

Abstract
The plasmodial surface anion channel (PSAC) is an unusual small-conductance ion channel induced on erythrocytes infected with plasmodia, including parasites responsible for human malaria. Although broadly available inhibitors produce microscopic clearance of parasite cultures at high concentrations and suggest that PSAC is an antimalarial target, they have low affinity for the channel and may interfere with other parasite activities. To address these concerns, we developed a miniaturized assay for PSAC activity and carried out a high-throughput inhibitor screen. Approximately 70,000 compounds from synthetic and natural product libraries were screened, revealing inhibitors from multiple structural classes including two novel and potent heterocyclic scaffolds. Single-channel patch-clamp studies indicated that these compounds act directly on PSAC, further implicating a proposed role in transport of diverse solutes. A statistically significant correlation between channel inhibition and in vitro parasite killing by a family of compounds provided chemical validation of PSAC as a drug target. These new inhibitors should be important research tools and may be starting points for much-needed antimalarial drugs.
Plasmodium falciparum malaria is a leading infectious disease burden on global health, with more than 500 million people infected and more than 1 million deaths each year. Estimates of the economic burden on developing countries are also astounding. Despite ongoing trials, a globally useful vaccine is still years away. Aside from preventative measures, such as insecticide-treated bed nets (Maxwell et al., 2002), treatment of malaria cases with drugs targeting one or more parasite activities is the only meaningful option at present. Evolving resistance to nearly all available antimalarial agents renders this option imperfect and has motivated workers to identify new parasite targets and small molecule inhibitors. Despite this drive, less than 1% of the ∼5600 proteins encoded by the parasite have been explored as therapeutic targets.
Most of the clinical sequelae of malaria result from blood-stage parasites that invade, grow, and replicate asexually in erythrocytes. For this reason, identification and characterization of novel blood stage parasite activities represents an important approach to antimalarial drug discovery and development. The plasmodial surface anion channel (PSAC), an unusual ion channel on the infected erythrocyte membrane, is an important new target. Although the genetic basis of this channel remains unknown, studies from several groups suggest that it may play an essential role in intracellular parasite survival. Consistent with this hypothesis, transport studies, including patch clamp, reveal strict conservation in all plasmodia (Lisk and Desai, 2005). One possibility is that PSAC functions in uptake of nutrient solutes from serum and may also help to discharge parasite metabolic waste products (Desai et al., 2000). An important example of a nutrient requiring uptake is isoleucine, the only amino acid that cannot be obtained through hemoglobin degradation (Liu et al., 2006; Martin and Kirk, 2007). The vitamin pantothenic acid is also essential for parasite growth and requires uptake (Saliba et al., 1998).
Biochemical studies suggest this channel has a number of unusual functional properties not present in known mammalian channels. For example, PSAC efficiently excludes the small Na+ ion even though it mediates transport of bulky solutes of various sizes and charge (Cohn et al., 2003). Another is that patch-clamp studies reveal a surprisingly small single-channel conductance for a channel permeant to large organic ions. Other examples include atypical fast-flickering ion channel gating (Desai et al., 2000) and an unexpected interaction between permeating solutes and known inhibitors (Lisk et al., 2007; Bokhari et al., 2008). It should therefore be possible to identify specific inhibitors suitable for clinical use.
A conceptual advantage of PSAC over intraerythrocytic parasite targets is its surface location on infected erythrocytes. Because inhibitors do not need to cross multiple lipid membranes to reach this target, this location relaxes drug design constraints on inhibitor membrane permeability. The location also largely excludes parasite resistance resulting from energy-dependent extrusion of unmodified drugs, which has been implicated in treatment failures with chloroquine (Fidock et al., 2000) and possibly other antimalarial agents (Sanchez et al., 2008).
Although these advantages are now well recognized, development of antimalarial agents targeting parasite-induced transport has not been pursued because it is not known whether the channel serves an essential function for the parasite. Moreover, existing inhibitors have low specificity and affinity, making the drug discovery path unclear. Finally, some electrophysiological surveys have detected multiple distinct ion channels on infected erythrocytes (Staines et al., 2007), raising questions about the relative contributions of PSAC and other routes. To address these and other concerns, we have now developed a miniaturized assay for organic solute uptake by infected cells and performed a high-throughput screen for small-molecule inhibitors. Our studies identified novel compounds with improved affinities for transport inhibition, confirmed the central role of PSAC in the parasite-induced uptake of solutes, and validated this ion channel as an antimalarial drug target.
Materials and Methods
Reagents.
Compounds 1 through 9 were obtained from ChemDiv (San Diego, CA). All other reagents were obtained from Sigma Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Waltham, MA).
Osmotic Lysis.
Osmotic lysis experiments were performed as described previously (Wagner et al., 2003). Trophozoite-infected red blood cells were enriched by Percoll/sorbitol separation to >95% parasitemia. The cells were then washed in PBS (150 mM NaCl and 20 mM Na2HPO4, pH 7.5), and resuspended at 0.05 to 0.1% hematocrit in lysis solution (280 mM sorbitol, 20 mM Na-HEPES, and 0.1 mg/ml BSA, pH 7.4) supplemented with PSAC inhibitors as indicated in the figure legends. Experiments examining uptake of other organic solutes used the same protocol except for iso-osmotic replacement of sorbitol by the solute or Cl− salt. Osmotic swelling and lysis was then continuously followed by transmittance measurements at a wavelength of 700 nm through the cell suspension (DU640 spectrophotometer with Peltier temperature control; Beckman Coulter, Fullerton, CA). Each suspension was manually resuspended at 7-min intervals to minimize effects of cell settling. Control experiments indicated that uninfected erythrocytes do not undergo lysis in PBS or sorbitol with or without PSAC inhibitors.
High-Throughput Screen.
