

Abstract
In designing a synthetic scaffold for engineering soft, mechanically active tissues, desirable properties include elasticity, support of cell adhesion and growth, ease of processability, and responsiveness to in vivo remodeling. To achieve these properties, we have developed a family of thermoplastic elastomers, polyurethaneureas (PUs), that possess enzymatic remodeling capabilities in addition to simple hydrolytic lability. PUs were synthesized using either polycaprolactone or triblock copolymer polycaprolactone-b-poly(ethylene glycol)-b-polycaprolactone as the soft segment, 1,4-butanediisocyanate as the hard segment, and the peptide Ala-Ala-Lys as a chain extender. The synthesized PUs had high molecular weights, low glass transition temperatures (<−54 °C), and were flexible with breaking strains of 670–890% and tensile strengths of 15–28 MPa. Incubation in buffered saline without elastase for 8 weeks resulted in mass loss from 12% to 18% depending on soft segment composition. The degradation significantly increased (p < 0.05) in the presence of elastase, ranging from 19% to 34% with degradation products showing no cytotoxicity. To encourage cell adhesion, PUs were surface-modified with radio frequency glow discharge followed by coupling of Arg-Gly-Asp-Ser (RGDS). Endothelial cell adhesion was >140% of tissue culture polystyrene on PU surfaces and >200% on RGDS-modified surfaces. The synthesized PUs thus combine mechanical, chemical, and bioresponsive properties that might be employed in soft-tissue engineering applications.
Introduction
The design challenge in creating a synthetic scaffold for the engineering of soft tissue is manifold. For mechanically active soft tissues, such as arteries, the benefits of mechanical conditioning during tissue development are increasingly apparent.1–4 A synthetic scaffold for this type of tissue engineering should thus possess elasticity to permit stress transfer to the developing tissue and also degrade at a rate that will allow cellular components to gradually be exposed to the mechanical load and respond accordingly. Beyond these mechanical and degradation properties, it would be advantageous to be able to process the scaffold into appropriate architectures and incorporate bioactivity in the form of adhesion, growth, and differentiation signals.
There has been an increasing interest in the biomaterials community in developing hydrolytically labile elastomers for soft-tissue engineering applications. Several approaches have been taken, with the majority employing polyester functionality to present lability. These include thermoplastic or thermoset polyesters5–10 and polyurethanes with polyester-containing soft segments11–22 as well as cross-linked polymers with polyester functionality.23,24 We have been particularly interested in utilizing linear polyurethanes because of their inherent design flexibility and amenability to processing.11,12,25,26
While the vast majority of synthetic tissue engineering scaffolds degrade by simple hydrolysis, this is in contrast to the body's own temporary (and permanent) scaffolding, which is remodeled enzymatically. This cell-facilitated process proceeds with a complex physiological control not possible in purely hydrolytic systems. To begin to mimic the physiological pathways in a synthetic scaffold, the groups of Hubbell, West, and others have developed cross-linked hydrogel systems that incorporate peptide sequences in their backbones that are specific for some of the enzymes relevant to extracellular matrix remodeling in vivo, such as elastase, matrix metalloproteinases, and plasmin.27–31 These systems have been shown to be responsive to cell-based degradation. Our interest was to translate this general approach for enzymatic degradability to a thermoplastic elastomer system using a peptide sequence exhibiting elastase sensitivity.
The polymers that we report on here were based on poly(ester urethane) ureas and poly(ether ester urethane)ureas previously described.11,12 Briefly, these earlier polymers possessed soft segments of polycaprolactone (PCL) or triblock copolymers of PCL-poly(ethylene glycol)-PCL, hard segments of 1,4-butanediisocyanate, and were chain extended with putrescine. For polyurethanes with triblock soft segments, manipulation of the polyester/polyether ratio in the soft segment was shown to predictably impact the degradation rate in buffer.12 To add enzymatic lability, in this study we have replaced putrescine in the chain extension step with Ala-Ala-Lys (AAK), with the rationale being that elastase has specificity for cleavage between alanine residues30,32 and the terminal lysine creates a diamine structure.
By synthesizing and characterizing this new group of polyurethanes, we were able to investigate a number of hypotheses: specifically, whether introduction of the AAK sequence could impart elastase sensitivity in a polyurethane system and whether manipulating the polyester/polyether ratio in the soft segment of a polyurethane also containing AAK in the backbone would allow manipulation of the polymer degradation rate in buffer with and without elastase present. Furthermore, we were interested in demonstrating that the degradation products from these polyurethanes were not cytotoxic, that the polyurethanes supported cell adhesion and growth, and that the support of cell adhesion could be further augmented by surface modification with the cell adhesion peptide Arg-Gly-Asp-Ser (RGDS).
Experimental Section
Materials
Polycaprolactone diol (PCL, MW 2000) and poly(ethylene glycol) (PEG, MW 600 and 1000) were obtained from Aldrich and dried under vacuum for 24 h to remove residual water. ∈-Caprolactone (Aldrich) was dried over CaH2. Butyldiisocyanate (BDI, Fluka) and putrescine (Aldrich) were vacuum distilled before use. Stannous octoate (Sigma) and H-Ala-Ala-Lys-OH·HCl (AAK, BaChem Inc.) were used as received. Dimethyl sulfoxide (DMSO) was dried over 4 Å molecular sieves.
