosphate buffer, pH 7.4, and 200 mM NaCl. GABA (5 μM) was added to all screening assays and selected assays with GABA-free membranes to analyze its effect on [35S]TBPS binding. For experiments shown in Fig. 4, 1 μM GABA was used because it inhibited [35S]TBPS binding by ≈50%, whereas 5 μM GABA completely inhibited specific TBPS binding in QT-6 cells expressing recombinant α1β2 subunits of the GABAA receptors. Control binding was defined as binding observed in the presence of 0.5% Me2SO and the absence of steroid; all assays contained 0.5% Me2SO. Nonspecific binding was defined as binding observed in the presence of 200 μM picrotoxin and ranged from 12.4 to 32.6% of total binding. Assay tubes were incubated for 2 h at room temperature. A cell harvester (Brandel Inc., Gaithersburg, MD) was used for filtration of the assay tubes through GF/C glass fiber filter paper (Whatman, Maidstone, UK). Filter paper was rinsed with 4 ml of ice-cold buffer three times and dissolved in 4 ml of ScintiVerse II (Thermo Fisher Scientific, Waltham, MA). Radioactivity bound to the filters was measured by liquid scintillation spectrometry. Each data point was done in triplicate, and all experiments were performed at least three times. The average specific binding values of each triplicate were used for curve fitting and EC50 or IC50 is presented as the parameters of the curve fitting to the pooled data from the repeated experiments ± S.E.M.
Fig. 4.

Effects of 3α5αACN, 3α5β-18-norACN, and GABA on [35S]TBPS binding to α1β2 GABAA receptors expressed in QT-6 cells. A, in the absence of added GABA, 3α5αACN modulates [35S]TBPS binding to α1mycβ2FLAG receptors in a biphasic manner (EC50 = 28 ± 14 nM; IC50 = 537 ± 115 nM). In the presence of 1 μM GABA, enhancement is eliminated and 3α5αACN only inhibits TBPS binding (IC50 = 20 ± 9 nM). B, in the absence of GABA, 3α5β-18-norACN selectively enhances [35S]TBPS binding to α1mycβ2FLAG receptors (EC50 = 50 ± 16 nM). In the presence of 1 μM GABA, 3α5β-18-norACN partially inhibits [35S]TBPS binding (IC50 = 20 ± 9 nM). C, GABA modulates [35S]TBPS binding to α1mycβ2FLAG receptors in a biphasic manner (EC50 = 119 ± 1 nM; IC50 = 120 ± 1 nM).
The data from [35S]TBPS binding performed in the presence of GABA were fit to the Hill equation (eq. 1).
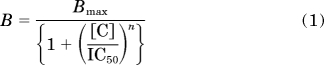 |
where B is TPBS bound in the presence of steroid at a given concentration, Bmax is control binding, [C] is steroid concentration, IC50 is the half-maximal inhibition, and n is the Hill coefficient.
The curves describing [35S]TBPS binding performed in the absence of GABA were fit to an equation (eq. 2) in which the term for enhanced binding is multiplied by the term for inhibition of binding:
where B is steroid bound, Z is the starting maximal binding in the absence of steroids; A is the amplitude of the enhancement, K1 is the half-maximal enhancement concentration, K2 is the half-maximal inhibition concentration, and [C] is steroid concentration. All fits were performed using SigmaPlot version 8 (SPSS Inc., Chicago, IL) and Prism (GraphPad Software Inc., San Diego, CA).
Xenopus laevis Oocyte Electrophysiological Methods.
Stage V and VI oocytes were harvested from sexually mature female X. laevis (Xenopus I, Northland, MI) under 0.1% tricaine (3-aminobenzoic acid ethyl ester) anesthesia. Oocytes were defolliculated by shaking for 20 min at 37°C in collagenase (2 mg/ml) dissolved in calcium-free solution containing 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES at pH 7.4. Capped mRNA encoding rat GABAA receptor α1, β2, and γ2L subunits was transcribed in vitro using the mMESSAGE mMachine kit (Ambion, Austin, TX) from linearized pBluescript vectors containing receptor coding regions. Subunit transcripts were injected in equal parts (20–40 ng of total RNA) 8 to 24 h after defolliculation. Oocytes were incubated up to 5 days at 18°C in ND96 medium containing 96 mM NaCl, 1 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 5 mM HEPES at pH 7.4, supplemented with pyruvate (5 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), and gentamicin (50 μg/ml). The cDNAs for the rat GABAA-receptor subunits were originally provided by A. Tobin [University of California, Los Angeles (α1)], P. Malherbe [F. Hoffman-La Roche, Basel, Switzerland (β2)], and C. Fraser [National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD (γ2L)].
GABA currents were measured with an OC725 two-electrode voltage-clamp amplifier (Warner Instruments, Hamden, CT) 2 to 5 days after RNA injection in a bath of unsupplemented ND96 medium. Intracellular recording pipettes had a resistance of ∼1 MΩ when filled with 3 M KCl. Compounds were simultaneously coapplied with GABA using a gravity-flow perfusion system. Holding potential was −70 mV, and peak current during 20-s drug applications was used for quantification. Data were acquired and analyzed with pCLAMP software (Molecular Devices, Sunnyvale, CA). Statistical differences were determined using a two-tailed Student's t test.
Tadpole and Mouse Anesthetic Assay.
