

Abstract
Prolonged ethanol exposure causes central nervous system hyperexcitability that involves a loss of GABAergic inhibition. We previously demonstrated that long-term ethanol exposure enhances the internalization of synaptic GABAA receptors composed of α1β2/3γ2 subunits. However, the mechanisms of ethanol-mediated internalization are unknown. This study explored the effect of ethanol on surface expression of GABAA α1 subunit-containing receptors in cultured cerebral cortical neurons and the role of protein kinase C (PKC) β, γ, and ε isoforms in their trafficking. Cultured neurons were prepared from rat pups on postnatal day 1 and maintained for 18 days. Cells were exposed to ethanol, and surface receptors were isolated by biotinylation and P2 fractionation, whereas functional analysis was conducted by whole-cell patch-clamp recording of GABA- and zolpidem-evoked responses. Ethanol exposure for 4 h decreased biotinylated surface expression of GABAA receptor α1 subunits and reduced zolpidem (100 nM) enhancement of GABA-evoked currents. The PKC activator phorbol-12,13-dibutyrate mimicked the effect of ethanol, and the selective PKC inhibitor calphostin C prevented ethanol-induced internalization of these receptors. Ethanol exposure for 4 h also increased the colocalization and coimmunoprecipitation of PKCγ with α1 subunits, whereas PKCβ/α1 association and PKCε/α1 colocalization were not altered by ethanol exposure. Selective PKCγ inhibition by transfection of selective PKCγ small interfering RNAs blocked ethanol-induced internalization of GABAA receptor α1 subunits, whereas PKCβ inhibition using pseudo-PKCβ had no effect. These findings suggest that ethanol exposure selectively alters PKCγ translocation to GABAA receptors and PKCγ regulates GABAA α1 receptor trafficking after ethanol exposure.
Long-term ethanol exposure causes central nervous system hyperexcitability and tremor that has been partly attributed to a loss of GABAA α1 receptor subunit expression in vivo (Kralic et al., 2002, 2005; for review, see Kumar et al, 2009). This effect of ethanol is recapitulated in cultured cortical neurons in vitro (Sanna et al., 2003; Sheela Rani and Ticku, 2006). The reduction of GABAA α1 subunit protein has been determined to involve the internalization of GABAA α1 subunit receptors in the cerebral cortex (Kumar et al., 2003) and the hippocampus (Liang et al., 2007), where a reduction in surface expression of these receptors has been demonstrated. Furthermore, ethanol exposure produces a reduction in sensitivity to the α1 GABAA receptor-selective modulator zolpidem in hippocampal neurons both in vivo (Liang et al., 2004) and in vitro (Sanna et al., 2003), consistent with the interpretation that ethanol promotes α1 subunit-containing GABAA receptor internalization.
GABAA receptors are heteropentameric ligand-gated ion channels that mediate inhibition in the central nervous system. The α1 subunit receptors are the most abundant subtype of synaptic GABAA receptor, expressed widely throughout the brain, and are generally composed of two α1, two β2 or β3, and one γ2 subunit that confer sensitivity to benzodiazepines (Sieghart and Sperk, 2002). Global knockout of the GABAA receptor α1 subunit results in increased seizure susceptibility and essential-like tremor (Kralic et al., 2002). Previous studies have demonstrated that trafficking of α1 subunit-containing GABAA receptors is critical for synaptic efficacy and can be regulated by many factors, including PKC-mediated phosphorylation (Chapell et al., 1998; Connolly et al., 1999; Poisbeau et al., 1999; Brandon et al., 2000).
The mechanisms by which ethanol promotes the internalization of α1 subunit-containing GABAA receptors are not yet elucidated. Ethanol induces GABAA α1 receptor association with adaptor-complex 2 and internalization into clathrin-coated vesicles (CCVs) in cerebral cortex (Kumar et al., 2003). Ethanol has also been shown to alter the expression and translocation of various PKC isoforms (Kumar et al., 2006), but the role of these isoforms in receptor internalization is unknown. Studies with PKCε and PKCγ knockout mice have demonstrated the potential relevance of these PKC isoforms to ethanol-mediated plasticity (Hodge et al., 1999; Bowers et al., 2001; Proctor et al., 2003; Choi et al., 2008). Mutant mice lacking PKCε are more sensitive to the short-term effects of ethanol, and the deletion of PKCε attenuates ethanol withdrawal-associated seizures in mice (Olive et al., 2001). In contrast, PKCγ knockout mice are resistant to the short-term intoxicating effects of ethanol and fail to develop ethanol tolerance. Therefore, it seems that these isoforms of PKC are important for ethanol-mediated behavioral effects and may be involved in ethanol-induced plasticity.
Twelve isoforms of PKC exhibit cell- and region-specific distribution in the brain. Recent studies show that PKCβ, -γ, and -ε are associated with GABAA receptors (Brandon et al., 1999; Kumar et al., 2002). PKC has been shown to alter surface expression of both recombinant GABAA receptors and native receptors in cultured neurons (Chapell et al., 1998) by endocytosis into CCVs (Kittler et al., 2000). However, other evidence suggests that PKC prevents receptor recycling back to the surface (Connolly et al., 1999). To understand the mechanisms of ethanol effects on surface expression of GABAA receptors, it is critical to determine the role of various PKC isoforms because this knowledge may be useful to modulate trafficking of α1 subunit-containing GABAA receptors.
The present study focused on the role of PKC isoforms on ethanol-induced alterations of GABAA α1 receptor expression in cultured cortical neurons. Trafficking of GABAA receptors was assessed by determination of α1 GABAA receptor peptide levels on the cell surface by biotinylation of surface proteins. To establish that internalization of α1 subunit peptides represented internalization of α1 subunit-containing GABAA receptors, we also explored the effects of ethanol on zolpidem (100 nM) enhancement of GABA evoked Cl− currents to assess the functional consequences of changes in surface expression of α1 subunit receptors. We explored the effects of ethanol on PKCβ, -γ, and -ε expression and their interactions with GABAA α1 subunits by coimmunoprecipitation and dual fluorescence confocal microscopy. Furthermore, using PKCγ-specific siRNA and a PKCβ inhibitor, we determined the role of these PKC isoforms on ethanol-induced loss of α1 subunit-containing GABAA receptors.
Materials and Methods
Cultured Cerebral Cortical Neurons.
Experiments were conducted in accordance with National Institutes of Health Guidelines under Institutional Animal Care and Use Committee-approved protocols. Rat pups were decapitated and the brains removed on postnatal day 1. The cerebral cortex was isolated from the brain stem, hippocampus, and olfactory bulb, and the meninges were removed from the cortical tissue. The tissue was then placed in a solution of papain, l-cysteine and DNase in CO2-independent medium and minced into small pieces of 1 to 2 mm2. The tissue was incubated at 37°C for 30 min and then dissociated by trituration into a growth medium consisting of Dulbecco's modified Eagle's medium supplemented with 10% horse serum and Penicillin-Streptomycin (Pen-Strep; 10,000 U/ml; final concentration in flasks, 50 units; Invitrogen, Carlsbad, CA). Cells were plated on poly-d-lysine–coated flasks (Corning Life Sciences, Lowell, MA), and glass coverslips (Carolina Biological Supply Company, Burlington, NC) in 12-well plates at a density of 1.0 to 1.5 × 106 live cells per well. Healthy neurons grew processes within 24 h after plating. Glial cells were retained in the culture to encourage neuron survival and synapse formation. After 3 days in culture, cells were fed with serum-free medium (Dulbecco's modified Eagle's medium supplemented with B27) containing Pen-Strep (50 units) to prevent glial cell overgrowth. Pen-Strep was removed from the cultures on day 14. Rat cortical neurons were maintained in vitro for at least 18 days before experimentation to allow expression of mature subtypes of PKC isozymes and GABAA receptors.