Synthesis of Polyurethaneureas
The triblock copolymer polycaprolactone-b-poly(ethylene glycol)-b-polycaprolactone (PCL-b-PEG-b-PCL) was synthesized by ring-opening polymerization of ∈-caprolactone initiated by poly(ethylene glycol).12 Stannous octoate was used as a catalyst, and the polymerization was carried out at 120 °C for 24 h under a nitrogen atmosphere. The products obtained were washed with ethyl ether and hexane, and then dried in a vacuum oven. The polymers were characterized by 1H NMR. Triblock copolymers with variable PEG and PCL molecular weights were synthesized (Table 1).
Table 1.
Block Length of PCL-b-PEG-b-PCL Triblock Copolymers
| polymer | feed ratio (molCL/molPEG) | block length of PCL-b-PEG-b-PCL |
|---|---|---|
| PCL-b-PEG1000-b-PCL | 18.0/1 | 1000–1000–1000 |
| PCL-b-PEG600-b-PCL | 18.0/1 | 1000–600–1000 |
| PCL-b-PEG600-b-PCL | 27.1/1 | 1500–600–1500 |
Polyurethaneureas (PUs) were synthesized using a two-step solution polymerization (Scheme 1).11,12 The synthesis was carried out in a 250 mL three-necked round-bottom flask. The stoichiometry of the reaction was 2:1:1 of BDI/PCL or PCL-b-PEG-b-PCL/ Ala-Ala-Lys. A 15 wt % solution of BDI in DMSO was continuously stirred with a 25 wt % solution of diol in DMSO. Stannous octoate was then added. This mixture was allowed to react at 75 °C for 3.5 h. A solution of AAK in DMSO was heated to 50 °C, and triethylamine at twice the AAK molar concentration was added to aid dissolution. This solution was subsequently added to the prepolymer solution, and the reaction was continued for 18 h at 75 °C. The polymer solutions were precipitated in distilled water. Polymers were dried under vacuum at 50 °C for 24 h. The polymers were then redissolved in DMSO, centrifuged, precipitated in distilled water, and vacuum-dried. The polymers are abbreviated as PU/CEXCYAAK, where CEXCY refers to the PCL-b-PEG-b-PCL triblock copolymer, X refers to the molecular weight of PEG divided by 100, and Y refers to the total molecular weight of the triblock copolymer divided by 100.
Scheme 1.
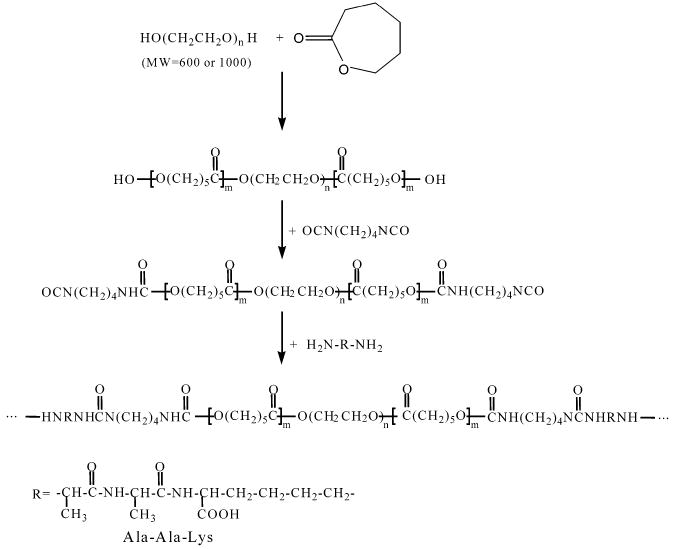
Synthesis of Elastase-Sensitive Polyurethaneureas
PU films were cast from a 3 wt % solution in dimethylformamide (DMF) onto a poly(tetrafluoroethylene) (PTFE) dish, and solvent was evaporated at room temperature. The films were then dried under vacuum for 2 days at 50 °C to obtain a film approximately 60 μm thick. This film was cut into pieces 4 × 0.5 cm2 for characterization and biodegradation experiments.
Polymer Characterization
Polymer molecular weight was determined by gel permeation chromatography (GPC, Waters Breeze System, Waters 1515 HPLC Pump, Waters 2414 differential refractometer), using monodisperse polystyrene standards for calibration. Measurements were made at room temperature with 1-methyl-2-pyrrolidinone as a solvent. The injection volume was 100 μL, and the polymer concentration was approximately 15 mg/mL.