Assays for neuroactive steroid-induced loss-of-righting reflex (LRR) in X. laevis tadpoles and in BALB/c mice were performed as described previously (Covey et al., 2000). In brief, groups of 10 early prelimb-bud stage X. laevis tadpoles (Nasco, Fort Atkinison, WI) were placed in 100 ml of oxygenated Ringer's buffer containing varying concentrations of 3α5β-18-norACN. After 3 h of equilibration the tadpoles were assessed for the LRR and loss-of-swimming reflex (LSR) behavioral endpoints. LRR was defined as failure of the tadpole to right itself within 5 s after being flipped by a smooth glass rod. LSR was defined as failure to initiate swimming after being flipped by a smooth glass rod. Concentration-response curves were fit to the Hill equation using SigmaPlot version 8.0. For the mouse assay, BALB/C mice were injected intravenously through a tail vein with various doses of 3α5β-18-norACN in an 8% ethanol, 16% Cremophor EL (Sigma-Aldrich) solution. LRR was defined as inability of mice to right themselves within 5 s after being placed in a prone position. LRR time was measured from the moment mice displayed LRR until they were able to right themselves.
Results
Structures.
The structures of the neuroactive steroid analogs used in this study are shown in Fig. 1. The structural variables were the 5α- versus 5β-configuration, and the presence or absence of the C18 and/or C19 methyl groups. Structural variables were examined both in steroids with an acetyl group at carbon 17 (naturally occurring neurosteroids) and with a carbonitrile substitution at carbon 17.
Fig. 1.

Structures of two series of neurosteroid analogs based on either a 5α-reduced or a 5β-reduced steroid backbone. Structural variables include methyl groups at C18 (R2) and C19 (R1) and acetyl versus carbonitrile groups at C17 (R3).
Neuroactive Steroid Modulation of [35S]TBPS Binding in Rat Brain Membranes.
The effects of neurosteroids on [35S]TBPS binding were examined in the presence of 5 μM GABA (Fig. 2, A and B) and in membranes depleted of GABA (Fig. 2, A and B). Because complete removal of GABA is difficult, experiments were also done examining the effects of one neurosteroid, 3α5αACN, in the presence of GABAzine, a competitive GABA antagonist (Fig. 2A).
Fig. 2.

Effects of neuroactive steroids on specific [35S]TBPS binding to rat brain membranes in the presence and absence of exogenous GABA. 3α5αACN (A) and 3α5βACN (C) modulate [35S]TBPS binding in a biphasic manner in the absence of GABA, whereas only the inhibition phase is observed in the presence of 5 μM GABA. In the presence of GABAzine (A), both the stimulatory and inhibitory effects of 3α5αACN are observed. B, the same data as in A plotted as femtomoles of specific [35S]TBPS binding per milligram of membrane protein. D, compound 3α5β-18-norACN is a preferential NS1 site agonist. In the absence of GABA, 3α5β-18-norACN selectively enhances [35S]TBPS binding with minimal inhibition at higher concentrations. 3α5β-18-norACN partially inhibits TBPS binding in the presence of 5 μM GABA. Data shown are the means of triplicate determinations for representative experiments.
3α5αACN inhibited [35S]TBPS binding, in the presence of 5 μM GABA, with an IC50 value of 46 ± 4 nM and a Hill slope of 0.96, consistent with inhibition of a single class of binding sites (Majewska et al., 1986). In the absence of GABA, 3α5αACN showed two-component behavior with enhancement of [35S]TBPS binding at low concentration (EC50 = 40 ± 35 nM) and inhibition of binding at higher concentrations (IC50 = 1.4 ± 0.4 μM) (Fig. 2A). We henceforth refer to the high-affinity (enhancement of TBPS binding) site as NS1 and the low-affinity site (inhibition of TBPS binding) as NS2. It is important to note that the addition of 5 μM GABA not only eliminated enhancement of [35S]TBPS binding but also remarkably reduced baseline [35S]TBPS binding (Fig. 2B). In rat brain membranes, 5 μM GABA reduced TBPS binding (in the absence of steroid) by amounts varying from 50 to 85%. In the presence of GABAzine, baseline TBPS binding was reduced (presumably by antagonizing small amounts of residual GABA) but both the enhancing effects (EC50 = 50 ± 22 nM) and inhibitory effects (IC50 = 8 ± 2 μM) of 3α5αACN on [35S]TBPS binding were observed (Fig. 2A). It is noteworthy that the IC50 (NS2 effect) value for 3α5αACN was shifted to the right in the presence of GABAzine.
3α5βACN also inhibited [35S]TBPS binding, in the presence of GABA, with an IC50 value of 63 ± 7 nM and a Hill slope of 0.9. In the absence of GABA, 3α5βACN enhanced [35S]TBPS binding at low concentrations (EC50 = 29 ± 36 nM) and partially inhibited it at higher concentrations (IC50 = 10.7 ± 3.6 μM) (Fig. 2C). The endogenous neurosteroids (acetyl at C17) exhibited behavior that was qualitatively similar to the carbonitrile series; in the absence of GABA, 3α5αP stimulated [35S]TBPS binding, with an EC50 value of 54 ± 39 nM, and inhibited it, with an IC50 = 3.3 ± 0.7 μM; 3α5βP had an EC50 = 8 ± 4 nM and an IC50 = 38.2 ± 10.7 μM (data not shown in the figure).
Compound 3α5β-18-norACN (previously referred to as B285; Akk et al., 2004), a 5β-reduced steroid lacking the 18-methyl group, modulated [35S]TBPS binding in a distinct pattern (Fig. 2D). In the absence of GABA, 3α5β-18-norACN enhanced [35S]TBPS binding, with an EC50 of 67.9 ± 11.1 nM. However, it showed barely discernible inhibition of [35S]TBPS binding even at a concentration of 10 μM. In the presence of 5 μM GABA, 3α5β-18-norACN partially inhibited [35S]TBPS binding (IC50 = 49 ± 4 nM), with a Hill slope of 1.27 and a minimal binding of 41 ± 1% of control binding. There is no additional effect of 3α5β-18-norACN between concentrations of 0.3 and 10 μM. These data show that 3α5β-18-norACN preferentially acts at the NS1 site and has low potency and/or efficacy at the NS2 site.