Ethanol and Drug Exposure of Cultured Cerebral Cortical Neurons.
Cultured cells were exposed to ethanol using a vapor chamber for the maintenance of high ethanol concentrations in the tissue culture medium. At the beginning of the ethanol exposure period, cells were fed by replacing one third of the tissue culture medium with fresh medium containing ethanol (final concentration, 50 mM). Stable ethanol levels were maintained up to 5 days in the medium by placing the cells in a plastic vapor chamber containing a beaker with 200 ml of 50 mM ethanol in distilled water. Control cells were fed with media that did not contain ethanol and were placed in a vapor chamber with a beaker containing water. After ethanol exposure, cells were removed from the vapor chamber and washed twice with ice-cold phosphate-buffered saline (PBS), and the P2 fraction was prepared as described below for Western blot analysis of PKC isoforms and GABAA receptor subunits using specific antibodies. For immunohistochemistry, cells on coverslips were fixed with 4% paraformaldehyde, washed twice with PBS, and stored at 4°C. Calphostin C and PDBu were purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in 0.1% dimethyl sulfoxide.
For inhibition of specific PKC isoforms, we used PKCγ siRNA and PKCβ pseudosubstrate to selectively reduce PKC isozyme expression in cultured cerebral cortical neurons. Three different siRNA sequence pairs were obtained from Invitrogen and used simultaneously to inhibit PKCγ (PRKCCRSS332451, PRKCCRSS332452, and PRKCCRSS332453). The sequences are as follows: pair 1, 5′-GGAGGAGGGCGAGUAUUACAAUGUA-3′ and 5′-UACAUUGUAAUACUCGCCCUCCUCC-3′; pair 2, 5′-UCGGCAUGUGUAAAGAGAAUGUCUU-3′ and 5′-AAGACAUUCUCUUUACACAUGCCGA-3′; and pair 3, 5′-CCUGCAAUGUCAAGUCUGCAGCUUU-3′ and 5′-AAAGCUGCAGACUUGACAUUGCAGG-3′.
ClustalW alignment indicates that the sequences were to nucleotides 1058 to 1082, 1714 to 1738, and 407 to 431, respectively, of the rat protein kinase Cγ mRNA sequence (Gen Bank Accession number NM_012628.1). The specificity of all siRNA sequences were BLAST-analyzed to make sure no other unwanted targets would be potentially down-regulated. For transfection, cerebral cortical neurons were cultured for 14 days as described above, and siRNA for PKCγ or scrambled siRNA was transfected using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. Selective PKCγ siRNA or scrambled siRNA were mixed in 200 μl of OptiMEM I low-serum media (Invitrogen) with 6 μl of Lipofectamine reagent. Complexes were allowed to form for 20 min then added to cells (6 pmol). Cells were subjected to gentle rocking by hand and incubated at 37°C for 24 to 72 h before collection of cells. Experiments were initiated 67 to 68 h after transfection because inhibition of PKCγ was maximal at 72 h. PKCβ pseudosubstrate was obtained from Tocris (Ellisville, MO) and used to block PKCβ activity (final concentration, 0.1 μM). PKCβ pseudosubstrate was applied with ethanol at the beginning of experiment and after 2 h to maintain inhibition for 4 h of ethanol exposure.
Whole Cell Voltage-Clamp Recordings.
Standard whole-cell voltage-clamp recordings were made with glass electrodes, fire-polished to a resistance of 3 to 5 MΩ and filled with internal solution (150 mM KCl, 3.1 mM MgCl2, 15 mM HEPES, 5 mM K-ATP, 5 mM EGTA, and 15 mM phosphocreatine, adjusted to pH 7.4 with KOH). The recording chamber was perfused with external solution (145 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 5 mM sucrose, and 10 mM glucose, adjusted to pH 7.4 with NaOH). The sodium-channel blocker tetrodotoxin (0.5 μM; Sigma, St. Louis, MO), was included in the perfusion solution. Drugs were diluted in external solution and applied using a U-tube apparatus. This technique allowed a brief cellular application and rapid removal of drugs. The interval between applications was at least 1 min. Recordings were performed at room temperature (22–23°C). The membrane potential was held at −60 mV using a patch-clamp amplifier (Axopatch 1D; Molecular Devices, Sunnyvale, CA), and data were collected with Clampex 10.2 software (Molecular Devices).
Concentration-response curves were determined to select the proper concentration of GABA to detect enhancement of currents by 100 nM zolpidem (Sigma). The logEC50 for GABA was 0.91 ± 0.08 (8.19 μM), and the Hill coefficient was 1.3 ± 0.3. The EC10 was estimated to approximately 1.49 μM. On the basis of these data, we employed a GABA concentration of 1 μM, a value close to the EC10, to investigate zolpidem enhancement of GABA responses. For each neuron, GABA was applied two to three consecutive times to obtain a stable baseline. One hundred nanomolar zolpidem was coapplied with 1 μM GABA for 8 s to study enhancement of GABA currents. For each neuron, the percentage potentiation of 1 μM GABA current by 100 nM zolpidem was calculated as follows: % potentiation = (current in presence of GABA + zolpidem ÷ current in presence of GABA alone) · 100. The percentage potentiation for each experimental group was compared statistically using the Student's t test or ANOVA as appropriate. A p value < 0.05 was considered statistically significant.
Tissue Preparation.
P2 membrane fractions from cultured cortical neurons were prepared by homogenization, low-speed centrifugation in 0.32 M sucrose, and centrifugation of the supernatant at 12,000g for 20 min (Kumar et al., 2002). The pellet was resuspended in PBS with phosphatase inhibitor cocktail I (proprietary mixture of Microcystin LR, cantharidin, and bromotetramisole; Sigma-Aldrich) and stored at −80°C. Protein was quantified using the bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA).
Biotinylation of Surface Receptors.
Surface expression of receptors was determined in cultured cells using a biotin labeling kit (Pierce Chemical) as described by the manufacturer. Cerebral cortical cells were cultured on poly-d-lysine–coated flasks for 18 days. On day 19, cells were incubated with ethanol and/or PKC modulators or vehicle. The cells were washed twice with ice-cold PBS followed by addition of sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (10 ml in each flask; Pierce Chemical) diluted with ice-cold PBS. The cells were gently mixed with biotin reagent on a rocker for 30 min at 4°C. Unbound biotin was inactivated with quenching solution (Pierce Chemical). The cells were then scraped and transferred to a 50-ml conical tube and washed three times by adding Tris-buffered saline and centrifuging at 500g for 5 min. After washing, lysis buffer provided in the kit (500 μl) was added and cells were sonicated on ice for five 1-s bursts. Biotin-labeled proteins and flowthrough (cytosolic proteins) were separated with NeutrAvidin slurry (Pierce Chemical) as described by the manufacturer. The biotinylated (surface) proteins were eluted from the beads by incubation for 60 min at room temperature with an equal volume of Laemmli SDS-polyacrylamide gel electrophoresis sample buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 0.05% mercaptoethanol, and 0.05% bromphenol blue). The samples were then subjected to gel electrophoresis and Western blotting.
Immunoprecipitation.