Fourier transform infrared (FTIR) spectra were obtained at room temperature using a Nicolet FTIR spectrometer. A 5% solution of polymer in DMF was placed directly onto NaCl plates with subsequent evaporation of DMF at 50 °C. The spectra did not show evidence of residual solvent. Differential scanning calorimetry (DSC) was performed in a Thermal Analyst 2000 (TA Instruments) DSC 2910 differential scanning calorimeter. Scanning rates of 20 °C/min were used over a temperature range of −100 to 200 °C. Data from the first run were used to characterize PU thermal properties. The glass transition temperature (Tg), melting temperature (Tm), and melting enthalpy (ΔHm), on the basis of the total PU weight and the PCL fraction, were analyzed with software supplied with the DSC 2910 differential scanning calorimeter. The inputs for this curve integration were the temperature boundaries for the endotherm. X-ray diffraction (XRD) measurements on cast films were conducted on a Scintag ×2 diffraction system with Peltier detector. The tube was operated at 45 kV and 40 mA. The step size for each measurement was 0.02 deg, and time per step was 1 s. PEG (MW 1000) and PCL (MW 2000) were used as control samples.
The water absorption ratio was measured after 24 h immersion in water at room temperature and defined as the normalized difference of the wet mass (w2) and dry mass of the film (w1).20
Six independent measurements were performed. Water contact angles (n = 10 for each polymer type) were measured by a sessile drop method on a VCA2000 contact angle instrument (AST Products).
Tensile properties were measured for PU films, where cast films 60 μm in thickness were cut with an ASTM D638-98 die. Testing was conducted in an Instron testing machine equipped with a 5 lb load cell. A cross-head speed of 10 mm/min was used. Four samples were evaluated for each polymer composition.
Polyurethaneurea Degradation
To quantify PU degradation, dry films (4 × 0.5 cm2) were weighed and immersed in 10 mL phosphate-buffered saline (PBS; pH = 7.4) alone or with 0.3 mg/mL elastase (Sigma, Type I, from porcine pancreas). The degradation was conducted at 37 °C in a water bath with the solution exchanged every 7 days. Samples were taken at intervals, rinsed with water, and weighed after drying in a vacuum for 2 days. The weight remaining was calculated as
where w1 and w3 are the weights of films before and after degradation, respectively.
Synthesis and Degradation of Model mPEG Peptide Conjugates
To verify the sensitivity of the AAK peptide to elastase, triblock polymer segments were synthesized with either AAK or control peptide, Gly-Gly-Lys (GGK), flanked by methyl-PEG (mPEG). The mPEG-AAK-mPEG conjugate was synthesized in a 250 mL three-neck flask equipped with argon inlet and outlet, and mPEG-succinimidyl propionate (mPEG-SPA, Mn = 5268, PDI = 1.03, Nektar Therapeutics) added at 4.5 g (0.854 mmol) with 50 mL DMSO. This was followed by the slow addition of a solution of 0.139 g (0.427 mmol) AAK in DMSO. The reaction was conducted at room temperature for 24 h. The solution was concentrated under reduced pressure and then precipitated with ether. The polymer was dried under vacuum for 2 days. The mPEG-GGK-mPEG conjugate was synthesized with the same method as mPEG-AAK-mPEG, except that 0.111 g (0.427 mmol) H-Gly-Gly-Lys-OH acetate salt (BaChem, Inc.) was used.
The degradation of the two conjugates was conducted in the presence of 0.3 mg/mL elastase (Sigma, Type I, from porcine pancreas) or 0.3 mg/mL collagenase (Sigma, Type I, from Clostridium histolyticum). mPEG-AAK-mPEG or mPEG-GGK-mPEG (30 mg) was placed in a 15 mL conical tube; 10 mL elastase or collagenase solution was then added. The solution was kept at 37 °C for 24 h. After the incubation period, the solution was lyophilized, absolute ethanol was added, and the solution was then filtered with a 0.45 μm pore size PTFE filter. The ethanol was evaporated, and the polymer was dried under vacuum. The dried polymer was then dissolved in buffer (aqueous solution containing 0.1 M sodium nitrate and 0.01% sodium azide) at a concentration of 2 mg/mL. The molecular weight of the polymer was then measured by GPC (Viscotek Triple Detector Array model 300).
Cytotoxicity Assay of Degradation Products
The cytotoxicity of products from PU degradation was assessed as described previously.11 Human umbilical vein endothelial cells (HUVECs, Cambrex, at passage two) were seeded at a density of 1.5 × 105 cells/mL into a 96-well tissue culture polystyrene plate. The culture medium (EGM-2 endothelial cell medium-2 supplemented with 10% FBS) and degradation solution at 4 weeks were mixed 10:1, and 220 μL of the solution was obtained. For control experiments, culture medium was used alone. After 48 and 96 h of HUVEC culture, cell viability was measured (n = 4) using a colorimetric tetrazolium salt (MTT) assay for mitochondrial activity.11,12
Surface Immobilization of RGDS Peptide
The polymers were surface-modified using radio frequency glow discharge (RFGD) under an ammonia atmosphere to introduce reactive groups for bonding RGDS peptide. The polymer films were placed into the reactor (PLASMOD benchtop model, March Corp.) for 2 min at 100 W and 13.6 MHz under ammonia gas at 3 × 10−3 Torr. The cell adhesion peptide RGDS (Sigma) was immobilized onto the surface of PUs modified with RFGD (PU-NH2) using glutaraldehyde as a spacer (Scheme 2). PU-NH2 polymers were immersed in 30% glutaraldehyde for 12 h, rinsed thoroughly in DI water, and then immersed in a solution of 1 mg/mL RGDS in PBS for 12 h. Both reactions occurred at room temperature. The film was then extensively rinsed with PBS. Using the same method, Arg-Gly-Glu-Ser (RGES) peptide was immobilized onto the film surface as a control for the RGDS-modified PU surface when evaluating cell adhesion.