Interactions of 3α5β-18-norACN and 3α5αACN at the NS1 and NS2 Sites.
To determine whether the minimal effect of 3α5β-18-norACN on NS2 results from poor binding or low efficacy, we examined the interaction of 3α5β-18-norACN and 3α5αACN. In Fig. 3A, the experiments were conducted simultaneously using the same membrane preparation and the same radioligand stock to allow comparison of the absolute amount (femtomoles per milligram of protein) of binding. The effect of 3α5αACN (in the absence of GABA) on [35S]TBPS binding was examined in the presence of 3α5β-18-norACN. 3α5β-18-norACN (3 μM), a concentration that provides maximum 3α5β-18-norACN effect (Fig. 2D), occluded the enhancing action of 3α5αACN at NS1, maximally enhancing [35S]TBPS binding and preventing any further enhancement by 3α5αACN (Fig. 3A). This indicates that 3α5β-18-norACN is a full agonist at the NS1 site. The presence of 3 μM 3α5β-18-norACN also produced a modest change in the IC50 value of 3α5αACN at NS2 (1.4 ± 0.4 μM without 3α5β-18-norACN and 5.8 ± 4.9 μM in the presence of 3α5β-18-norACN). To further probe the actions of 3α5β-18-norACN at NS2, the inhibitory effects of 3α5αACN on [35S]TBPS binding were examined in the presence of various concentrations of 3α5β-18-norACN (1, 3, 10, and 30 μM). As shown in Fig. 3B, 1 μM 3α5β-18-norACN enhanced and 10 μM 3α5αACN inhibited [35S]TBPS binding. Increasing concentrations of 3α5β-18-norACN added to 10 μM 3α5αACN significantly increased [35S]TBPS binding (p < 0.05, analysis of variance followed by Tukey's multiple comparison test of the means), presumably by antagonizing the inhibitory effect of 10 μM 3α5αACN. These data indicate that 3α5β-18-norACN binds to both sites NS1 and NS2 but has low efficacy at NS2.
Fig. 3.

3α5β-18-norACN occludes enhancement and antagonizes inhibition of [35S]TBPS binding by 3α5αACN in GABA-depleted rat brain membranes. A, [35S]TBPS binding (femtomoles per milligram of protein) data demonstrates that 3 μM 3α5β-18-norACN occludes the enhancement action of 3α5αACN at site NS1 and modestly right-shifts its inhibitory effect at site NS2. B, 3α5β-18-norACN antagonizes the inhibitory effect of 10 μM 3α5αACN on [35S]TBPS binding in a concentration-dependent manner.
Effects of Neuroactive Steroids on [35S]TBPS Binding in Heterologously Expressed GABAA Receptors.
To confirm that the NS1 and NS2 sites both reside on a single pentameric GABAA receptor, steroid modulation of [35S]TBPS binding was examined in cell membranes expressing defined combinations of GABAA receptor subunits. These studies used α1β2 heteropentamers. Based on the poor ability of β2 subunits to form homopentamers (Bracamontes and Steinbach, 2008), this combination maximizes the likelihood of working with homogeneous populations of GABAA receptors. Membranes from QT-6 cells expressing α1β2 GABAA receptor subunits were modulated by 3α5αACN in a manner very similar to that observed in rat brain membranes. In the absence of GABA, 3α5αACN stimulated TBPS binding at low concentrations (EC50 = 28 ± 14 nM) and inhibited at higher concentrations (IC50 = 537 ± 115 nM). In the presence of GABA (1 μM), 3α5αACN inhibited TBPS binding, with an IC50 value of 20 ± 9 nM and a Hill slope of 1 (Fig. 4A). 3α5β-18-norACN appeared to be a selective NS1 agonist in α1β2 receptors (Fig. 4B); in the absence of GABA, 3α5β-18-norACN enhanced [35S]TBPS binding, with an EC50 value of 50 ± 16 nM, and in the presence of 1 μM GABA, it partially (65% inhibition) inhibited TBPS binding, with an IC50 value of 20 ± 9 nM. We also examined the concentration-dependent effects of GABA on [35S]TBPS binding in QT-6 cells expressing α1β2 GABAA receptor subunits. Consistent with previous studies (Pregenzer et al., 1993), GABA enhances TBPS binding at low concentrations and inhibits it at higher concentrations (Fig. 4C).
Structural Requirements for Steroids Providing Low Efficacy at the NS2 Site.
To determine which structural properties of 3α5β-18-norACN cause it to have low efficacy at the NS2 site, [35S]TBPS binding (in the presence of 5 μM GABA) was performed with all of the compounds shown in Fig. 1. The data were fit to a one-component inhibition curve. In the C17-carbonitrile series (Table 1), all steroids lacking the 18-methyl group failed to completely inhibit [35S]TBPS binding (minimal binding >10%); in contrast all steroid containing the 18-methyl group completely inhibited TBPS binding. This effect of the 18-nor compounds was much more pronounced in the 5β-reduced steroids than in the 5α-reduced steroids. Absence of the 19-methyl group affected neither minimal binding nor the Hill slope. In the C17-acetyl series (Table 2), a similar effect of the 18-nor and 5β-reduced configurations was observed. It is noteworthy that pregnanolone (3α5βP) almost completely inhibited [35S]TBPS binding but did so with a Hill slope of 0.65, suggesting the possibility of two-component inhibition. These data indicate that the absence of the 18-methyl group and the 5β-reduced configuration both contribute to lack of neurosteroid efficacy at the NS2 site.
Electrophysiological Effects of 3α5β-18-norACN.