Immunoprecipitation was conducted as described previously (Kumar et al., 2002). In brief, 135 μg of P2 fraction protein was solubilized at 4°C in 1% (w/v) Triton X-100, 1% (w/v) sodium deoxycholate, and 0.1% (w/v) SDS, 140 mM NaCl, and 10 mM Tris-HCl, pH 7.5, and protease inhibitors. Dynabeads (125 μl) were washed three times with 0.5 ml of 0.1 M phosphate buffer with 0.1% BSA, pH 8.1, and resuspended in 100 μl of 0.1 M phosphate buffer. GABAA receptor α1 subunit-specific antibody (rabbit polyclonal; Werner Sieghart) (14.5 μl, 628 μg/μl) was conjugated to Dynabeads (Invitrogen) by incubation for 1 h at room temperature with agitation, and then washed once with 0.1 M phosphate buffer/0.1% BSA and twice with 0.2 M triethanolamine, pH 8.2. Antibodies were cross-linked with protein A in 1 ml of 0.2 M triethanolamine and 5 mg of dimethyl pimelimidate cross-linker to prevent coelution of antibody with receptor. The reaction was incubated for 30 min at room temperature with constant rotation and washed three times with PBS/0.1% BSA.
Solubilized receptors and antibody conjugated to Dynabeads were mixed and incubated overnight at 4°C in a final volume of 500 μl of solubilizing buffer. Receptor-antibody beads were then washed three times with PBS. After the final wash, the receptor-antibody-bead complex was resuspended in 50 μl of 1× SDS and boiled for 5 min. Beads were separated from the immunoprecipitate by exposure to a magnet for 2 min, and the immunoprecipitate was collected and denatured with 1 μl of 5 M dithiothreitol, subjected to Western blot analysis, and immunoblotted with various antibodies using standard immunoblotting procedures.
Because the expression of α1 subunits is altered in P2 membrane fractions after ethanol (4 h) exposure, it was necessary to conduct immunoprecipitation experiments under conditions of limited antibody so that identical amounts of α1 subunit receptors would be isolated from both control and experimental groups. Because the concentration of antibody determines the number of receptors immunoprecipitated, it was possible to determine whether ethanol exposure altered the association of PKCβ and -γ with an equivalent sample of α1 subunit receptors from each experimental group of cells. We confirmed that an equivalent amount of α1 subunits was immunoprecipitated across experimental groups. Furthermore, the signal intensity for PKCβ and γ was normalized to the signal intensity for the α1 subunit in the α1 immunoprecipitate to rule out the possibility of nonequivalent immunoprecipitation efficiency between experimental groups (for explanation see Kumar et al., 2002). Coimmunoprecipitation of PKCε with GABAA α1 subunit receptors is not detected with this method (Kumar et al., 2002). Therefore, colocalization of PKCε with the GABAA receptor α1 subunit was assessed by dual fluorescence confocal microscopy.
Western Blot Analysis.
The various subcellular fractions were analyzed by Western blot analysis under conditions of protein linearity (Kumar et al., 2003). P2 fractions were subjected to SDS-PAGE using Novex Tris-Glycine gels (8–16%) and transferred to polyvinylidene difluoride membranes (Invitrogen). The membranes with transferred proteins were probed with α1 (Thermo Fisher Scientific), PKCβ, PKCγ, and PKCε antibodies (BD Biosciences, San Jose, CA). Guinea pig α1 subunit antibody (provided by Jean-Marc Fritschy, University of Zurich, Zurich, Switzerland) was used for Western blot analysis of the α1 subunit immunoprecipitate to avoid interference with the rabbit α1 subunit antibody used for immunoprecipitation. Blots were subsequently exposed to a second primary antibody directed against β-actin, to verify equivalent protein loading and transfer. Bands were detected by enhanced chemiluminescence (GE Healthcare, Chalfont, St. Giles, Buckinghamshire, UK), exposed to X-ray films under nonsaturating conditions, and analyzed by densitometric measurements using NIH Image 1.57 (http://rsb.info.nih.gov/nih-image/). All comparisons were made within blots. Statistical analysis was conducted using the Student's t test or one-way ANOVA.
Dual Label Confocal Fluorescence Microscopy.
Staining and microscopy were performed as described by Essrich et al. (1998) with some modifications. Coverslips with cortical neurons (paraformaldehyde fixed) were used for staining and confocal microscopy. Antibody dilution curves were performed to determine the optimum concentration of primary and secondary antibody for staining. Specific labeling was determined by staining in the absence of primary antibodies and by monitoring fluorescence detection in every channel to exclude bleed-through. In brief, coverslips were washed with PBS and permeabilized with 0.1% Triton X-100 (w/v) in PBS containing 10% normal horse serum (Vector Laboratories, Burlingame, CA) for 30 min at room temperature. After washing, the cells were stained with anti-PKCβ (mouse, 1:75 dilution; BD Biosciences, San Jose, CA), anti-PKCε (mouse, 1:100 dilution; BD Biosciences), or anti-PKCγ (mouse, 1:75 dilution; BD Biosciences) and GABAA α1 antibody (rabbit, 1:400 dilution; Novus Biologicals, Littleton, CO) overnight at 4°C. The following morning, coverslips were washed with PBS (containing 0.1% Triton X-100 and 10% serum) followed by incubation with the fluorescent-coupled secondary antibodies, Alexa Fluor 488 goat anti-mouse IgG (1:200 dilution) and Alexa Fluor 594 goat anti-rabbit IgG (1:200 dilution; Invitrogen) for 1 h at room temperature. Coverslips were washed and mounted onto glass slides with mounting medium (Fluoromount-G; Southern Biotechnology Associates, Birmingham, AL) and examined under a confocal laser scanning microscope.
Visualization Was Performed with the Use of a Leica SP2 Laser Scanning Confocal Microscope.
(Wetzlar, Germany). The microscope was used at 40× magnification to identify relevant GABAA receptor subunits (α1, 594, Red Color) using the objective lens and 594 filter. Next, the view field was scanned in both 594 (anti-rabbit-red) and 488 (anti-mouse-green). Images were saved for analysis by an observer (K.N.B.) blind to the experimental conditions. Approximately 20 to 27 cells expressing GABAA receptor α1 subunits were identified on each coverslip (five per quadrant). Each cell was then scored for colocalization of the PKC isozyme labeled. The total number of α1 subunit/PKC colocalized cells were normalized to total number of α1 subunit-stained cells on the same coverslip and presented as a ratio. Colocalization of each PKC isozyme with GABAA α1 subunit receptors was determined in independent experiments from five coverslips (approximately 30 cells/coverslip).
Results
Ethanol and PKC Activator PDBu Alter the Cell-Surface Expression of GABAA Receptor α1 Subunits.
Ethanol exposure (50 mM) for 4 h produced a maximal decrease in expression of GABAA receptor α1 subunit (40.2 ± 15%, n = 5, p < 0.05) in the P2 membrane fraction of cerebral cortical neurons compared with control values (Fig. 1A). The effect of ethanol was dose-dependent, and 4-h ethanol (25 mM) exposure did not significantly decrease GABAA receptor α1 subunits in the P2 fraction (Fig. 1B). Biotinylation of surface proteins confirmed the decrease in cell-surface expression of α1 subunits after 4 h of ethanol exposure (Fig. 1C, 38.0 ± 14%, n = 4, p < 0.05). Furthermore, unlabeled cytosolic α1 subunit protein levels were increased by 111.1 ± 35% (Fig. 1D, n = 4, p < 0.05). PKC activators have been shown to internalize GABAA receptors in recombinant cell expression systems (Chapell et al., 1998) and primary cultures (Connolly et al., 1999). We used the PKC activator PDBu to determine whether PKC has similar activity in our primary cell culture. PDBu exposure for 1 h reduced the expression of α1 subunits by 37.3 ± 7% in the P2 fraction (Fig. 2A, n = 4, p < 0.05). Likewise, biotinylation of surface receptors revealed approximately the same decrease in GABAA receptor α1 subunits (Fig. 2B, 44.88 ± 16%, p < 0.05, n = 4), whereas cytosolic α1 subunit protein levels were increased by 126.5 ± 45% (Fig. 2C, n = 4, p < 0.05, in duplicate). Therefore, ethanol induces the internalization of GABAA receptor α1 subunits and this effect is mimicked by PKC activation using PDBu.