Scheme 2.
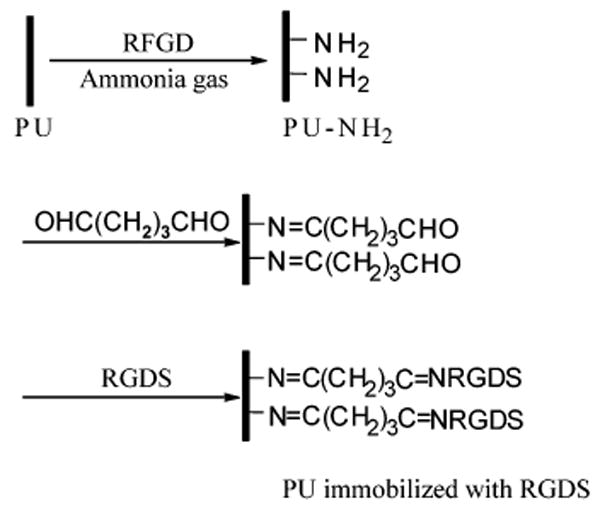
Covalent Immobilization of RGDS onto a PU Film
Endothelial Cell Adhesion and Growth
The ability of PU films to support endothelial cell adhesion was evaluated for PU/CE0C20AAK and PU/CE6C26AAK before and after RGDS and RGES modification. PU samples were cut and sterilized by immersion in 70% ethanol for 6 h. After rinsing thoroughly with PBS, samples were fit into the bottom of a 96-well tissue culture plate. HUVECs were seeded at a density of 1.5 × 105 cells/mL. To evaluate the total cell adhesion after 24 h of cell seeding, cell viability was quantified using the MTT assay (n = 4 per polymer type). Cell adhesion was defined in a normalized fashion as the ratio of optical absorption for the test polymer surfaces relative to the tissue culture plate.
To evaluate cell growth, the culture medium (EGM-2 supplemented with 10% FBS) was replaced every second day. Randomly selected wells were destructively evaluated with the MTT cell viability assay each day for 5 days (n = 4 per polymer type per day). For control purposes, HUVECs were cultured on tissue culture polystyrene 96-well plates.
Statistical Methods
Data are expressed as mean ± standard deviation. Statistical comparisons were performed by unpaired students t-testing if only two groups were being compared and by ANOVA with post hoc Neuman–Keuls testing for evaluations of three or more groups.
Results
Bulk Polymer Characterization
The FTIR spectrum (Figure 1) confirmed the structure of PU/CE6C26AAK. The ester groups showed the pronounced carbonyl peak at 1733 cm−1; the shoulder peak at lower wavelength was attributed to hydrogen bond associations of the urethane, urea, or amide groups. The urethane, urea, and amide groups also showed absorption at 3350 cm−1. The absorption of amide I and amide II appear at 1670 and 1560 cm−1, respectively. The absorbance at 1110 cm−1 was attributed to ether in the soft segment. The absence of absorbance at 2267 cm−1 indicated that there were no unreacted isocyanate groups in the PU.
Figure 1.
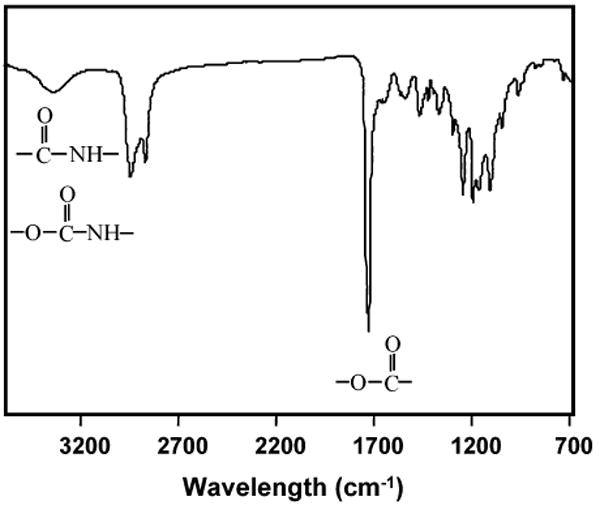
FTIR spectrum of PU/CE6C26AAK.
The typical 1H NMR spectrum of a synthesized PU is shown in Figure 2. The PCL-b-PEG-b-PCL segment demonstrated the characteristic peaks of methyl protons of the PCL block (peaks a, b, c, and d); the ethylene oxide protons of the PEG segment were seen as peaks e and f. The hard segment showed the characteristic peaks of the BDI derivative (peaks k and c) and AAK (peaks g, h, i, j, k, and c).
Figure 2.