The above osmotic lysis assay was used for high-throughput screening after miniaturization to a 384-well format with the following modifications. Infected erythrocytes were enriched, washed, and suspended in PBS at 1% hematocrit before dispensing 20 μl/well into 384-well plates with a microplate dispenser (Matrix Wellmate; Thermo Fisher Scientific), yielding 2 × 106 trophozoite-infected cells/well. Compounds from the available screening collections were then dispensed by a 100-nl pin transfer of a 10 mM DMSO stock into separate wells and incubated with the cells for a minimum of 20 min. Sorbitol transport and osmotic lysis were then initiated by addition of 80 μl of the lysis solution. The final compound screening concentration was therefore 10 μM. Plates were immediately vortexed and then incubated at room temperature before reading absorbance without resuspension of cells at 30 min, 2 h, and 18 h (700 nm light, Synergy HT, BioTek). Although parasite viability is quickly lost in saline incubations at room temperature, our previous studies have shown that PSAC-mediated osmotic fragility of infected cells does not require live parasites (Alkhalil et al., 2004). This observation is consistent with the unchanged single-channel properties for many ion channels upon excision of cell-attached patches or on reconstitution in artificial bilayers. We also found that readings after the 18-h time-point were additionally useful for distinguishing among the most potent hits.
Plates run in duplicate revealed highly reproducible results for all compounds. Negative control wells in each plate received DMSO by pin transfer. With each experiment, separate positive control plates were identically run except that the lysis solution was replaced by PBS supplemented with 2 mM furosemide. The activity of each screening compound was calculated at each reading time-point according to the equation % block(t) = 100 · (Acpd − Āneg)/(Āpos − Āneg), where Acpd represents the absorbance from the compound well and Āneg and Āpos represent the mean absorbances of negative and positive control wells run in parallel. At each time-point, this parameter represents a quantitative measure of activity against PSAC-mediated transport.
As described previously (Zhang et al., 1999), the Z′ statistic was used to evaluate assay signal-to-noise and was calculated according to the equation Z′ = 1 − 3 · (σneg + σpos)/(Āpos − Āneg), where σneg and σpos represent the S.D. of control absorbance measurements. According to this equation, a perfect noise-free assay yields a Z′ of 1.0; assays having values >0.5 are generally considered robust and suitable for single replicate high-throughput screening.
Electrophysiology.
Cell-attached patch-clamp recordings on trophozoite-stage infected red blood cells were obtained in symmetric bath and pipette solutions of 1000 mM choline chloride, 115 mM NaCl, 10 mM MgCl2, 5 mM CaCl2, 20 mM sodium-HEPES, pH 7.4. This hypertonic recording solution improves the signal-to-noise ratio for single PSAC detection; chloride concentration dependences of single-channel currents reveal saturation of flux only at higher concentrations (Alkhalil et al., 2004). When used, inhibitors were present at identical concentrations in both pipette and bath compartments. Pipettes were fabricated from quartz capillaries by pulling to tip diameters <0.5 μm with resistances of 1 to 3 MΩ in the recording solution. Seal resistances were >100 GΩ. Membrane holding potential was 0 mV; pulses to voltages between −100 mV and +100 mV were applied using Clampex 9.0 software and an Axopatch 200B amplifier in voltage clamp mode (Molecular Devices, Sunnyvale, CA). Acquired data were filtered at 5 kHz with an eight-pole Bessel filter, digitized at 100 kHz (Digidata 1322A; Molecular Devices), and recorded. The single-channel traces in Fig. 3E were subjected to postfiltering at 3 kHz and a 10-fold decimation to maintain manageable size of the graphic; detection of events was not affected.
Fig. 3.

Potent inhibition by furoquinolines results from direct action on PSAC. A, histogram showing distribution of percentage block at 20 min for 87 furoquinoline derivatives, calculated according to the equation % block(t) = 100 · (Acpd − Āneg)/(Āpos − Āneg). B, structure of compound 9, a more potent furoquinoline derivative. C, dose responses for inhibition of sorbitol-induced osmotic lysis by compounds 1 and 9 (▿ and ●, respectively) measured as in Fig. 1A and normalized to 1.0 without inhibitor. Each symbol represents the mean ± S.E.M. of up to nine measurements. Solid curves represent best fits to y = K0.5/(K0.5 + x). D and E, single PSAC recordings in the presence of indicated concentrations of compounds 1 or 9 (D and E, respectively). Whereas each trace in D is 800 ms long, those in E are 9 s long. In each panel, membrane potential is −100 mV for all traces; horizontal dashes represent the closed channel level. Vertical scale bar, 3 pA for D and E; horizontal scale bar, 150 ms for D, and 1500 ms for E. F, closed-channel dwell time distributions from recordings as in E without or with 100 nM compound 9 (black and red histograms, respectively). The distributions are presented on a log-log scale and are normalized to the total number of closed channel events to facilitate visual estimation of blocking rate constants, as described previously (Sigworth and Sine, 1987). The broad peak at 100 to 1000 ms in the distribution with 9 corresponds to inhibitor-induced block events because they are absent from control recordings. Events shorter than ∼20 ms correspond to intrinsic closings, which are not affected by addition of 9.
Analyses of single-channel recordings were carried out using custom software, as described previously (Desai, 2005). Compound 9's affinity was determined by measuring open probability from up to eight independent single-channel patches at each concentration (total of 160 to 760 s of recording at each concentration). The code estimates open probability based on a threshold midway between the closed and open states. Dwell time distributions were determined from up to 360 s of recordings from single-channel patches using a script that detects mid-threshold crossings, uses linear interpolation of adjacent sample times, and corrects for a Gaussian filter rise time of 66.4 μs.
Growth Inhibition Studies.