Fig. 1.

Ethanol exposure alters GABAA receptor α1 subunit surface expression in cerebral cortical neurons. Cortical neurons were exposed to ethanol (50 mM for 1–6 h) followed by preparation of P2 fractions and biotinylation of surface proteins. Biotin-labeled (surface) and flow-through (cytosolic) proteins were isolated with NeutrAvidin slurry (Pierce Chemical). A, representative Western blots of α1 subunit in the P2 fraction; C and D, representative Western blots of α1 subunit on surface and cytosol, respectively. A, ethanol exposure for 1 h did not alter α1 subunit expression, whereas ethanol exposure for 4 h produced maximal decrease in expression of subunits in the P2 fraction. B, ethanol exposure (25 mM) for 4 h had no effect on α1 subunit expression, whereas 50 mM ethanol decreased α1 subunit expression by 40% in the P2 fraction of cells. After biotinylation of surface receptors, ethanol exposure for 4 h decreased α1 subunit level on cell surface (C) and increased α1 subunit protein levels in cytosol (D). Data represent mean ± S.E.M. *, p < 0.05.
Fig. 2.
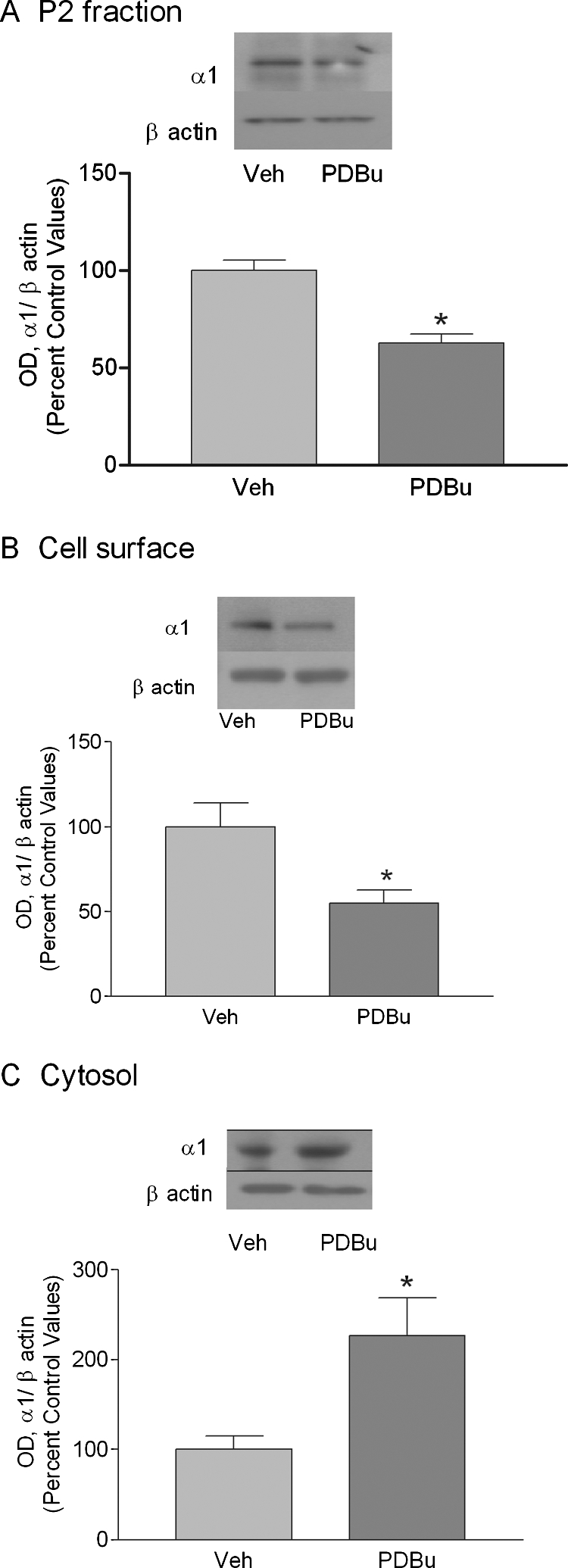
PDBu alters GABAA receptor α1 subunit surface expression in cultured cerebral cortical neurons. Cortical neurons were exposed to PDBu (100 nM for 60 min) followed by preparation of P2 fraction and biotinylation of surface proteins as described in the methods. A, Western blot analysis of P2 fraction of cortical neurons shows a decrease in α1 subunit after PDBu exposure. B and C, purification of surface (B) and cytosolic (C) receptors by biotinylation confirms increased internalization of GABAA receptor α1 subunits. Data represent mean ± S.E.M. *, p < 0.05, Student's t test.
Ethanol Exposure Reduces Zolpidem Enhancement of GABA-Evoked Cl− Currents.
The response to application of 1 μM GABA in cortical neurons was not significantly different in neurons treated with vehicle versus ethanol for 4 h (vehicle, 133.1 ± 26.50 pA; 4-h ethanol-treated neurons, 187.1 ± 43.94 pA). A low dose of zolpidem (100 nM) selective for α1 subunit-containing GABAA receptors was chosen to study the effects of ethanol exposure on α1 subunit-containing GABAA receptors. This dose of zolpidem enhanced responses elicited by 1 μM GABA (∼EC10) in cultured cortical neurons, consistent with prior studies (Liang et al., 2004). As shown in Fig. 3, this enhancement was significantly reduced in neurons exposed to ethanol (50 mM) for 4 h [percentage potentiation in vehicle-treated neurons, 218.2 ± 18.31 (n = 14); neurons treated with ethanol for 4 h, 144.4 ± 14.63 (n = 12), p = 0.0052]. These data support the conclusion that that the fraction of α1-containing receptors at the cell-surface is significantly reduced by ethanol exposure in vitro, similar to prior studies on the effects of ethanol exposure in vivo (Kumar et al., 2003; Liang et al., 2004).
Fig. 3.

Ethanol exposure reduces zolpidem potentiation of GABA-induced currents in whole-cell patch-clamp recordings of cerebral cortical neurons. Cortical neurons were exposed to vehicle or ethanol (50 mM) for 4 h. Representative electrophysiological responses to the application of 1 μM GABA, coapplication of 1 μM GABA with 100 nM zolpidem and subsequent washout with 1 μM GABA after vehicle (A) and ethanol (B) exposure (50 mM) for 4 h. C, zolpidem (100 nM) potentiation of the 1 μM GABA response in cortical neurons was significantly diminished in ethanol-treated versus vehicle-treated neurons. Data represent the mean ± S.E.M. **, p < 0.001.
PKC Inhibitor Calphostin C Prevents Ethanol-Induced Internalization of α1 Subunit-Containing Receptors.
Cultured cortical cells were exposed to vehicle, ethanol, and ethanol combined with calphostin C (0.3 μM). Calphostin C was added 15 min before the start of ethanol exposure. These doses have been shown to inhibit PKC activity and to alter GABAA receptor function in cortical synaptoneurosomes (Kumar et al., 2005). Calphostin C alone did not alter the expression of α1 subunits in the P2 fraction (Fig. 4A). Ethanol exposure for 4 h decreased the expression of GABAA receptor α1 subunit expression by 48.2 ± 8% (n = 4, p < 0.05) in the P2 fraction of cultured cortical neurons. Calphostin C completely inhibited the ethanol-mediated decrease in GABAA receptor α1 subunits (Fig. 4B).