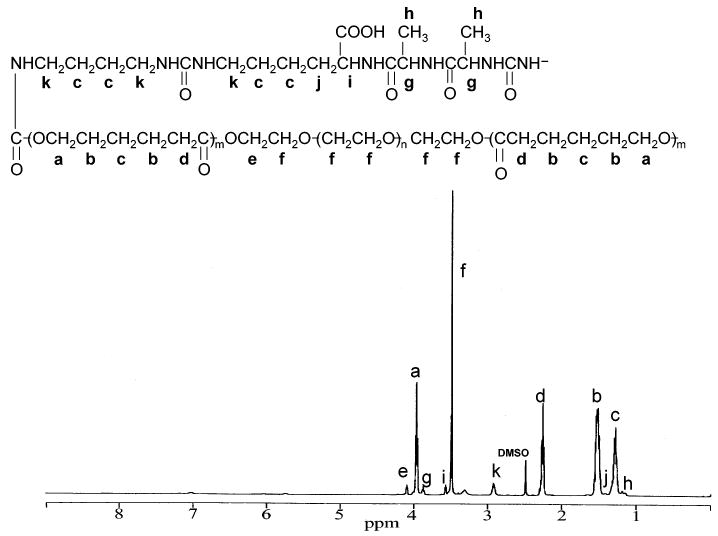
1H NMR spectrum of polyurethaneurea.
PU molecular weights as determined by GPC are shown in Table 2. Number average molecular weights were around 21 kD, and weight average molecular weights varied from 34 to 40 kD. The synthesized PUs dissolved completely in the 1-methyl-2-pyrrolidinone solvent used in GPC analyses and were greater than 99% soluble in DMF and DMSO, suggesting minimal polymer cross-linking.
Table 2.
Molecular Weight of Polyurethaneureas before Degradation
| polymer | Mw | Mn | Mw/Mn |
|---|---|---|---|
| PU/CE10C30AAK | 34 500 | 21 200 | 1.6 |
| PU/CE0C20AAK | 39 700 | 22 000 | 1.8 |
| PU/CE6C26AAK | 35 600 | 20 900 | 1.7 |
| PU/CE6C36AAK | 36 700 | 22 700 | 1.6 |
Thermal Properties
DSC curves are shown in Figure 3, and the parameters listed in Table 3. The PUs exhibited soft segment transitions over the temperature range of −100 to 200 °C. None of the PUs showed any hard segment transitions or PEG crystalline and melting transitions. All of the PUs had glass transition temperatures <−54 °C. The glass transition temperature decreased with increasing soft segment molecular weight. By comparing the PUs with PCL block molecular weight of 2000 but different PEG lengths, it was found that introduction of PEG into the soft segment decreased the glass transition temperature. Similarly, the PCL block melting temperature and melting enthalpies of the PU with PEG molecular weight of 1000 were higher than PUs with PEG molecular weight of 600 and 0 with the same PCL length. When the central PEG block was uniform, increasing the PCL block length increased the melting temperature and melting enthalpies.
Figure 3.
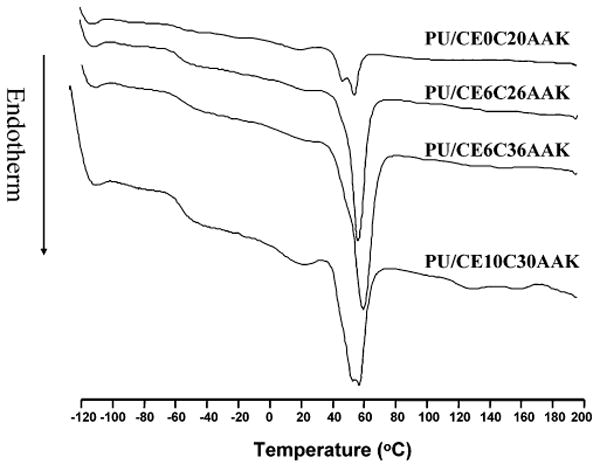
DSC heating curves for four different polyurethaneureas.
Table 3.
Thermal Properties of the Polyurethaneureas
| polymer | Tg (°C) | Tm (°C) | ΔHm (J/g PU) | ΔHm (J/g PCL)a |
|---|---|---|---|---|
| PU/CE0C20AAK | −54.3 | 53.7 | 16.3 | 20.9 |
| PU/CE6C26AAK | −55.6 | 56.3 | 32.6 | 51.6 |
| PU/CE6C36AAK | −56.7 | 59.4 | 43.4 | 60.3 |
| PU/CE10C30AAK | −56.4 | 57.4 | 38.8 | 69.2 |
Enthalpy normalized to the amount of PCL in the PU.
PUs were further characterized by XRD (Figure 4) to aid in interpreting the melting endotherms from the DSC results. XRD spectra showed characteristic peaks at 19.4 2θ and 23.7 2θ for PEG and 21.7 2θ and 24.0 2θ for PCL. All of the PUs showed only peaks that were characteristic of PCL.
Figure 4.