In vitro parasite killing by identified transport inhibitors was performed using a SYBR Green I-based fluorescence assay for parasite nucleic acid in 96-well format. Parasite cultures were synchronized by incubation in 5% sorbitol before seeding at 0.5% parasitemia and 2% hematocrit in RPMI 1640 medium without phenol red, but supplemented with 25 mM HEPES, 5% serum, 50 mg/l hypoxanthine, and PSAC inhibitors at indicated concentrations. Cultures were maintained for 3 days at 37°C in 5% O2/5% CO2/95% N2. The plates were then subjected to freeze-thaw before addition of SYBR Green I at twice the manufacturer's recommended final concentration (Invitrogen, Carlsbad, CA), incubation in the dark for 30 min, and measurement of fluorescence (excitation/emission wavelengths, 485/528 nm). Standard curves using serial dilutions of parasite cultures revealed a linear relationship between fluorescence and parasitemia over the range used in our study (not shown).
For each PSAC inhibitor, growth inhibitory IC50 values were estimated from measurements at a minimum of eight concentrations after subtraction of background fluorescence in matched cultures killed by 20 μM chloroquine.
Results
HTS Assay Development and Optimization.
Sorbitol, a poorly metabolized sugar alcohol, has high permeability into trophozoite-stage infected erythrocytes and negligible uptake by uninfected cells. Basic research labs have capitalized on this difference with routinely used protocols for synchronization of in vitro parasite cultures and for enrichment of mature infected cells on density gradients. It has also been extensively used to detect and characterize the parasite-induced permeability with a transmittance-based assay that tracks decreasing turbidity of infected cells as they undergo osmotic lysis in sorbitol (Fig. 1A; Wagner et al., 2003). This kinetic assay yields estimates of sorbitol permeability and affinity of various inhibitors that are concordant with those from single-channel and whole-cell patch-clamp under conditions that isolate PSAC activity (Alkhalil et al., 2004; Desai et al., 2005; Lisk et al., 2006).
Fig. 1.

A miniaturized osmotic lysis assay for PSAC inhibitors suitable for high-throughput screening. A, continuous transmittance measurements through suspensions of enriched infected cells in buffered sorbitol using a 1-ml cuvette format. Curves show osmotic lysis kinetics in sorbitol with indicated concentrations of furosemide (micromolar) normalized to 100% lysis without inhibitor at 1 h. Infected cells do not undergo lysis in PBS (red trace) because of their very low Na+ permeability (Cohn et al., 2003). B, assay adapted to 384-well format with a vertical optical path. Symbols represent mean ± S.E.M. absorbance values for infected cells in sorbitol with indicated concentrations of furosemide (micromolar). Uninfected cells without furosemide are shown as a control (red circles). A single 384-well plate having 48 wells for each condition was read at 15-min intervals without cell resuspension. Note that increasing furosemide concentrations slow the kinetics of osmotic lysis without affecting the endpoint absorbance plateau, corresponding to 100% lysis. Weak inhibition (e.g., by 2 μM furosemide) is easily detected with readings in the first hour, whereas conditions producing potent block can be distinguished from each other at later time-points.
To facilitate identification of improved inhibitors, we miniaturized this transmittance assay into 384-well format. Although erythrocytes settle rapidly in this small geometry format, we determined that vertical transmittance of light at a wavelength of 700 nm nevertheless increases upon lysis. This presumably reflects the greater scattering of light by intact cells, which contain locally high concentrations of hemoglobin. Our studies determined that the magnitude of the lysis-associated transmittance change has a strong and complex dependence on hematocrit. We therefore empirically optimized hematocrit by comparing transmittances through infected cells lysed in sorbitol to those of intact cells in physiological saline. Under the optimized conditions (see Materials and Methods), Z′ factors calculated from positive and negative control wells were typically >0.7, indicating suitability for high-throughput screening with a low rate of false positives and false negatives (Zhang et al., 1999).
Because lysis of individual cells is irreversible and results from accumulation of a threshold amount of sorbitol as previously quantified (Wagner et al., 2003), it should be possible to estimate relative inhibitor efficacy using a single defined concentration of each compound in a plate read at multiple time-points. We tested this prediction with furosemide, a well characterized PSAC inhibitor with a K0.5 of 2.7 μM (Desai et al., 2005; Desai, 2005). These studies revealed that increasing furosemide concentrations monotonically retarded the kinetics of osmotic lysis without affecting the plateau transmittance that corresponds to completed lysis of infected cells (Fig. 1B). Using this kinetic dose response, transmittance measurements at selected time-points can be used to estimate furosemide concentration in a blinded solution. Conversely, furosemide's known K0.5 and a standard curve from its dose response in this format can also be used to estimate relative inhibitory activities of each screening hit with multiple timed measurements at a single screening concentration. This approach successfully maximized the information content of our screen while minimizing the effort- and reagent-intensive culturing of parasites.
These microplate-based osmotic lysis experiments also determined that infected cells continue to accumulate sorbitol and accrue transmittance changes associated with osmotic lysis for prolonged durations despite use of nonphysiological conditions that do not support parasite growth (8 h in Fig. 1B). The transmittance changes reflect channel-mediated uptake rather than nonspecific cell lysis because they can be inhibited by PSAC inhibitors including furosemide and because uninfected cells are osmotically stable in sorbitol (Fig. 1B, red symbols). This observation is consistent with the biochemical stability of PSAC activity in killed parasite cultures (Alkhalil et al., 2004). We have also observed unchanged single-channel properties in cell-attached patch-clamp of infected cells killed by storage at 4°C for 24 h (not shown).
Identification of a Diverse Collection of Inhibitors.