The ability of 3α5β-18-norACN, 3α5αACN, and 3α5βACN to potentiate GABA-elicited (2 μM) currents and to directly activate GABAA receptors was examined in X. laevis oocytes expressing α1β2γ2L GABAA receptor subunits. Figure 5A shows superimposed traces of representative currents elicited by 2 μM GABA alone (the lowest amplitude trace) and 2 μM GABA plus 0.1, 1, or 10 μM 3α5β-18-norACN, 3α5βACN, or 3α5αACN. The concentration-response curves (Fig. 5B) demonstrate that 3α5αACN (Emax = 14 ± 21) has modestly higher efficacy than 3α5β-18-norACN (Emax = 10 ± 0.3) or 3α5βACN (Emax = 7.8 ± 1.6) in potentiating GABA-elicited currents. However, there is no statistical difference among them. Two-way analysis of variance indicated that 0.3, 1, and 3 μM 3α5αACN had higher potentiation than 3α5β-18-norACN and 3α5βACN. However, there was no difference among these three neurosteroids at 10 μM. 3α5β-18-norACN, 3α5βACN, and 3α5αACN have similar potency for potentiation of GABA responses, with EC50 values of 0.6 ± 0.1, 1 ± 0.4, and 0.2 ± 0.4 μM, respectively. These results indicate that there is not a major difference in potency or efficacy among 3α5αACN, 3α5β-18-norACN, and 3α5βACN in potentiating GABA-elicited currents.
Fig. 5.

Neuroactive steroids potentiate GABA currents in X. laevis oocytes expressing α1β2γ2L GABAA receptor subunits. A, superimposed traces of representative currents elicited by 2 μM GABA alone (lowest amplitude trace)) and 2 μM GABA plus 0.1, 1 or 10 μM 3α5β-18-norACN, 3α5βACN, or 3α5αACN. B, the concentration-response curves for 3α5β-18-norACN, 3α5βACN, and 3α5αACN indicate that these three neuroactive steroids have similar potency and efficacy as potentiators of GABA-elicited currents.
The ability of 3α5β-18-norACN, 3α5αACN, and 3α5βACN to directly activate GABAA receptors in the absence of GABA was also examined. To decrease differences among oocytes, the direct gating currents were normalized to currents elicited by 2 μM GABA in the same cell. Figure 6A shows representative traces of steroid-elicited currents in comparison with 2 μM GABA. As shown in Fig. 6B, 3α5β-18-norACN elicited very small GABA currents compared with 3α5αACN; 30 μM 3α5β-18-norACN gated a current 1.7 ± 0.2% as large as that elicited by 2 μM GABA. Based on this low-efficacy concentration-response curve (Fig. 6B, bottom), the observed EC50 value of 3α5β-18-norACN for direct gating was 1.6 ± 0.4 μM. 3α5βACN at 30 μM elicited currents equal to 5.0 ± 0.2% of the 2 μM GABA currents, with an EC50 value of 3.3 ± 0.2 μM. 3α5αACN showed much higher gating efficacy; 30 μM 3α5αACN elicited currents were as large as 34 ± 2.4% of 2 μM GABA. The EC50 value of 3α5αACN could not be accurately determined because maximal effect was not achieved at concentrations that maintained solubility.
Fig. 6.

Neurosteroids directly activate GABAA receptors in X. laevis oocytes expressing α1β2γ2L GABAA receptor subunits. A, superimposed traces of representative currents elicited by 10 μM 3α5β-18-norACN, 3α5βACN, and 3α5αACN. B, concentration-response curves for direct activation of the GABAA receptors by 3α5β-18-norACN, 3α5βACN, and 3α5αACN. Curves for 3α5β-18-norACN and 3α5βACN are enlarged in the bottom panel, with EC50 values equal to 1.6 ± 0.4 and 3.3 ± 0.2 μM, respectively. 3α5αACN has significantly higher maximal gating than 3α5βACN or 3α5β-18-norACN (p < 0.001), but there is no significant difference between 3α5βACN and 3α5β-18-norACN (p > 0.05; n = 4 oocytes tested at each concentration). C, representative GABAA receptor activation traces elicited by 10 μM 3α5αACN, 30 μM 3α5β-18-norACN, or a mixture of 10 μM 3α5αACN with 30 μM 3α5β-18-norACN. D, 30 μM 3α5β-18-norACN significantly antagonizes the direct activation of GABAA receptors by 10 μM 3α5αACN (***, p < 0.001; n = 4).
The low efficacy of 3α5β-18-norACN, coupled with its relatively high apparent potency as a direct activator of GABAA currents suggested that it might antagonize the actions of more efficacious neurosteroids at the site mediating direct activation. To test this idea, we examined the ability of 10 μM 3α5αACN to directly activate currents in the presence and absence of 30 μM 3α5β-18-norACN. The currents elicited by 10 μM 3α5αACN and 30 μM 3α5β-18-norACN were 0.40 ± 0.05 and 0.03 ± 0.01 μA, respectively (Fig. 6C). 3α5β-18-norACN at 30 μM dramatically decreased the current elicited by 10 μM 3α5αACN. In the presence of 3α5β-18-norACN, the current elicited by 10 μM 3α5αACN was 0.10 ± 0.02 μA (Fig. 6D, ***, p < 0.001 versus 3α5αACN alone). These results are consistent with competition between 3α5β-18-norACN and 3α5αACN at the direct activation site.
Anesthetic Effects of 3α5β-18-norACN in Tadpoles and Mice.