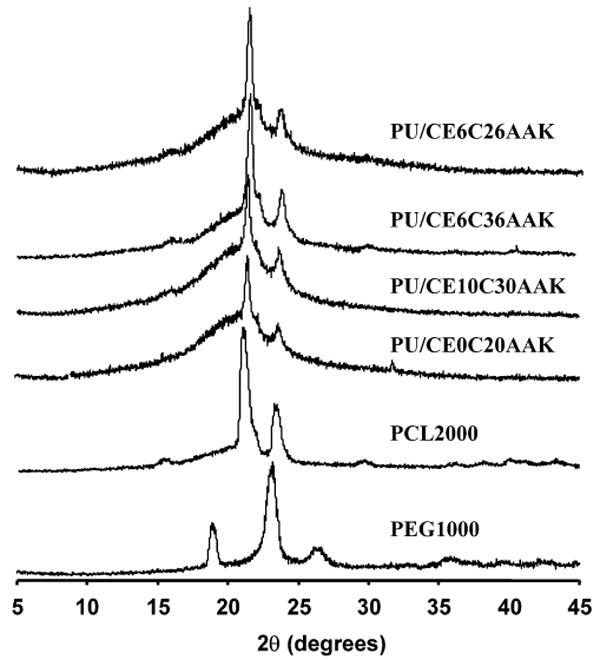
X-ray diffraction (XRD) spectra for PUs and relevant controls of PEG (MW 1000) and PCL (MW 2000).
Mechanical Properties
Measured mechanical parameters are summarized in Table 4. The polymers were highly flexible with tensile strengths ranging from 15 to 28 MPa and breaking strains from 670% to 890%. When comparing PUs having the same PCL molecular weight of 2000 but different PEG lengths, it was found that as the PEG segment increased the tensile strength decreased (p < 0.01). The breaking strain showed no significant differences between PUs with PEG MW of 0 and 600 (p > 0.5). The PU with PEG molecular weight of 1000 in the backbone had a significantly lower breaking strain than that without PEG (p < 0.02). When the central PEG length was 600, increasing the PCL length from 2000 to 3000 did not change the tensile strength or the breaking strain significantly.
Table 4.
Mechanical Properties of Polyurethaneureas
| polymer | tensile strength (MPa) | breaking strain (%) |
|---|---|---|
| PU/CE0C20AAK | 28 ± 4 | 830 ± 80 |
| PU/CE6C26AAK | 20 ± 3 | 890 ± 120 |
| PU/CE6C36AAK | 21 ± 2 | 720 ± 60 |
| PU/CE10C30AAK | 15 ± 2 | 670 ± 170 |
Polymer Swelling Properties and Surface Hydrophilicity
Water absorption was measured to determine polymer bulk hydrophilicity, as this was expected to have a substantial impact on polymer degradation (Figure 5). When the PCL block had the same length, the increase of PEG length increased water absorption (p < 0.02). In comparison of PU/CE6C26AAK with PU/CE6C36AAK, the increase in PCL length decreased water absorption (p < 0.01).
Figure 5.
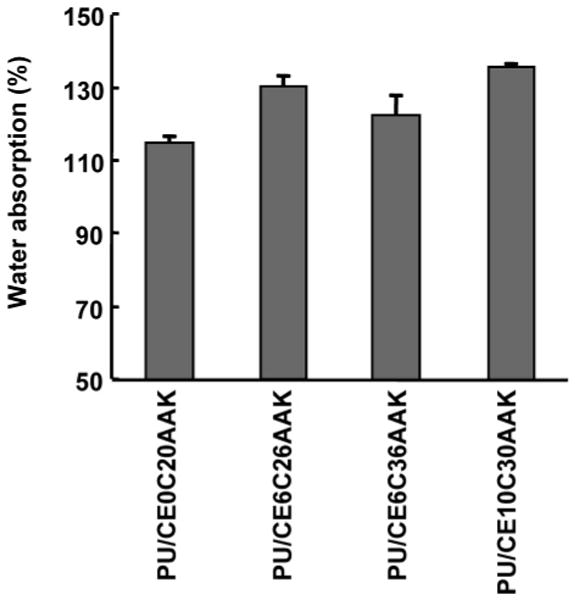
Water absorption of polyurethaneureas measured after 24 h immersion in water at room temperature.
Surface hydrophilicity was characterized by the static water contact angle. The results in Figure 6 show that PU hydrophilicity increased as the hydrophilic PEG molecule was incorporated into the backbone when the PCL was 2000 MW (p < 0.001). When the PU had the same PEG length but longer PCL length in the soft segment, the contact angle increased (p < 0.001).
Figure 6.