We then used the optimized assay to execute a high-throughput screen of approximately 70,000 compounds from commercial, synthetic, and natural product libraries under an investigator-initiated open access facility at the Broad Institute (Tolliday et al., 2006). Each compound was tested at a 10 μM final concentration along with DMSO alone control wells in each screening plate. Osmotic lysis was initiated by addition of sorbitol and subsequently tracked with readings at t = 30 min, 2 h, and 18 h. With each experiment, we also included one or more plates where sorbitol was replaced with buffered NaCl solution containing furosemide as a zero lysis control. For each reading time-point, we defined a normalized statistic [% block(t)] to quantify the relative level of inhibition by each test compound (see Materials and Methods). Figure 2A shows the distribution of this statistic for NaCl and DMSO controls superimposed upon those of all compounds tested for the 30-min and 2-h reads. Figure 2B shows the results for the 18-h read. These distributions reveal several important points about the screen. First, the two types of control wells were tightly clustered and well separated at each time-point, indicating good reproducibility and a visual correlate of the assay's high Z′ statistic. Second, there was a spectrum of compound activities observed in our screen. Although difficult to see in these distributions, the majority of compounds were without measurable effect. There were also a large number of relatively weak inhibitors: a conservative threshold of ≥50% block at the first time-point yielded 6512 hits or a high hit rate of ∼9%. The same threshold at the 2- and 18-h time-points yielded 1936 and 27 hits, corresponding to hits rates of 2.7 and 0.03%, respectively. Third, we achieved increasing assay stringency with consecutive readings as indicated by the sigmoidal profile of the distributions in Fig. 2, A and B. For example, the subset of compounds producing >90% inhibition at the first read averaged 75 ± 18 and 25 ± 20% inhibition at the second and third time-points, respectively. By comparison with standard curves generated from experiments with furosemide, we calculated that compounds producing 50% inhibition at 30 min, 2 h, and 18 h have K0.5 values of ∼ 10, 0.5 to 1.0, and < 0.2 μM, respectively. As confirmed below with single-channel patch-clamp studies, our screen permitted estimation of inhibitor affinity for hits spanning a broad range of activities in a high-throughput format. In keeping with the guidelines for screening at the Broad Institute, the structures and activities of all compounds we screened are publicly available at http://chembank.broadinstitute.org/.
Fig. 2.

Distribution of small molecule inhibitory activities at each time-point in the high-throughput screen. Percentage block calculated according to the equation % block(t) = 100 · (Acpd − Āneg)/(Āpos − Āneg) for individual wells in 384-well plates containing each of ∼70,000 compounds (yellow or blue dots), DMSO alone (red dots), or PBS + 2 mM furosemide (green dots). A shows values for each well at 30 min and 2 h, whereas B shows the values from the 18-h read against the 30-min read. Forty-nine compounds containing a furoquinoline scaffold are shown as blue dots. Note the marked separation of positive and negative controls and the sigmoidal profile in each panel, reflecting the higher stringency of late readings.
A Novel Furoquinoline Scaffold Is Highly Effective against Organic Solute Uptake and Confirms a Primary Role for PSAC.
Although some hits were from natural product libraries and may contain mixtures of compounds with unknown structures, many hits were from commercial libraries with high purity and known structures. Additional hits came from the Broad Institute's in-house diversity-oriented syntheses (Tolliday et al., 2006). Starting with compounds having known structures, we selected 16 compounds available for secondary evaluation and obtained a 100% retest rate using cuvette-based osmotic lysis experiments in our National Institute of Allergy and Infectious Diseases laboratory. Representative compounds, along with estimates of K0.5 values for inhibition of sorbitol uptake, are shown in Table 1. Each of these compounds also inhibited in vitro parasite growth (IC50 values in Table 1).
TABLE 1.
High-affinity chemical entities identified by the high-throughput screen along with their K0.5 values for inhibition of sorbitol uptake and IC50 values for in vitro parasite growth inhibition.
Values are presented as mean ± S.E.M.
| Compound | K0.5 | IC50 | |
|---|---|---|---|
| nM | μM | ||
| 1 | 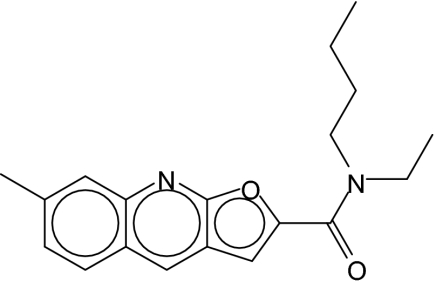 |
87 ± 9 | 6.0 ± 1.0 |
| 2 |  |
84 ± 6 | 2.2 ± 0.4 |
| 3 |  |
33 ± 3 | 4.7 ± 2.4 |
| 4 |  |
124 ± 8 | 15.2 ± 1.5 |
| 5 |  |
81 ± 13 | 13.9 ± 1.0 |
| 6 | 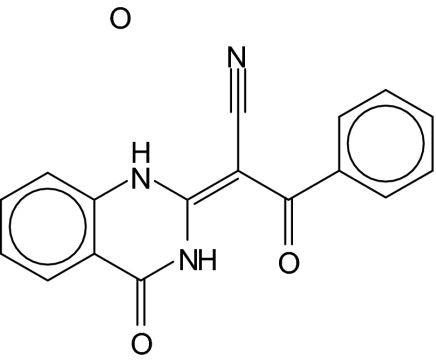 |
83 ± 6 | 6.4 ± 0.4 |
| 7 | 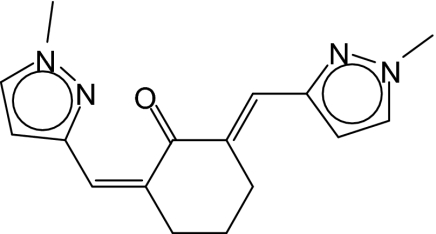 |
98 ± 7 | 3.4 ± 0.9 |
| 8 |  |
44 ± 9 | 6.6 ± 1.3 |
To examine the structural basis of inhibition, we selected a tricyclic furoquinoline scaffold that was consistently active in the high-throughput screen (Fig. 2, blue symbols; compound 1, Table 1) and obtained an additional 87 derivatives. Although these derivatives were distinct from compounds in the primary screen, all 87 compounds produced significant inhibition of sorbitol uptake at 1 μM (Fig. 3A), confirming that the furoquinoline moiety is responsible for inhibition by these compounds. Several of these derivatives were significantly more effective than the parent compound, presumably because they carry substituents that enhance interactions with critical sites on the channel. One compound, 9, had a K0.5 of 6 nM for inhibition of sorbitol uptake, making it the most potent inhibitor of parasite-induced permeability known to date (Fig. 3, B and C).