Fig. 4.

PKC inhibitor calphostin C blocks ethanol-mediated internalization of GABAA receptor α1 subunit. Cultured cortical neurons were exposed to vehicle (Veh), ethanol, and ethanol (EtOH) with calphostin C (Cal). P2 fractions were prepared and analyzed by Western blot analysis. In a separate set of experiments, whole-cell patch-clamp recordings were carried out from cultured cortical neurons. A, effect of calphostin C alone on expression of GABAA receptor α1 subunits. B, ethanol exposure for 4 h decreased the expression of α1 subunits and this was blocked by calphostin C. *, p < 0.05, Newman-Keuls test, compared with control. Data represent mean ± S.E.M. C, calphostin C alone had no significant effect on zolpidem enhancement of 1 μM GABA currents in neurons treated with ethanol for 4 h compared with vehicle-treated neurons. D, coexposure of neurons with calphostin C and ethanol for 4 h prevented the decrease in zolpidem potentiation of 1 μM GABA evoked currents that was found in neurons that were exposed to ethanol alone for 4 h.
We then employed calphostin C in electrophysiological studies to determine the involvement of PKC in ethanol-induced alterations in zolpidem enhancement of GABA evoked Cl− conductance. First, we examined the effect of calphostin C alone (0.3 μM, 4 h) on currents elicited by application of 1 μM GABA in cortical neurons. Calphostin C alone had no significant effect on the amplitude of 1 μM GABA currents (vehicle, 114.3 ± 20.24 pA; calphostin C for 4 h, 152.7 ± 25.48 pA; Fig. 4C). To investigate the involvement of PKC in the effects of ethanol on activation of benzodiazepine responses, we incubated the cells with 50 mM ethanol in presence or absence of 0.3 μM calphostin C for 4 h. We then examined zolpidem (100 nM) potentiation of GABA (1 μM) current in these treatment groups. As shown in Fig. 4D, calphostin C ablated the decrease in zolpidem enhancement that was seen in neurons treated with ethanol alone for 4 h [percentage potentiation in vehicle neurons, 211.1 ± 15.50 (n = 10); neurons treated with 50 mM ethanol + 0.3 μM calphostin C for 4 h, 191.6 ± 15.58 (n = 6), p > 0.5]. These data further support the requirement for PKC activity in ethanol-induced internalization of α1 subunit GABAA benzodiazepine receptors in cultured cortical neurons.
Ethanol Increases PKC Isoform Expression and Selectively Increases the Association of PKCγ with GABAA Receptor α1 Subunits.
Because the effect of ethanol on GABAA α1 receptor internalization was mimicked by PKC activation and blocked by PKC inhibition, we investigated whether specific PKC isozymes were involved in these effects. Ethanol exposure for 1 h increased PKCβ, PKCγ, and PKCε peptide expression in the P2 membrane fraction of cortical neurons. In contrast, ethanol exposure for 4 h increased PKCβ and PKCγ but not PKCε protein levels (Table 1). To determine whether these isozymes were translocated and bound to GABAA α1 subunit receptors, we employed immunoprecipitation studies to determine their association. For this study, intact GABAA α1 subunit receptors were immunoprecipitated from the P2 fraction using a GABAA receptor α1 subunit-specific antibody. The immunoprecipitate was denatured, separated by SDS-PAGE, and probed with PKCγ or PKCβ antibodies. Ethanol exposure for 4 h increased the coimmunoprecipitation of PKCγ with GABAA receptor α1 subunits by 92.88 ± 24% (p < 0.05, n = 4) but did not alter the coimmunoprecipitation of PKCβ with GABAA receptor α1 subunits (Fig. 5, A and B). Furthermore, ethanol exposure for 1 h did not alter association of PKCβ or PKCγ with α1 subunit-containing GABAA receptors (Table 2).
TABLE 1.
PKC isoforms expression in P2 fraction of cultured cortical neurons
The values are mean percentage ± S.E.M. change from four experiments performed in duplicate. EtOH was used at a 50 mM final concentration.
| PKCβ | PKCγ | PKCε | |
|---|---|---|---|
| % | |||
| EtOH 1 h | ↑ 60.8 ± 24* | ↑ 78.1 ± 27* | ↑ 24.3 ± 10* |
| EtOH 4 h | ↑ 51.1 ± 22* | ↑ 52.3 ± 19 | ↓ 13.6 ± 29 |
p < 0.05 compared with control.
Fig. 5.

Ethanol exposure for 4 h increases the association of PKCγ with α1-containing GABAA receptors in cultured cortical neurons. Receptors in the P2 fraction of control (lane 1) and ethanol-exposed cells (lane 2) were immunoprecipitated with α1 subunit antibody. Immunoprecipitated receptors were separated by SDS-PAGE and immunoblotted with PKCγ or PKCβ antibodies. The same blot was probed by α1 subunit antibody. PKCγ or PKCβ signal intensity was normalized to GABAA receptor α1 subunit signal intensity from the Western blots of the immunoprecipitate. A, association of PKCγ with α1-containing GABAA receptors was increased 92.88 ± 23.60% in cultured neurons after 4 h ethanol exposure. B, there was no change in PKCβ association with α1-containing receptors after 4-h ethanol exposure. Data represent mean ± S.E.M. *, p < 0.05, Student's t test.
TABLE 2.
Coimmunoprecipitation of PKC isoforms with GABAA α1 subunit receptors after ethanol (50 mM) exposure for 1 h
Values are mean ± S.E.M. from four experiments performed in duplicate.
| Association of | Vehicle | EtOH |
|---|---|---|
| PKCβ | 100.0 ± 9 | 102.9 ± 9 |
| PKCγ | 100.0 ± 11 | 104.8 ± 13 |
We also employed dual fluorescence confocal microscopy to visualize the colocalization of PKCβ, -γ, or -ε with GABAA receptors that contain α1 subunits (Fig. 6). In ethanol-naive cells, GABAA α1 subunits were colocalized with PKCβ in 44 ± 5% of cells, with PKCγ in 30.34 ± 2% of cells and PKCε in 45.68 ± 2% of cells (data not shown). Ethanol exposure for 4 h increased the association of PKCγ with GABAA receptor α1 subunits by 70.2 ± 15% (p < 0.05, n = 5 coverslips/group, 309 α1-labeled cells counted) (Fig. 7A) but did not alter the colocalization of PKCβ (n = 4 coverslips/group, 377 α1-labeled cells counted) or PKCε (n = 5 coverslips/group, 361 α1-labeled cells counted) (Fig. 7, B and C) with GABAA receptor α1 subunits. Furthermore, ethanol exposure for 1 h did not alter the association of PKCβ (n = 5 coverslips/group, 306 α1-labeled cells counted), PKCγ (n = 5 coverslips/group, 300 α1-labeled cells counted), or PKCε (n = 5 coverslips/group, 313 α1-labeled cells counted) with GABAA receptor αl subunits (Table 3).
Fig. 6.

PKC isoforms are colocalized with GABAA receptor α1 subunits in ethanol-naive cells. Cortical neurons were stained with anti-α1 and PKCβ, PKCγ, or PKCε antibodies for confocal microscopy. A, D, and G, α1-labeled cells; B, E, and H, PKCβ-, -γ-, and -ε-labeled cells, respectively. C, F, and I are merged images of dual immunostaining of α1 subunit and the PKC isoform. The yellow color in merged panels (arrow) indicates colocalization of the two proteins.