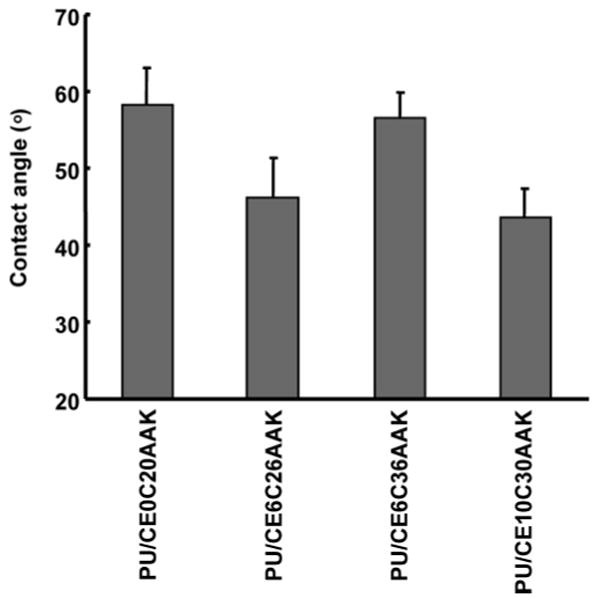
Static water contact angle of polyurethaneureas (n = 10 for each PU type).
Polyurethaneurea Degradation
To assess the degradability of AAK-containing PUs, the samples were incubated in PBS with or without elastase at 37 °C. As shown in Figure 7, when PU films were incubated in PBS without elastase, the mass loss of PEG containing PUs was higher than non-PEG-containing PU when the PCL length was 2000 (p < 0.05) at day 56. For PUs with a PEG block length of 600 MW, there was no difference between PCL blocks of 2000 and 3000 MW at the same time point.
Figure 7.
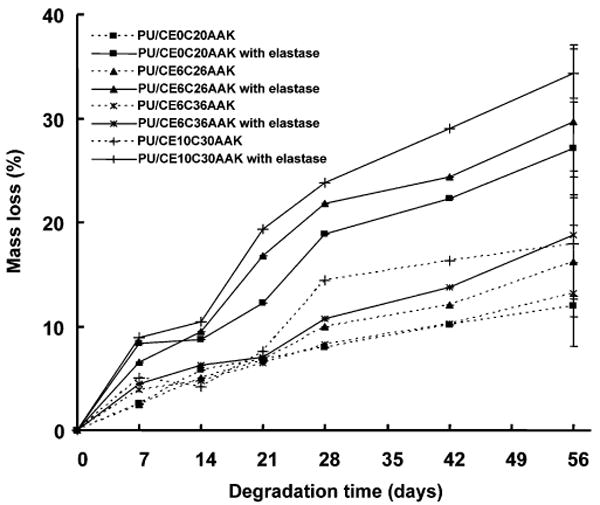
PU mass loss during incubation in an aqueous buffer with or without 0.3 mg/mL elastase. Error bars are only shown for the last time point for clarity.
When the PUs were incubated in elastase solution, the mass losses of all AAK-containing polymers were significantly higher at 56 days than corresponding samples incubated in PBS without elastase (p < 0.03). When comparing the degradation of PUs with the same PCL length of 2000 MW but different PEG lengths, the PU with PEG length of 1000 degraded more at 56 days than those with PEG length of 600 and without PEG (p < 0.02). The PU/CE6C36AAK had the lowest mass loss of all samples in elastase, although it had PEG 600 in the soft segment.
To further confirm the sensitivity of the AAK sequence to elastase, mPEG-AAK-mPEG and mPEG-GGK-mPEG conjugates were synthesized. The conjugates were incubated in PBS, elastase solution, or collagenase solution with molecular weights measured before and after incubation. Since mPEG is generally resistant to cleavage in water, elastase, and collagenase, a drop in molecular weight by approximately 50% would result if the peptide sequence were enzymatically cleaved. The observed molecular weight changes are listed in Table 5. For mPEG-AAK-mPEG, the molecular weight was close to the synthesized mPEG-AAK-mPEG after 24 h incubation in PBS; however, the molecular weight was almost halved after 24 h in elastase solution. A 24 h incubation of mPEG-AAK-mPEG in collagenase solution did not reduce the original molecular weight. Replacing AAK with GGK in the conjugate as a control revealed no sensitivity to elastase.
Table 5.
Number Average Molecular Weight (Mn) Change of mPEG-AAK-mPEG and MPEG-GGK-mPEG Conjugates Before and After Enzyme Incubationa
| day 1 | ||||
|---|---|---|---|---|
| polymer | day 0 | PBS | elastase | collagenase |
| mPEG-AAK-mPEG | 10 600b | 10 100 | 5400c | 11 400 |
| mPEG-GGK-mPEG | 10 600d | 9980 | 9730 | |
Mn of mPEG-SPA used to synthesize conjugates 5268, PDI = 1.03.
PDI = 1.06.
PDI = 1.14.
PDI = 1.05.
Cytotoxicity of Degradation Products
HUVEC viability on tissue culture polystyrene was measured for culture in medium supplemented at a 1:10 ratio with four-week biodegradation solutions from PU/CE0C20AAK or PU/CE6C26AAK. Data were normalized to HUVECs cultured in nonsupplemented medium. HUVECs cultured in PU/CE0C20AAK degradation solution supplemented medium had slightly higher viability (115%) at day 2 relative to cells cultured in the control medium (p < 0.02). HUVECs cultured in medium supplemented with PU/CE6C26AAK degradation solution had similar viability (102%) to nonsupplemented medium at day 2 (p = 0.69).