We next evaluated the effect of these furoquinolines on parasite-induced Cl− currents with single-channel patch-clamp and found high-level inhibition of PSAC activity at concentrations consistent with their effects on sorbitol uptake (Fig. 3, D and E). With compounds 1 and 9, inhibition was evident as long periods of inactivity in single PSAC recordings. As illustrated with a condensed timescale in Fig. 3E, these block durations were especially long with compound 9, suggesting that its improved efficacy is due to longer residence in a shared site on the channel. Figure 3F tallies the durations of these long block events and demonstrates that they are absent from recordings without inhibitor.
We used open probability determinations to estimate an inhibitory K0.5 of 18 ± 9 nM for compound 9 from single-channel recordings at 0, 20, and 100 nM (n = 8, 3, and 4 patches, respectively). Precise quantification of single-channel inhibition is complicated for compounds that produce block of very long durations. One error arises because of the relatively small number of closing events that can be captured in achievable recording durations; another results from underestimation of inhibition, because events from two channel molecules in a patch may be incorrectly attributed to a single channel in the setting of an inhibitor that imposes long block durations. Taking into consideration these limitations, this estimate is in good agreement with the value of 6 nM obtained for this inhibitor in macroscopic sorbitol uptake measurements (Fig. 3C). Thus, patch-clamp studies confirm that the sorbitol-induced osmotic lysis assay can quantify inhibitor potency in a high-throughput format.
Because the furoquinoline scaffold was not nonspecifically identified in other screens (see below) and because of quantitatively concordant inhibition of sorbitol uptake and patch-clamp currents by compound 9, these studies support PSAC as the primary parasite-induced channel responsible for both Cl− and sorbitol transport across the infected erythrocyte membrane.
Recent functional studies suggest that existing inhibitors of this channel have reduced effectiveness against a subset of permeating solutes (Lisk et al., 2007). Solutes exhibiting this effect are recognized and transported through PSAC by a distinct mechanism (Bokhari et al., 2008). To explore whether these improved inhibitors are susceptible to similar interactions with permeating solutes, we examined the efficacy of compound 9 against uptake of three solutes that exhibit these interactions—phenytrimethylammonium, proline, and arabinose (Fig. 4). Compound 9 was significantly less effective at inhibiting the uptake of these solutes than of sorbitol, alanine, or hydroxyproline, which do not exhibit these interactions. We also found that reducing the temperature in these experiments from 37°C to 15°C largely ameliorated the reduced efficacy of 9. It is noteworthy that both the list of solutes exhibiting this effect and its relatively steep temperature dependence parallel previous findings with furosemide and other weak inhibitors. Thus, inhibitors with diverse chemical structures and a broad range of PSAC affinities are susceptible to these interactions.
Fig. 4.

Compound 9 exhibits previously identified solute-inhibitor interactions. Osmotic lysis kinetics in indicated solutions without (black trace) or with 500 nM compound 9 (green and red traces). Lysis kinetics were tracked at 37°C (black and green traces) or at 15°C (red traces). Note that compound 9 is less effective at inhibiting uptake of phenyltrimethylammonium (PhTMA+), proline, and arabinose than of sorbitol, alanine, and hydroxyproline at 37°C; this difference is largely abolished at 15°C. In the absence of inhibitors, this temperature reduction has only a modest effect on lysis kinetics without inhibitors (Bokhari et al., 2008).
Validation of PSAC as a Drug Target with a Family of Derivatives Exhibiting a Spectrum of Efficacies.
To examine whether inhibition of PSAC interferes with parasite growth, we searched for and identified a family of compounds exhibiting a spectrum of channel affinities. In contrast to those carrying the furoquinoline scaffold, compounds carrying a dibenzothiazepinone scaffold did not uniformly produce high-level inhibition in our high-throughput screen (compound 2, Table 1). We then obtained 72 derivatives that share this scaffold but carry various substitutions at positions 8 and 10 on the scaffold. For each compound, we performed dose-response experiments using the osmotic lysis assay to estimate K0.5 for inhibition of PSAC. Because adsorption of some compounds by serum may adversely affect their PSAC affinities under in vitro culture conditions, these transport studies were performed in the presence of serum. pK0.5 were calculated from these values and are graphically presented with each compound's in vitro growth inhibitory pIC50 in Fig. 5. With serum present at identical concentrations in transport and growth studies, there was a significant correlation between PSAC inhibition and in vitro parasite growth inhibition (Spearman's rank correlation coefficient, rs, of 0.424; P < 0.0005 for the null hypothesis of statistical independence). Compounds producing high-level channel inhibition (K0.5 < 200 nM) were invariably effective against parasite growth with IC50 values uniformly below 3 μM. Although the relationship between channel activity and parasite growth is complex and nonlinear, the observed correlation provides chemical validation of PSAC as an antimalarial target.
Fig. 5.

Chemical validation of PSAC as an antimalarial target with a family of dibenzothiazepinone derivatives. In vitro parasite growth inhibition pIC50 values for each of 72 dibenzothiazepinone derivatives is plotted against pK0.5 values determined from dose responses for inhibition of osmotic lysis in the sorbitol lysis solution supplemented with 5% human serum. pIC50 and pK0.5 were calculated as −log(IC50) and −log(K0.5) in molar units, respectively; the most effective compounds are at the top right of the plot. Compound 2, the parent compound for this family, is shown as a star. Although some derivatives have additional “off-target” effects (e.g., compounds to the upper left of the profile), there is a significant correlation between channel inhibition and parasite killing.
Specificity of These Scaffolds for PSAC.