To test the anesthetic effects of a relatively selective NS1 agonist, two behavioral endpoints, LRR and LSR were examined in X. laevis prelimb-bud stage tadpoles exposed to varying concentrations of 3α5β-18-norACN. 3α5β-18-norACN caused LRR, with an EC50 value of 164 ± 40 nM (Fig. 7A). Our previous work showed that the EC50 values for LRR by 3α5αACN, 3α5αP, 3α5βACN, and 3α5βP in tadpoles were 70 ± 10, 390 ± 40, 80 ± 13, and 61 ± 4 nM, respectively (Wittmer et al., 1996; Covey et al., 2000). They are not statistically significantly different compared with 3α5β-18-norACN. 3α5β-18-norACN at 1.0 μM caused no LSR. However, 3 and 10 μM μM 3α5β-18-norACN caused LSR in all tadpoles. The ability of 3α5β-18-norACN to anesthetize mice was also examined. 3α5β-18-norACN produced loss of righting reflex at a threshold dose of ≈9 mg/kg i.v. This is similar to the threshold dose of 4 mg/kg for 3α5αACN to produce loss of righting reflex (Wittmer et al., 1996). The duration of loss of righting reflex was dose-dependent (Fig. 7B).
Fig. 7.

Anesthetic effects of 3α5β-18-norACN in X. laevis tadpoles and BALB/c mice. A, compound 3α5β-18-norACN caused LRR and LSR in tadpoles. Points on the tadpole concentration-response curves represent 10 to 20 animals, scored quantally. The EC50 value for LRR was 164 ± 40 nM (S.E.). 3α5β-18-norACN at 0.3 μM has no effect on LSR, whereas 1 and 3 μM 3α5β-18-norACN produce LSR in all the tadpoles. B, intravenous injection of 3α5β-18-norACN produced dose-dependent LRR (sleep times) in mice. Points ± S.E. on the mouse dose-response curve represent the average sleep time for three or four animals and were fit to a straight line.
Discussion
This study demonstrates that compound 3α5β-18-norACN (previously named B285; Akk et al., 2004), a 5β-reduced steroid lacking the 18-methyl group, binds to steroid sites NS1 and NS2 on GABAA receptors, acting as an agonist at the NS1 site and as a weak partial agonist at the NS2 site. Both the 5β-reduced configuration and the absence of the 18-methyl group contribute to the low efficacy of 3α5β-18-norACN at the NS2 site. 3α5β-18-norACN also selectively potentiates GABA-elicited currents but produces minimal direct activation of GABAA receptor currents; it appears to be a weak partial agonist at the site mediating direct activation of GABAA receptors, as it reduces the direct activation of GABAA currents elicited by 3α5αACN.
Action of Neuroactive Steroids on TBPS Binding.
[35S]TBPS (a cage convulsant that binds at the picrotoxin site on GABAA receptors) binding is a useful reporter for the actions of allosteric modulators of GABAA receptors. In well washed brain membranes and in recombinant GABAA receptors, 3α5αACN enhances [35S]TBPS binding at low concentrations and inhibits it at higher concentrations (Figs. 2 and 4), consistent with previous observations of similar actions of allopregnanolone [(3α,5α)-3-hydroxypregnan-20-one], pregnanolone, and alphaxalone (Davies et al., 1997; Srinivasan et al., 1999). We have designated the neuroactive steroid binding site mediating enhancement as NS1 and the inhibitory site as NS2 to distinguish them from sites A and B described using single channel recording (Akk et al., 2004, 2009; Bracamontes and Steinbach, 2009). Sites NS1 and NS2 do not represent steroid binding sites on distinct GABAA receptors differing in subunit composition, because both sites are observed in recombinant α1β2 GABAA receptors (Davies et al., 1997) (Fig. 4A), a combination in which neither subunit expresses well as a homomeric receptor (Bracamontes and Steinbach, 2008).
The biphasic actions of neuroactive steroids on [35S]TBPS binding can be explained using a conceptual model (Fig. 8) in which there are two GABA binding sites and two classes of neuroactive steroid binding sites (NS1 and NS2). It is important to note that the stoichiometry of neurosteroid binding and TBPS binding is not addressed in this model. The stoichiometry of TBPS (picrotoxin) binding to GABAA receptors is not precisely known. Although there may be multiple NS1 and/or NS2 sites on a pentameric GABAA receptor, we have not included NS1 or NS2 stoichiometry in our model, thus making the implicit assumption that occupancy of a single NS1 or NS2 site is sufficient to produce the full effect. Finally, our model assumes that as GABA concentration is increased, GABA sequentially occupies its two binding sites. This implies that at low GABA concentration monoliganded receptors will predominate, whereas diliganded receptors will be the principal species at high GABA concentrations. The above-mentioned assumptions about TBPS and neurosteroid (NS1 and NS2) stoichiometry and GABA site ligation are limitations of our model, which need to be validated before this model can be considered more than a conceptual framework that describes our data.
Fig. 8.

Model of neurosteroid and GABA modulation of [35S]TBPS binding. In the absence of bound GABA or neurosteroid, the receptor (R) can bind [35S]TBPS. When the receptor is monoliganded with GABA (RG) or site NS1 is occupied (RS1), its affinity for TBPS is increased, resulting in increased [35S]TBPS binding. In receptors that are monoliganded with both GABA and S1 (RS1G), TBPS binding is partially inhibited. This partial inhibition is most apparent when 3α5β-18-norACN occupies site NS1, because it has minimal efficacy at site NS2. When receptors are biliganded at either both GABA sites (RGG) or both steroid sites (RS1S2), TBPS binding is completely inhibited.