Fig. 7.

Ethanol increases PKCγ, but not PKCβ or PKCε, colocalization with GABAA receptor α1 subunit in cultured cortical neurons. Cell were exposed to ethanol for 4 h and stained with anti-α1 and PKC antibodies. Immunostaining of α1- and PKC-labeled cells were examined under confocal microscope. The total number of α1/PKC colocalized cells were normalized to total number of α1-stained cells on the same coverslip and presented as percentage control values. Ethanol exposure for 4 h increased PKCγ association with GABAA receptor α1 subunit by 70.17 ± 15% (A). In contrast, ethanol exposure did not alter the association of PKCβ or PKCε with GABAA receptor α1 subunit compared with controls (B and C). Data represent mean ± S.E.M. *, p < 0.05, Student's t test.
TABLE 3.
Colocalization of PKC isoforms with GABAA receptor α1 subunit after 1-h ethanol (50 mM) exposure
Data represent the percentage change in the ratio of the number of α1/PKC colocalized cells to the total number of GABAA receptor α1 subunit-stained cells.
| Association with GABAA α1 | Vehicle | EtOH |
|---|---|---|
| % | ||
| PKCβ | 100.0 ± 11 | 95.4 ± 16 |
| PKCγ | 100.0 ± 11 | 112.4 ± 16 |
| PKCε | 100.0 ± 8 | 116.2 ± 4 |
PKCγ, but Not PKCβ, Inhibition Reduces Ethanol-Mediated Internalization of GABAA Receptor α1 Subunits.
Ethanol exposure for 4 h increased the association of PKCγ with GABAA receptor α1 subunits and internalization of GABAA receptor α1 subunits. Therefore, we hypothesized that the internalization of GABAA receptor α1 subunit was PKCγ-dependent. PKCγ siRNA transfection was used to inhibit the effect of ethanol on PKCγ expression in cultured cortical neurons. Selective PKCγ siRNA reduced PKCγ expression by 74.36 ± 7% (p < 0.05, n = 3, in duplicate) in the P2 fraction of cortical neurons compared with control. However, PKCγ siRNA transfection of cortical neurons had no effect on expression of PKCβ and PKCε isoforms (Fig. 8A.). Likewise, PKCγ siRNA transfection did not alter expression of GABAA receptor α1 subunits. However, the effect of ethanol (4 h) on GABAA receptor α1 subunit expression was blocked by selective PKCγ siRNA transfection (Fig. 8B).
Fig. 8.

PKCγ RNA interference transfection inhibits ethanol-induced internalization of GABAA receptor α1 subunit in cultured cortical cells. Cerebral cortical neurons were transfected with scrambled or PKCγ siRNA. The P2 fraction was analyzed by SDS-PAGE analysis and probed with PKCγ, -β, and -ε antibodies. A, PKCγ-specific, but not scrambled, siRNA decreased the expression of PKCγ by 76.09 ± 1.087%, whereas the expression of PKCβ and -ε were not altered. B, selective PKCγ siRNA inhibits ethanol (50 mM)-induced internalization of GABAA receptor α1 subunit. *, p < 0.05 compared with vehicle, Newman-Keuls test.
Ethanol exposure did not increase the association of PKCβ with GABAA α1 subunit receptors but increased PKCβ expression in the P2 fraction. Therefore, to further verify the role of PKCβ in ethanol-mediated internalization of α1 subunits, we used PKCβ pseudosubstrate peptide to block the activity of PKCβ. PDBu exposure was used as a positive control to evaluate the effectiveness of PKCβ pseudosubstrate. PDBu increased the expression of PKCβ in P2 fraction by 103.2 ± 27% (p < 0.05, ANOVA, n = 4). PKCβ pseudosubstrate completely inhibited the effect of PDBu on PKCβ expression, demonstrating inhibition of PKCβ expression in the cells. PKCβ pseudosubstrate alone did not alter PKCβ expression. (Fig. 9A). Furthermore, PKCβ pseudosubstrate had no effect on the ethanol-mediated decrease in GABAA receptor α1 subunits (Fig. 9B).
Fig. 9.

PKCβ inhibition does not prevent ethanol-induced internalization of GABAA receptor α1 subunit. Cultured cortical cells were exposed to PDBu and or PKCβ pseudosubstrate. A, PDBu increased the PKCβ expression in P2 fraction by 203 ± 26%. The PDBu-induced increase in PKCβ expression was blocked by PKCβ pseudosubstrate. PKCβ pseudosubstrate alone did not affect PKCβ expression. B, ethanol exposure for 4 h reduced the expression of GABAA receptor α1 subunit and PKCβ pseudosubstrate had no effect on GABAA receptor α1 subunit expression after ethanol exposure. *, p < 0.05 compared with vehicle, Newman-Keuls test.
Discussion
The present study demonstrates that ethanol exposure in vitro increases internalization of GABAA receptor α1 subunits in a time-dependent manner. The effect of ethanol on GABAA receptor α1 subunit surface expression is PKC-dependent, because it is inhibited by the PKC inhibitor calphostin C. In addition, electrophysiological measurement of zolpidem enhancement of GABA-mediated currents confirms that surface expression of α1-containing receptors are significantly reduced after ethanol exposure, and this effect is also dependent upon PKC. Ethanol exposure increases PKCβ, -ε, and -γ isoform expression, but ethanol-induced association with GABAA receptor α1 subunits is selective for PKCγ isoforms. PKCγ, but not PKCβ, inhibition prevented ethanol-induced internalization of GABAA receptor α1 subunits. Thus, these data suggest that decreased expression of α1 subunits at the cell surface after ethanol exposure is mediated by PKCγ activity. The effect of PKCγ siRNA on zolpidem enhancement of GABA-evoked currents could not be determined because individual cells that were transfected could not be identified. However, it seems unlikely that the preservation of surface expression by PKCγ siRNA would lack functional significance, because the PKC inhibitor calphostin C altered the effect of ethanol on zolpidem responses.
The regulation of GABAA receptor α1 subunit surface expression seems to be a conserved mechanism both in vitro and in vivo. The effects of ethanol exposure on GABAA α1 subunit receptors in cultured cerebral cortical neurons mimics the effects of long-term ethanol exposure in vivo, because α1 subunits are reduced after ethanol exposure in both experimental paradigms. The ethanol-induced reduction in GABAA receptor α1 subunits are thought to contribute to ethanol dependence. Studies have shown that α1 knockout mice exhibit hyperexcitability marked by increased seizure susceptibility to bicuculline (Kralic et al., 2002), as well as withdrawal-like tremor (Kralic et al., 2005). Furthermore, other groups have shown that ethanol exposure also reduces GABAA receptor α1 subunit expression in hippocampus (Liang et al., 2004) and hippocampal cultured neurons (Sanna et al., 2003), where electrophysiological studies have also demonstrated a loss of zolpidem enhancement of GABA responses, suggesting similar effects of ethanol on the trafficking of hippocampal α1 subunit receptors. Together, these data suggest that loss of α1-containing GABAA receptors plays an important role in the etiology of ethanol dependence and withdrawal (Biggio et al., 2007). Therefore, further understanding of the mechanisms that regulate cell surface expression of these α1 subunit-containing receptors could have profound therapeutic relevance for treatment of alcohol abuse and alcoholism.