We then examined whether these inhibitors are specific for PSAC and whether they are suitable starting points for antimalarial drug discovery. To this end, we searched for activities of compounds 1 and 2 against other targets screened at the Broad Institute using the public ChemBank database (Seiler et al., 2008). ChemBank stores raw values associated with each screen as well as a number of statistical parameters that are designed to evaluate each screen's signal-to-noise ratio, the reproducibility of measurements, and whether individual compounds are considered hits. According to this database, compounds 1 and 2 have been used for screening in 18 and 20 separate projects, respectively. Statistical criteria used by ChemBank reproducibly identified both as “standard hits” in our PSAC inhibitor screens. Compound 1 was judged to be a standard hit in one other project, but this was not clearly reproducible (positive in 6/24 trials against that target). Compound 2 was judged to be a standard hit in two other projects, but again neither was clearly reproducible (positive in 2/6 and 2/18 trials each). Thus, these compounds do not appear to be nonspecific inhibitors of diverse targets.
Compounds 1 and 2 also meet all of Lipinski's rule of five criteria (Lipinski et al., 2001), suggesting that they are sufficiently drug-like for exploration as starting points for antimalarial drug discovery targeting PSAC.
Discussion
We describe a high-throughput screen that successfully identified potent inhibitors of malaria parasite-induced transport across the erythrocyte membrane. The screen was based on selective osmotic lysis of infected erythrocytes in the sugar alcohol sorbitol. Compounds that inhibit parasite-induced sorbitol uptake abrogate osmotic lysis and were detected in our screen as wells with higher absorbance. Statistical measures indicate that our screen had high reproducibility with clear separation of positive and negative controls. Because osmotic lysis of individual cells is an irreversible process resulting from accumulation of a threshold amount of sorbitol (Wagner et al., 2003), sequential readings of each plate permitted estimation of inhibitory K0.5 values over almost 4 orders of magnitude in a high-throughput format.
Our assay and screening algorithm has a number of important conceptual and technical advantages. Most importantly, our approach combines the advantages of target- and cell-based screens. Target-based screens, typically carried out with recombinant purified protein, generally have high confidence that identified hits will directly interact with the target in question. Their main disadvantage is that the compound may fail to be active against the target in the context of living cells; important examples of this include inability of the compound to access its intracellular target because of low membrane permeability and reduced stability of the compound as a result of cellular metabolism (Payne et al., 2007). Cell-based screens overcome this limitation by identifying activity in intact cells. However, most cell-based screens are plagued by the identification of hits that act against multiple unrelated targets. A major limitation of most cell-based screens, therefore, is that downstream studies to definitively identify the target of key hits may be both arduous and unsuccessful (An and Tolliday, 2009). Our screen overcomes these various limitations by using a cell-based assay that is specific for a single parasite activity.
Another common problem with cell-based screens arises from the observation that some compounds in nearly all screening collections have detergent-like activity. Weak detergent activity is often sufficient to produce a false positive in many screens. Our screen is not subject to this concern because the osmotic lysis assay identifies hits based on their abilities to retard or abolish infected cell lysis. Thus, compounds having detergent activity may produce cell lysis and be incorrectly classified as negatives, but they cannot yield false positive results in the screen presented here.
We also used simple and inexpensive absorbance measures, avoiding false positive detection of autofluorescent compounds as commonly seen in fluorescence-based assays. Because compounds that significantly absorb light of 700 nm wavelength might be detected as false positives in our screen, we screened several collections of compounds in the osmotic lysis buffer without addition of infected cells and did not find a single compound that altered absorbance significantly. This may reflect the generally poor absorbance of small molecules at near-infrared wavelengths, physicochemical limitations on absorption by compounds present at relatively low screening concentrations, or the much larger absorbance change associated with lysis of erythrocytes. Indeed, all of the screening hits shown here as well as a number of weaker hits examined in secondary studies seem to specifically target PSAC activity.
The results of our high-throughput screen provide several important insights into parasite biology. One important unanswered question is the number of distinct ion channels required to account for the diverse collection of organic and inorganic solutes with increased permeability after infection. Some studies have suggested that there may be separate ion channels for various subsets of the solutes (Ginsburg and Stein, 2005; Bouyer et al., 2006; Akkaya et al., 2009); in this scenario, the combined activities of these channels would then be responsible for the overall profile of parasite-induced host cell permeability. Although quantitative examination of the effects of inhibitors on solute transport should be able to determine whether there are multiple separate routes or a single shared route, previous transport inhibitors were unable to address this debate because they had relatively low affinities and uncertain specificities (Staines et al., 2007). Our study provides evidence for a single, shared ion channel because high-affinity inhibitors identified through a sorbitol permeability screen are also potent inhibitors of amino acid, sugar, and organic cation uptake (Fig. 4). Our single-channel studies add Cl− to this list of solutes and implicate PSAC as the common mechanistic basis of the parasite-induced permeabilities. Nevertheless, definitive resolution of the relative contributions of various proposed channels will be possible only when the responsible genes are identified.
We present two novel scaffolds that interact directly with one or more sites on the channel. Although medicinal chemistry to systematically evaluate derivatives of these scaffolds is pending, the furoquinoline scaffold was almost invariably associated with high-level channel inhibition, and one derivative, compound 9, is the most potent reversible inhibitor of PSAC identified thus far. In contrast, the dibenzothiazepinone scaffold was also highly active but was more susceptible to adverse effects of substitutions on the scaffold. A more rigorous examination of structure-activity relationships around this scaffold may provide a better understanding of how these molecules achieve block. Our analyses of single-channel recordings with both scaffolds suggest that the most potent inhibitors induce very long block durations (≥1 s) without reducing open-channel amplitudes, suggesting an allosteric mechanism of action.