In the absence of bound GABA or neurosteroid, the receptor (R) can bind [35S]TBPS (Fig. 8). When the receptor is monoliganded with GABA (RG) or site NS1 is occupied (RS1), its affinity for TBPS is increased, resulting in increased [35S]TBPS binding (Figs. 2A and 4, A and C) (Pregenzer et al., 1993; Lüddens and Korpi, 1995). For neurosteroids, the NS1-mediated increase in TBPS binding results from an increase in the receptor's affinity for TBPS, because TBPS binding curves performed in the presence of low concentrations of ACN (100 nM) demonstrate a lower Kd without an increase in Bmax compared with binding curves performed in the absence of neurosteroid (data not shown). When the receptor is monoliganded with GABA and site NS1 is occupied (RS1G), [35S]TBPS binding is partially inhibited. This partial inhibition is most apparent when 3α5β-18-norACN occupies NS1, because it has minimal efficacy at NS2 (Figs. 2D and 4B). Finally, when the receptor is either diliganded with GABA (RGG) or both NS1 and NS2 are occupied (RS1S2), the receptors are unable to bind [35S]TBPS.
Action of 3α5β-18-norACN on TBPS Binding.
3α5β-18-norACN behaves as a selective NS1 site agonist. In the absence of GABA (RS1 in Fig. 8), it enhances TBPS binding at low concentrations and minimally inhibits TBPS binding at higher concentrations (Fig. 2D). 3α5β-18-norACN also occludes the NS1 actions of 3α5αACN, indicating that these two ligands compete for binding at site NS1 and have similar efficacy (Fig. 3A). In contrast, although 3α5β-18-norACN alone produces no NS2 site effect, increasing concentrations of 3α5β-18-norACN antagonize the actions of 3α5αACN as an NS2 site agonist (Fig. 3B); this suggests that 3α5β-18-norACN occupies the NS2 site but has minimal efficacy.
In the presence of 5 μM GABA (Fig. 2), both the NS1 and NS2 sites contribute to complete inhibition of TBPS binding. 3α5β-18-norACN only partially inhibits TBPS binding indicating that it lacks the NS2 site effect (Figs. 2D and 4B; RS1G in Fig. 8). An 18-methyl group and a 5α-reduced configuration were identified as important contributors to agonist efficacy at the NS2 site. Neurosteroids lacking the 18-methyl and/or having a 5β-reduced configuration can bind to the NS2 site but have minimal agonist activity. Collectively, these data confirm that the NS1 and NS2 sites are nonidentical and indicate the feasibility of developing selective agonists and antagonists for these distinct steroid binding sites. Several other steroid analogs with the 5β-reduced configuration, including the 3,20-pregnanediols and (3α,5β)-3,21-dihydroxypregnan-20-one, have also been shown to partially inhibit [35S]TBPS binding in the presence of GABA and have been classified as partial agonists (Belelli and Gee, 1989; Morrow et al., 1990; Hawkinson et al., 1996; Xue et al., 1997). These compounds may also be selective ligands for either the NS1 or NS2 sites.
Electrophysiological Action of 3α5β-18-norACN.
At a macroscopic level, 3α5β-18-norACN potentiates GABA-elicited currents with potency and efficacy similar to that of 3α5αACN (Fig. 5). 3α5β-18-norACN shows minimal efficacy as a direct activator of GABAA receptors (α1β2γ2L) expressed in X. laevis oocytes (Fig. 6). Furthermore, 3α5β-18-norACN antagonizes the direct activation elicited by 3α5αACN, suggesting that it is a weak partial agonist at the site mediating direct activation of GABAA receptors.
Relationship between Neurosteroid Binding Sites Identified in Radioligand Binding and Electrophysiological Assays.
Two distinct concentration-dependent effects of neurosteroids are observed in [35S]TBPS binding assays. We have interpreted these data as indicative of two distinct (NS1 and NS2) neurosteroid binding sites on the GABAA receptor. Neurosteroids also produce two distinct concentration-dependent effects in electrophysiological assays: potentiation of GABA-responses at low neurosteroid concentrations and direct activation of the GABAA receptor at high concentrations. Site-directed mutagenesis studies indicate that these electrophysiological effects are mediated by two neurosteroid binding sites, one site that mediates potentiation and one site that mediates direct activation (Hosie et al., 2006). Although the relationship between the sites observed in radioligand binding studies and electrophysiological studies is not defined, it is simple and attractive to consider that the two assays are describing the same sites with NS1 corresponding to the potentiation site and NS2 corresponding to the direct activation site. 3α5β-18-norACN provides some evidence in support of this hypothesis: the concentrations of 3α5β-18-norACN that activate NS1 correspond closely with those that produce potentiation of GABA-elicited currents. 3α5β-18-norACN is also a poor agonist at the NS2 site in the TBPS binding assay and a weak direct activator of GABAA currents. Finally, 3α5β-18-norACN prevents the actions of 3α5α-ACN as an NS2 agonist in TBPS binding and as a direct activator of GABAA currents. Although these data support the hypothesis that the NS1 and NS2 sites are synonymous with the potentiating and direct activating sites, more extensive studies are required to confirm these assignments. Specifically, parallel examination of a larger set of neurosteroids in [35S]TBPS binding assays and whole cell electrophysiological assays in both wild-type GABAA receptors and receptors in which the potentiation and activation sites have been mutated would provide a more thorough test of this hypothesis.
Akk and colleagues have provided evidence for multiple neurosteroid binding sites (A, B1, and B2) in single channel electrophysiological studies using recombinant α1β2γ2-subunit GABAA receptors (Akk et al., 2004; Li et al., 2007). Their studies were performed with 50 μM GABA, a concentration at which most receptors are diliganded with GABA and thus not observable with [35S]TBPS binding. In our model, the neurosteroid effects they observe would correspond to states in which the receptor is diliganded with GABA and sites NS1, NS2, or both are occupied. None of these sites could be observed with [35S]TBPS binding. There is thus no basis for correlating the multiple neurosteroid binding sites characterized by single channel electrophysiology with sites NS1 and NS2 demonstrated in this study.
Anesthetic Action of 3α5β-18-norACN.