The observation that ethanol exposure diminishes zolpidem potentiation of GABA-evoked Cl− currents in the cortical cultured neurons supports the conclusion that ethanol reduces the surface expression of synaptic α1 subunit-containing GABAA benzodiazepine receptors. This result is also consistent with previous in vivo studies that found reductions in benzodiazepine sensitivity in ethanol dependence (Buck and Harris, 1990; Kumar et al., 2003; Sanna et al., 2003; Liang et al., 2004), suggesting that type 1 GABAA benzodiazepine (α1,β2/3,γ2) receptor internalization is the mechanism for ethanol-induced reductions in behavioral benzodiazepine sensitivity (cross-tolerance) in alcohol dependence (Cagetti et al., 2003).
The effects of ethanol on GABAA receptor surface expression in these studies did not involve withdrawal from ethanol. This is consistent with our studies on the effects of ethanol on α1 subunit-containing GABAA receptors in the cerebral cortex (Kumar et al., 2003) and with the effects of ethanol on α1 subunit expression in cerebral cortical and hippocampal cultured neurons (Sheela Rani and Ticku, 2006). Other studies suggest that ethanol-induced internalization of α1 receptors may occur more rapidly (Liang et al., 2007), less rapidly (Matthews et al., 1998), or may require withdrawal from ethanol (Cagetti et al., 2003). The reason for these discrepancies is unclear but may be related to different ethanol doses, brain regions, and the intervals after ethanol exposure. Indeed, ethanol does not produce the same effects on PKC isoform expression in hippocampus as in cerebral cortex (Kumar et al., 2006), and this could result in differential effects of ethanol on receptor trafficking in these regions. Further studies on the mechanisms of ethanol trafficking of GABAA receptors are needed.
Surface expression of GABAA receptors involves a highly regulated process of synthesis, endocytosis, recycling, and degradation. PKC has been shown to alter both endocytosis (Terunuma et al., 2008) and recycling of GABAA receptors. In the present study, despite the increases in PKCβ, -γ, and -ε expression, ethanol exposure for 1 h did not alter the surface expression of GABAA receptor α1 subunits. This may indicate that there was no endocytosis of α1 subunits or that receptor recycling compensated for endocytosis of the receptors. Previous studies demonstrated that ethanol exposure to rats for 1 h in vivo did not alter the expression of α1 subunit receptors in CCVs or alter association of α1 subunits with adaptin-α, which is required for GABAA receptor internalization (Kumar et al., 2003), suggesting that endocytosis does not occur at this point. Alternatively, it is possible that ethanol activation of other kinases, such as cAMP-dependent protein kinase (Dohrman et al., 2002) or tyrosine kinases (Marutha Ravindran and Ticku, 2006), influence the surface expression of GABAA receptor α1 subunits. Systematic study of these possibilities is needed to further understand effects of ethanol on GABAA receptor trafficking.
Association of kinases with receptor targets and subsequent phosphorylation may directly alter receptor conformation and channel conductance, whereas indirect actions may produce changes in receptor subunit composition at the membrane surface by altering the normal trafficking of receptors. In the present study, ethanol exposure for 1 h increased PKCβ, -γ, and -ε expression in the membrane fraction of cultured cortical neurons without altering their association with GABAA α1 subunits. Therefore, it is possible that these PKC isoforms are targeted to other subtypes of GABAA receptors and/or other ion channels. Translocation of PKC from cytosol to the membrane requires distinct mechanisms and intermediary proteins such as AKAP and RACK (Mochly-Rosen, 1995; Wong and Scott, 2004), and these proteins are also altered by ethanol exposure (He et al., 2002; Ron, 2004). Therefore, it is likely that ethanol effects on such transporting molecules determine the translocation of PKC to GABAA receptors. These intermediary proteins are also affected by ethanol and may determine the localization of specific PKC isoforms on the cell surface after ethanol exposure.
Ethanol exposure for 4 h selectively increased PKCγ colocalization and coimmunoprecipitation with GABAA receptor α1 subunits. Furthermore, surface expression of GABAA α1 receptors was decreased after 4 h of ethanol exposure suggesting that PKCγ is necessary for ethanol regulation of α1 receptor internalization. Previous studies have shown that in vivo long-term ethanol exposure (2 weeks) increases endocytosis of α1 subunit-containing GABAA receptors into CCVs of cerebral cortex (Kumar et al., 2003), but this effect is accompanied by a decrease in PKCγ coimmunoprecipitation with GABAA α1 receptors (Kumar et al., 2002). It is possible that ethanol effects on PKCγ association with GABAA receptors are transient, whereas continual ethanol effects on neurons during longer ethanol exposure periods may account for this observed difference. Alternatively, the potential role of other signaling pathways, endogenous molecules, and neurocircuitry that differ in vivo versus in vitro may account for this difference.
Selective PKC activity has also been suggested to play a role in GABAA receptor function after ethanol exposure in vivo and in vitro. For example, mice lacking the gene for PKCγ show a significant reduction in ethanol potentiation of muscimol-stimulated Cl− influx compared with responses in wild-type mice (Harris et al., 1995). In contrast, ethanol and flunitrazepam potentiation of muscimol-stimulated Cl− uptake is greater in microsacs from PKCε-null mutant compared with wild-type control mice (Hodge et al., 1999). In our study, ethanol exposure did not alter PKCβ or -ε association with GABAA α1 receptors after ethanol exposure. Although PKCβ and PKCε may not be responsible for ethanol-induced receptor trafficking, these kinases clearly play a role in receptor activity. For example, PKCε has been shown to alter phosphorylation of GABAA receptor γ2 subunit and receptor function after ethanol exposure (Qi et al., 2007). In addition PKCβ has been shown to phosphorylate GABAA receptor β1 subunits (Brandon et al., 1999). Thus, ethanol exposure may alter the association of these PKC isoforms with other subunits of GABAA receptors and thereby regulate the trafficking of distinct receptor subtypes. Further studies are needed to explore these possibilities.
The present work demonstrates that PKCγ plays an essential role in the regulation of the surface expression of α1 subunit-containing GABAA receptors and highlights the possibility of modulating GABAergic activity through selective targeting of PKC isoforms. Altered GABAA receptor α1 subunit expression is implicated in anxiety, alcoholism, epilepsy, and many other neurological disorders. Therefore, an understanding of the mechanism of GABAA receptor α1 subunit trafficking could lead to new therapeutic approaches that aim to restore normal surface expression of GABAA receptors.
Acknowledgments
We thank Dr. Michael Chua for helpful discussions and Dr. Jean-Marc Fritschy (Institute of Pharmacology and Toxicology, University of Zurich, Zurich, Switzerland) and Dr. Werner Sieghart (Brain Research Institute, University of Vienna, Vienna, Austria) for generously providing antibodies. The confocal microscopy was conducted at the Michael Hooker Microscopy Facility, University of North Carolina.
This work was supported by the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grants AA015409 (to S.K.), AA11605 (to A.L.M.)].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.063016.
- CCV
- clathrin-coated vesicle
- PKC
- protein kinase C
- siRNA
- small interfering RNA
- Pen-Strep
- penicillin-streptomycin
- PBS
- phosphate-buffered saline
- ANOVA
- analysis of variance
- BSA
- bovine serum albumin
- PAGE
- polyacrylamide gel electrophoresis
- PDBu
- phorbol-12,13-dibutyrate.