Previous studies have surveyed derivatives of furosemide and dantrolene and achieved substantial improvements in PSAC affinity (Staines et al., 2004; Kang et al., 2005). Although those scaffolds also deserve consideration, our study makes several fundamental advances that should facilitate drug discovery against this target. First, we have identified a large number of diverse scaffolds by high-throughput screening (all available online at http://chembank.broadinstitute.org). Multiple, unrelated scaffolds are generally considered necessary because of attrition in hit-to-lead chemistry as a result of issues with chemical tractability for lead optimization, pharmacokinetic liabilities, specificity for the desired target, cost, and other factors (Zhao, 2007). Second, drug discovery will be assisted by our single-channel studies, which provide mechanistic insights into how derivatives may achieve greater potency. Finally, another advantage of our screen is that each of the hits described here is immediately available for basic research studies (ChemDiv, San Diego, CA).
Although multiple studies have suggested that inhibition of PSAC may adversely affect the intracellular parasite, evidence supporting this hypothesis has been based only on small numbers of relatively weaker inhibitors that may also interfere with other parasite activities (Kutner et al., 1987; Desai et al., 2005; Kang et al., 2005). These so-called “off-target effects” represent a major hurdle for drug discovery projects and are commonplace (Kell, 2006). Although it is difficult to definitively exclude off-target effects (Dorato and Engelhardt, 2005), the statistically significant correlation between PSAC inhibition and in vitro parasite growth inhibition observed for a large family of dibenzothiazepinone inhibitors supports an essential role of PSAC in intracellular parasite survival. Additional studies, possibly facilitated by these potent inhibitors, will be necessary to determine whether this essential role is related to nutrient acquisition (Desai et al., 2000) or other proposed functions (Brand et al., 2003; Lang et al., 2004; Mauritz et al., 2009).
Although they are effective at inhibiting in vitro parasite growth, these PSAC inhibitors had IC50 values for in vitro parasite growth inhibition generally higher than their K0.5 values for inhibition of sorbitol uptake. For example, the most active dibenzothiazepinone inhibited sorbitol uptake in serum-containing solutions with a K0.5 of 70 nM and was also the best in this class at parasite growth inhibition (Fig. 5); however, its IC50 for parasite growth inhibition was approximately 10-fold higher (670 nM). Similar or larger discrepancies have been seen with other parasite targets (Rosenthal et al., 1991; Baldwin et al., 2005; Freundlich et al., 2007); they have frequently been attributed to either poor membrane permeability of inhibitors or expression of the target enzyme at levels higher than minimally required for parasite growth. Two factors specific to PSAC may also contribute to this discrepancy in our studies. First, if PSAC-mediated uptake of organic solutes does function in nutrient acquisition, use of RPMI formulations, which contain supraphysiological concentrations of essential nutrients, may lead to reduced efficacy of these compounds in culture-based growth inhibition studies. Second, the reduced efficacy of these and other PSAC inhibitors against a subset of permeating solutes (Lisk et al., 2007; Bokhari et al., 2008; Fig. 4) may also contribute to the observed discrepancy if these solute-inhibitor interactions occur in parasite cultures. Inhibitors not susceptible to these interactions are being sought and may be better starting points for drug development against PSAC.
A major hurdle in both parasite cell biology and drug discovery targeting PSAC is the identification of the genetic basis of PSAC. Definitive identification has in part been hindered by uncertainties about whether the activity reflects proteins encoded by the parasite or human proteins selectively modified by the parasite (Staines et al., 2007). The availability of potent small-molecule ligands for PSAC now renders biochemical purification of the protein on an affinity column followed by peptide sequencing a viable approach to gene identification. Such an approach would not require assumptions about the origin or number of proteins involved in this important activity.
Acknowledgments
We thank Ian Bathurst, the Medicines for Malaria Venture Project Director, for these studies, and Michael Fay for help with statistical analysis. We thank the National Cancer Institute, the Initiative for Chemical Genetics, and the Chemical Biology Platform of the Broad Institute of Harvard and MIT, which provided support for high-throughput screening.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases and by the Medicines for Malaria Venture (MMV).
S.A.D. and A.D.P. are named inventors on a provisional patent application describing compounds presented in this article. The other authors have no competing interests to declare.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.062711.
- PSAC
- plasmodial surface anion channel
- PBS
- phosphate-buffered saline
- DMSO
- dimethyl sulfoxide.
References
- Akkaya C, Shumilina E, Bobballa D, Brand VB, Mahmud H, Lang F, Huber SM. (2009) The Plasmodium falciparum-induced anion channel of human erythrocytes is an ATP-release pathway. Pflugers Arch 457:1035–1047 [DOI] [PubMed] [Google Scholar]
- Alkhalil A, Cohn JV, Wagner MA, Cabrera JS, Rajapandi T, Desai SA. (2004) Plasmodium falciparum likely encodes the principal anion channel on infected human erythrocytes. Blood 104:4279–4286 [DOI] [PubMed] [Google Scholar]
- An WF, Tolliday NJ. (2009) Introduction: cell-based assays for high-throughput screening. Methods Mol Biol 486:1–12 [DOI] [PubMed] [Google Scholar]
- Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. (2005) High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem 280:21847–21853 [DOI] [PubMed] [Google Scholar]
- Bokhari AA, Solomon T, Desai SA. (2008) Two distinct mechanisms of transport through the plasmodial surface anion channel. J Membr Biol 226:27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer G, Egée S, Thomas SL. (2006) Three types of spontaneously active anionic channels in malaria-infected human red blood cells. Blood Cells Mol Dis 36:248–254 [DOI] [PubMed] [Google Scholar]