Compound 3α5β-18-norACN also provides a tool for understanding the biological actions of neurosteroids at the NS1 and NS2 sites. 3α5β-18-norACN produces loss-of-righting reflex in X. laevis tadpoles and in mice (Fig. 7). The loss-of-righting reflex and loss-of-swimming reflex effects of 3α5β-18-norACN have a similar concentration dependence to those of 3α5αACN and 3α5βACN (Wittmer et al., 1996; Covey et al., 2000). Because 3α5αACN, 3α5β-ACN, and 3α5β-18-norACN all have similar efficacy at NS1 and as potentiators of GABA-elicited currents and 3α5β-18-norACN has minimal efficacy at NS2 or as a direct activator of GABAA receptors, these data indicate that efficacy at NS1 and potentiation of GABA-elicited currents is sufficient for a neurosteroid to produce loss-of-righting reflex.
In summary, the behavior of 3α5β-18-norACN in radioligand binding and electrophysiological assays increases the evidence that there are two classes of neurosteroid binding sites on GABAA receptors that can be distinguished by selected neurosteroid ligands. The initial description of the structure-activity requirements for efficacy at these sites should provide impetus for the development of selective agonist and antagonists for the two neurosteroid sites. The development of such selective neurosteroid ligands will be a vital tool for elucidating the biological actions of endogenous neurosteroids and may provide useful clinical agents.
Acknowledgments
We thank Amanda Taylor and Ann Benz for technical help with the oocyte studies. J.H.S. is the Russell and Mary Shelden Professor of Anesthesiology and A.S.E. is the Henry E. Mallinckrodt Professor of Anesthesiology.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM P01-47969] (to A.S.E., J.H.S., C.F.Z., and D.F.C.).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.164079.
- TBPS
- t-butylbicyclophosphorothionate
- 3α5αACN
- (3α,5α,17β)-3-hydroxyandrostane-17-carbonitrile
- 3α5βACN
- (3α,5β,17β)-3-hydroxyandrostane-17-carbonitrile
- 3α5β-18-norACN
- (3α,5β,17β)-3-hydroxy-18-norandrostane-17-carbonitrile
- 3α5α-18-norACN
- (3α,5α,17β)-3-hydroxy-18-norandrostane-17-carbonitrile
- 3α5β-19-norACN
- (3α,5β,17β)-3-hydroxyestrane-17-carbonitrile
- 3α5α-19-norACN
- (3α,5α,17β)-3-hydroxyestrane-17-carbonitrile
- 3α5β-18,19-dinorACN
- (3α,5β,17β)-3-hydroxygonane-17-carbonitrile
- 3α5α-18,19-dinorACN
- (3α,5α,17β)-3-hydroxygonane-17-carbonitrile
- 3α5αP
- (3α,5α)-3-hydroxypregnan-20-one
- 3α5βP
- (3α,5β)-3-hydroxypregnan-20-one
- 3α5β-18-norP
- (3α,5β,17β)-3-hydroxy-18-norpregnan-20-one
- 3α5α-18-norP
- (3α,5α,17β)-3-hydroxy-18-norpregnan-20-one
- 3α5β-19-norP
- (3α,5β,17β)-3-hydroxy-19-norpregnan-20-one
- 3α5α-19-norP
- (3α,5α,17β)-3-hydroxy-19-norpregnan-20-one
- IR
- infrared
- Anal. Calcd
- analytical calculated:
- LRR
- loss-of-righting reflex
- LSR
- loss-of-swimming reflex
- R
- receptor
- RS1
- site NS1 is occupied
- RS1G
- receptor is monoliganded with GABA and site NS1 is occupied.
References
- Akk G, Bracamontes JR, Covey DF, Evers A, Dao T, Steinbach JH. (2004) Neuroactive steroids have multiple actions to potentiate GABAA receptors. J Physiol 558:59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Bracamontes J, Steinbach JH. (2009) Activation and modulation of concatemeric GABA-A receptors expressed in human embryonic kidney cells. Mol Pharmacol 75:1400–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JL, Harrison NL, Lange GD, Owen DG. (1987) Potentiation of gamma-aminobutyric-acid-activated chloride conductance by a steroid anaesthetic in cultured rat spinal neurones. J Physiol 386:485–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Gee KW. (1989) 5 alpha-pregnan-3 alpha,20 alpha-diol behaves like a partial agonist in the modulation of GABA-stimulated chloride ion uptake by synaptoneurosomes. Eur J Pharmacol 167:173–176 [DOI] [PubMed] [Google Scholar]
- Bracamontes JR, Steinbach JH. (2008) Multiple modes for conferring surface expression of homomeric beta1 GABAA receptors. J Biol Chem 283:26128–26136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracamontes JR, Steinbach JH. (2009) Steroid interaction with a single potentiating site is sufficient to modulate GABA-A receptor function. Mol Pharmacol 75:973–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Fletcher GH, Steinbach JH. (1995) Selection of stably transfected cells expressing a high level of fetal muscle nicotinic receptors. J Neurosci Res 40:606–612 [DOI] [PubMed] [Google Scholar]
- Cottrell GA, Lambert JJ, Peters JA. (1987) Modulation of GABAA receptor activity by alphaxalone. Br J Pharmacol 90:491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey DF, Nathan D, Kalkbrenner M, Nilsson KR, Hu Y, Zorumski CF, Evers AS. (2000) Enantioselectivity of pregnanolone-induced gamma-aminobutyric acid(A) receptor modulation and anesthesia. J Pharmacol Exp Ther 293:1009–1016 [PubMed] [Google Scholar]