References
- Biggio G, Concas A, Follesa P, Sanna E, Serra M. (2007) Stress, ethanol, and neuroactive steroids. Pharmacol Ther 116:140–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers BJ, Elliott KJ, Wehner JM. (2001) Differential sensitivity to the anxiolytic effects of ethanol and flunitrazepam in PKCgamma null mutant mice. Pharmacol Biochem Behav 69:99–110 [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Kittler JT, McDonald BJ, Sieghart W, Brown DA, Smart TG, Moss SJ. (2000) GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. J Biol Chem 275:38856–38862 [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Uren JM, Kittler JT, Wang H, Olsen R, Parker PJ, Moss SJ. (1999) Subunit-specific association of protein kinase C and the receptor for activated C kinase with GABA type A receptors. J Neurosci 19:9228–9234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck KJ, Harris RA. (1990) Benzodiazepine agonist and inverse agonist actions on GABAA receptor-operated chloride channels. II. Chronic effects of ethanol. J Pharmacol Exp Ther 253:713–719 [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW. (2003) Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol Pharmacol 63:53–64 [DOI] [PubMed] [Google Scholar]
- Chapell R, Bueno OF, Alvarez-Hernandez X, Robinson LC, Leidenheimer NJ. (1998) Activation of protein kinase C induces gamma-aminobutyric acid type A receptor internalization in Xenopus oocytes. J Biol Chem 273:32595–32601 [DOI] [PubMed] [Google Scholar]
- Choi DS, Wei W, Deitchman JK, Kharazia VN, Lesscher HM, McMahon T, Wang D, Qi ZH, Sieghart W, Zhang C, et al. (2008) Protein kinase Cdelta regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J Neurosci 28:11890–11899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, Moss SJ. (1999) Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. J Biol Chem 274:36565–36572 [DOI] [PubMed] [Google Scholar]
- Dohrman DP, Chen HM, Gordon AS, Diamond I. (2002) Ethanol-induced translocation of protein kinase A occurs in two phases: control by different molecular mechanisms. Alcohol Clin Exp Res 26:407–415 [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B. (1998) Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci 1:563–571 [DOI] [PubMed] [Google Scholar]
- Harris RA, McQuilkin SJ, Paylor R, Abeliovich A, Tonegawa S, Wehner JM. (1995) Mutant mice lacking the gamma isoform of protein kinase C show decreased behavioral actions of ethanol and altered function of gamma-aminobutyrate type A receptors. Proc Natl Acad Sci USA 92:3658–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He DY, Vagts AJ, Yaka R, Ron D. (2002) Ethanol induces gene expression via nuclear compartmentalization of receptor for activated C kinase 1. Mol Pharmacol 62:272–280 [DOI] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO. (1999) Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCε. Nat Neurosci 2:997–1002 [DOI] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovic JN, Brown DA, Smart TG, Moss SJ. (2000) Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J Neurosci 20:7972–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, Criswell HE, Osterman JL, O'Buckley TK, Wilkie ME, Matthews DB, Hamre K, Breese GR, Homanics GE, Morrow AL. (2005) Genetic essential tremor in gamma-aminobutyric acidA receptor alpha1 subunit knockout mice. J Clin Invest 115:774–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, Korpi ER, O'Buckley TK, Homanics GE, Morrow AL. (2002) Molecular and pharmacological characterization of GABAA receptor α1 subunit knockout mice. J Pharmacol Exp Ther 302:1037–1045 [DOI] [PubMed] [Google Scholar]
- Kumar S, Khisti RT, Morrow AL. (2005) Regulation of native GABAA receptors by PKC and protein phosphatase activity. Psychopharmacology 183:241–247 [DOI] [PubMed] [Google Scholar]
- Kumar S, Kralic JE, O'Buckley TK, Grobin AC, Morrow AL. (2003) Chronic ethanol consumption enhances internalization of α1 subunit-containing GABAA receptors in cerebral cortex. J Neurochem 86:700–708 [DOI] [PubMed] [Google Scholar]
- Kumar S, Lane BM, Morrow AL. (2006) Differential effects of systemic ethanol administration on protein kinase cε, γ and β isoform expression, translocation to membranes and target phosphorylation: reversal by chronic ethanol exposure. J Pharmacol Exp Ther 319:1366–1375 [DOI] [PubMed] [Google Scholar]
- Kumar S, Porcu P, Werner DF, Matthews DB, Diaz-Granados JL, Helfand RS, Morrow AL. (2009) The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology 205:529–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Sieghart W, Morrow AL. (2002) Association of protein kinase C with GABAA receptors containing α1 and α4 subunits in the cerebral cortex: selective effects of chronic ethanol consumption. J Neurochem 82:110–117 [DOI] [PubMed] [Google Scholar]
- Liang J, Cagetti E, Olsen RW, Spigelman I. (2004) Altered pharmacology of synaptic and extrasynaptic GABAA receptors on CA1 hippocampal neurons is consistent with subunit changes in a model of alcohol withdrawal and dependence. J Pharmacol Exp Ther 310:1234–1245 [DOI] [PubMed] [Google Scholar]
- Liang J, Suryanarayanan A, Abriam A, Snyder B, Olsen RW, Spigelman I. (2007) Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J Neurosci 27:12367–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marutha Ravindran CR, Ticku MK. (2006) Tyrosine kinase phosphorylation of GABAA receptor subunits following chronic ethanol exposure of cultured cortical neurons of mice. Brain Res 1086:35–41 [DOI] [PubMed] [Google Scholar]
- Matthews DB, Devaud LL, Fritschy JM, Sieghart W, Morrow AL. (1998) Differential regulation of GABAA receptor gene expression by ethanol in the rat hippocampus versus cerebral cortex. J Neurochem 70:1160–1166 [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D. (1995) Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science 268:247–251 [DOI] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Nannini MA, Camarini R, Messing RO, Hodge CW. (2001) Reduced ethanol withdrawal severity and altered withdrawal-induced c-fos expression in various brain regions of mice lacking protein kinase C-epsilon. Neuroscience 103:171–179 [DOI] [PubMed] [Google Scholar]
- Poisbeau P, Cheney MC, Browning MD, Mody I. (1999) Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J Neurosci 19:674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor WR, Poelchen W, Bowers BJ, Wehner JM, Messing RO, Dunwiddie TV. (2003) Ethanol differentially enhances hippocampal GABA A receptor-mediated responses in protein kinase C gamma (PKC gamma) and PKC epsilon null mice. J Pharmacol Exp Ther 305:264–270 [DOI] [PubMed] [Google Scholar]
- Qi ZH, Song M, Wallace MJ, Wang D, Newton PM, McMahon T, Chou WH, Zhang C, Shokat KM, Messing RO. (2007) Protein kinase Cε regulates γ-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of γ2 subunits. J Biol Chem 282:33052–33063 [DOI] [PubMed] [Google Scholar]
- Ron D. (2004) Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist 10:325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna E, Mostallino MC, Busonero F, Talani G, Tranquilli S, Mameli M, Spiga S, Follesa P, Biggio G. (2003) Changes in GABAA receptor gene expression associated with selective alterations in receptor function and pharmacology after ethanol withdrawal. J Neurosci 23:11711–11724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheela Rani CS, Ticku MK. (2006) Comparison of chronic ethanol and chronic intermittent ethanol treatments on the expression of GABA(A) and NMDA receptor subunits. Alcohol 38:89–97 [DOI] [PubMed] [Google Scholar]
- Sieghart W, Sperk G. (2002) Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr Top Med Chem 2:795–816 [DOI] [PubMed] [Google Scholar]
- Terunuma M, Xu J, Vithlani M, Sieghart W, Kittler J, Pangalos M, Haydon PG, Coulter DA, Moss SJ. (2008) Deficits in phosphorylation of GABAA receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J Neurosci 28:376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Scott JD. (2004) AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5:959–970 [DOI] [PubMed] [Google Scholar]