- Brand VB, Sandu CD, Duranton C, Tanneur V, Lang KS, Huber SM, Lang F. (2003) Dependence of Plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell Physiol Biochem 13:347–356 [DOI] [PubMed] [Google Scholar]
- Cohn JV, Alkhalil A, Wagner MA, Rajapandi T, Desai SA. (2003) Extracellular lysines on the plasmodial surface anion channel involved in Na+ exclusion. Mol Biochem Parasitol 132:27–34 [DOI] [PubMed] [Google Scholar]
- Desai SA. (2005) Open and closed states of the plasmodial surface anion channel. Nanomedicine 1:58–66 [DOI] [PubMed] [Google Scholar]
- Desai SA, Alkhalil A, Kang M, Ashfaq U, Nguyen ML. (2005) Plasmodial surface anion channel-independent phloridzin resistance in Plasmodium falciparum. J Biol Chem 280:16861–16867 [DOI] [PubMed] [Google Scholar]
- Desai SA, Bezrukov SM, Zimmerberg J. (2000) A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature 406:1001–1005 [DOI] [PubMed] [Google Scholar]
- Dorato MA, Engelhardt JA. (2005) The no-observed-adverse-effect-level in drug safety evaluations: use, issues, and definition(s). Regul Toxicol Pharmacol 42:265–274 [DOI] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naudé B, Deitsch KW, et al. (2000) Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6:861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundlich JS, Wang F, Tsai HC, Kuo M, Shieh HM, Anderson JW, Nkrumah LJ, Valderramos JC, Yu M, Kumar TR, Valderramos SG, Jacobs WR, Jr., Schiehser GA, Jacobus DP, Fidock DA, Sacchettini JC. (2007) X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. J Biol Chem 282:25436–25444 [DOI] [PubMed] [Google Scholar]
- Ginsburg H, Stein WD. (2005) How many functional transport pathways does Plasmodium falciparum induce in the membrane of its host erythrocyte? Trends Parasitol 21:118–121 [DOI] [PubMed] [Google Scholar]
- Kang M, Lisk G, Hollingworth S, Baylor SM, Desai SA. (2005) Malaria parasites are rapidly killed by dantrolene derivatives specific for the plasmodial surface anion channel. Mol Pharmacol 68:34–40 [DOI] [PubMed] [Google Scholar]
- Kell DB. (2006) Systems biology, metabolic modelling and metabolomics in drug discovery and development. Drug Discov Today 11:1085–1092 [DOI] [PubMed] [Google Scholar]
- Kutner S, Breuer WV, Ginsburg H, Cabantchik ZI. (1987) On the mode of action of phlorizin as an antimalarial agent in in vitro cultures of Plasmodium falciparum. Biochem Pharmacol 36:123–129 [DOI] [PubMed] [Google Scholar]
- Lang F, Lang PA, Lang KS, Brand V, Tanneur V, Duranton C, Wieder T, Huber SM. (2004) Channel-induced apoptosis of infected host cells-the case of malaria. Pflugers Arch 448:319–324 [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26 [DOI] [PubMed] [Google Scholar]
- Lisk G, Desai SA. (2005) The plasmodial surface anion channel is functionally conserved in divergent malaria parasites. Eukaryot Cell 4:2153–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisk G, Kang M, Cohn JV, Desai SA. (2006) Specific inhibition of the plasmodial surface anion channel by dantrolene. Eukaryot Cell 5:1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisk G, Scott S, Solomon T, Pillai AD, Desai SA. (2007) Solute-inhibitor interactions in the plasmodial surface anion channel reveal complexities in the transport process. Mol Pharmacol 71:1241–1250 [DOI] [PubMed] [Google Scholar]
- Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE. (2006) Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc Natl Acad Sci U S A 103:8840–8845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RE, Kirk K. (2007) Transport of the essential nutrient isoleucine in human erythrocytes infected with the malaria parasite Plasmodium falciparum. Blood 109:2217–2224 [DOI] [PubMed] [Google Scholar]
- Mauritz JM, Esposito A, Ginsburg H, Kaminski CF, Tiffert T, Lew VL. (2009) The homeostasis of Plasmodium falciparum-infected red blood cells. PLoS Comput Biol 5:e1000339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell CA, Msuya E, Sudi M, Njunwa KJ, Carneiro IA, Curtis CF. (2002) Effect of community-wide use of insecticide-treated nets for 3–4 years on malarial morbidity in Tanzania. Trop Med Int Health 7:1003–1008 [DOI] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40 [DOI] [PubMed] [Google Scholar]
- Rosenthal PJ, Wollish WS, Palmer JT, Rasnick D. (1991) Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J Clin Invest 88:1467–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba KJ, Horner HA, Kirk K. (1998) Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J Biol Chem 273:10190–10195 [DOI] [PubMed] [Google Scholar]
- Sanchez CP, Rotmann A, Stein WD, Lanzer M. (2008) Polymorphisms within PfMDR1 alter the substrate specificity for anti-malarial drugs in Plasmodium falciparum. Mol Microbiol 70:786–798 [DOI] [PubMed] [Google Scholar]
- Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, Brudz S, Sullivan JP, Muhlich J, Serrano M, et al. (2008) ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res 36:D351–D359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. (1987) Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J 52:1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staines HM, Alkhalil A, Allen RJ, De Jonge HR, Derbyshire E, Egée S, Ginsburg H, Hill DA, Huber SM, Kirk K, et al. (2007) Electrophysiological studies of malaria parasite-infected erythrocytes: current status. Int J Parasitol 37:475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staines HM, Dee BC, O'Brien M, Lang HJ, Englert H, Horner HA, Ellory JC, Kirk K. (2004) Furosemide analogues as potent inhibitors of the new permeability pathways of Plasmodium falciparum-infected human erythrocytes. Mol Biochem Parasitol 133:315–318 [DOI] [PubMed] [Google Scholar]
- Tolliday N, Clemons PA, Ferraiolo P, Koehler AN, Lewis TA, Li X, Schreiber SL, Gerhard DS, Eliasof S. (2006) Small molecules, big players: the National Cancer Institute's Initiative for Chemical Genetics. Cancer Res 66:8935–8942 [DOI] [PubMed] [Google Scholar]
- Wagner MA, Andemariam B, Desai SA. (2003) A two-compartment model of osmotic lysis in Plasmodium falciparum-infected erythrocytes. Biophys J 84:116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73 [DOI] [PubMed] [Google Scholar]
- Zhao H. (2007) Scaffold selection and scaffold hopping in lead generation: a medicinal chemistry perspective. Drug Discov Today 12:149–155 [DOI] [PubMed] [Google Scholar]