- Darbandi-Tonkabon R, Hastings WR, Zeng CM, Akk G, Manion BD, Bracamontes JR, Steinbach JH, Mennerick SJ, Covey DF, Evers AS. (2003) Photoaffinity labeling with a neuroactive steroid analogue. 6-azi-pregnanolone labels voltage-dependent anion channel-1 in rat brain. J Biol Chem 278:13196–13206 [DOI] [PubMed] [Google Scholar]
- Davies PA, Kirkness EF, Hales TG. (1997) Modulation by general anaesthetics of rat GABAA receptors comprised of alpha 1 beta 3 and beta 3 subunits expressed in human embryonic kidney 293 cells. Br J Pharmacol 120:899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Covey DF. (1996) An improved synthesis of 18-nor-17-ketosteroids and application of the method for the preparation of (3beta,5beta,13alpha)- and (3beta,5beta,13beta)-3-hydroxygonan-17-one. J Org Chem 61:7614–7616 [DOI] [PubMed] [Google Scholar]
- Han M, Zorumski CF, Covey DF. (1996) Neurosteroid analogues. 4. The effect of methyl substitution at the C-5 and C-10 positions of neurosteroids on electrophysiological activity at GABAA receptors. J Med Chem 39:4218–4232 [DOI] [PubMed] [Google Scholar]
- Hauser CA, Chesnoy-Marchais D, Robel P, Baulieu EE. (1995) Modulation of recombinant alpha 6 beta 2 gamma 2 GABAA receptors by neuroactive steroids. Eur J Pharmacol 289:249–257 [DOI] [PubMed] [Google Scholar]
- Hawkinson JE, Drewe JA, Kimbrough CL, Chen JS, Hogenkamp DJ, Lan NC, Gee KW, Shen KZ, Whittemore ER, Woodward RM. (1996) 3 alpha-Hydroxy-3 beta-trifluoromethyl-5 alpha-pregnan-20-one (Co 2-1970): a partial agonist at the neuroactive steroid site of the gamma-aminobutyric acidA receptor. Mol Pharmacol 49:897–906 [PubMed] [Google Scholar]
- Hawkinson JE, Kimbrough CL, Belelli D, Lambert JJ, Purdy RH, Lan NC. (1994a) Correlation of neuroactive steroid modulation of [35S]t-butylbicyclophosphorothionate and [3H]flunitrazepam binding and gamma-aminobutyric acidA receptor function. Mol Pharmacol 46:977–985 [PubMed] [Google Scholar]
- Hawkinson JE, Kimbrough CL, McCauley LD, Bolger MB, Lan NC, Gee KW. (1994b) The neuroactive steroid 3 alpha-hydroxy-5 beta-pregnan-20-one is a two-component modulator of ligand binding to the GABAA receptor. Eur J Pharmacol 269:157–163 [DOI] [PubMed] [Google Scholar]
- Hosie AM, Clarke L, da Silva H, Smart TG. (2009) Conserved site for neurosteroid modulation of GABAA receptors. Neuropharmacology 56:149–154 [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. (2006) Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444:486–489 [DOI] [PubMed] [Google Scholar]
- Hu Y, Zorumski CF, Covey DF. (1993) Neurosteroid analogues: structure-activity studies of benz[e]indene modulators of GABAA receptor function. 1. The effect of 6-methyl substitution on the electrophysiological activity of 7-substituted benz[e]indene-3-carbonitriles. J Med Chem 36:3956–3967 [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA. (2003) Neurosteroid modulation of GABAA receptors. Prog Neurobiol 71:67–80 [DOI] [PubMed] [Google Scholar]
- Li P, Bracamontes J, Katona BW, Covey DF, Steinbach JH, Akk G. (2007) Natural and enantiomeric etiocholanolone interact with distinct sites on the rat alpha1beta2gamma2L GABAA receptor. Mol Pharmacol 71:1582–1590 [DOI] [PubMed] [Google Scholar]
- Lüddens H, Korpi ER. (1995) GABA antagonists differentiate between recombinant GABAA/benzodiazepine receptor subtypes. J Neurosci 15:6957–6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. (1986) Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232:1004–1007 [DOI] [PubMed] [Google Scholar]
- Morrow AL, Pace JR, Purdy RH, Paul SM. (1990) Characterization of steroid interactions with gamma-aminobutyric acid receptor-gated chloride ion channels: evidence for multiple steroid recognition sites. Mol Pharmacol 37:263–270 [PubMed] [Google Scholar]
- Pregenzer JF, Im WB, Carter DB, Thomsen DR. (1993) Comparison of interactions of [3H]muscimol, t-butylbicyclophosphoro[35S]thionate, and [3H]flunitrazepam with cloned gamma-aminobutyric acidA receptors of the alpha 1 beta 2 and alpha 1 beta 2 gamma 2 subtypes. Mol Pharmacol 43:801–806 [PubMed] [Google Scholar]
- Srinivasan S, Sapp DW, Tobin AJ, Olsen RW. (1999) Biphasic modulation of GABA(A) receptor binding by steroids suggests functional correlates. Neurochem Res 24:1363–1372 [DOI] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. (1996) Endogenous subunits can cause ambiguities in the pharmacology of exogenous gamma-aminobutyric acidA receptors expressed in human embryonic kidney 293 cells. Mol Pharmacol 50:931–938 [PubMed] [Google Scholar]
- Wittmer LL, Hu Y, Kalkbrenner M, Evers AS, Zorumski CF, Covey DF. (1996) Enantioselectivity of steroid-induced gamma-aminobutyric acidA receptor modulation and anesthesia. Mol Pharmacol 50:1581–1586 [PubMed] [Google Scholar]
- Xue BG, Whittemore ER, Park CH, Woodward RM, Lan NC, Gee KW. (1997) Partial agonism by 3alpha,21-dihydroxy-5beta-pregnan-20-one at the gamma-aminobutyric acidA receptor neurosteroid site. J Pharmacol Exp Ther 281:1095–1101 [PubMed] [Google Scholar]