Endothelial Cell Adhesion and Growth
The number of adhered, viable HUVECs 24 h after cell seeding is presented in Figure 8 for unmodified PU/CE0C20AAK and PU/CE6C26AAK, for these two polymers with RGES attached following RFGD and glutaradehyde treatment, and for these two polymers with RGDS attached following RFGD and glutaradehyde treatment. Considering the adhered cell density onto a tissue culture polystyrene (TCPS) plate to be 100%, the adhesion on the unmodified PUs was about 130% (p < 0.01). For RGDS-modified surfaces, the 24 h cell adhesion was improved markedly to above 207% of that on tissue culture polystyrene (p < 0.001). Treatment with RGES had a slight effect on HUVEC adhesion with respect to unmodified PU surfaces (37% and 30% increases for PU/CE0C20AAK and PU/CE6C26AAK, respectively; p < 0.05), while RGDS-modified surfaces increased 24 h adhesion by 109% and 74% for PU/CE0C20AAK and PU/CE6C26AAK, respectively (p < 0.001 versus RGES).
Figure 8.
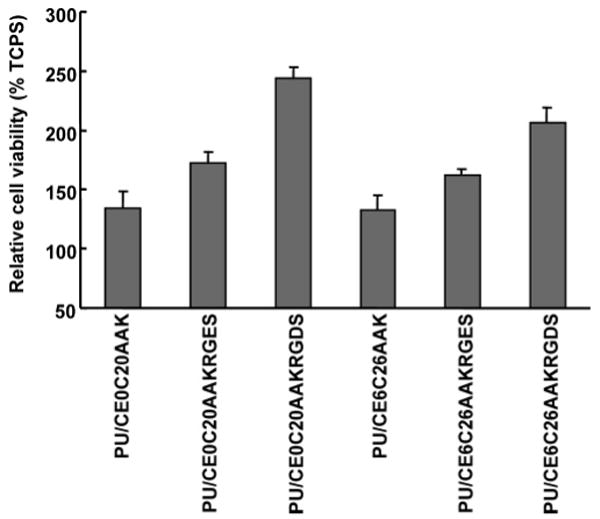
Endothelial cell adhesion on unmodified PU surfaces, as well as PU surfaces modified with RGES or RGDS peptide. Adhesion is presented relative to TCPS surfaces.
Proliferation of HUVECs on PU/CE0C20AAK and PU/CE6C26AAK with or without RGDS attachment was measured over time together with control TCPS surfaces (Figure 9). Nonmodified PUs had significantly higher cell numbers than tissue culture polystyrene at both time points. Similarly, the number of cells on RGDS-modified PUs was significantly higher than on unmodified PUs and TCPS at 3 and 5 days (p < 0.001). At day 3, the RGDS-modified PU/CE0C20AAK had higher cell number than RGDS-modified PU/CE6C26AAK. There was no difference between the RGDS-modified PUs at day 5.
Figure 9.
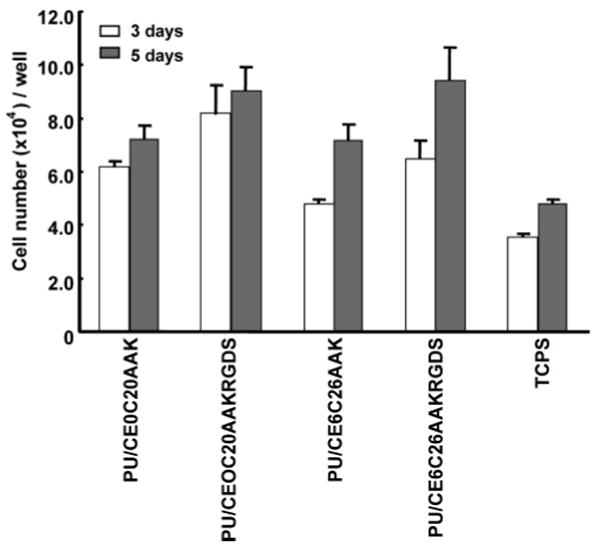
Endothelial cell growth on two PUs with and without immobilized RGDS after 3 and 5 day culture periods. Cell numbers on TCPS surfaces is presented for comparison.
Discussion
In synthesizing PUs with elastase-sensitive chain extenders and variable soft-segment chemistry to both control polymer hydrophilicity and putatively influence hydrolytic degradability, a range of polymer chemical and physical properties were obtained. All of the synthesized PUs had number average molecular weights around 20 kDa, high enough to impart adequate elastomeric behavior for the solid films.33 The PUs did not show any hard segment transitions inferring minimal phase separation structurally. There was also a lack of evidence of PEG crystallinity and melting (Figures 3 and 4; Table 4), probably because the PEG content of the polymers was too low to be able to form semicrystalline domains. This result agrees with our earlier findings for poly(ether ester)urethane ureas with similar soft segments but where putrescine was the chain extender.12 In Figure 3, there was a transition near the melting transition of the PCL block for PU/CE0C20AAK and PU/CE10C30AAK, respectively. The multiple melting peaks of the PCL block likely resulted from imperfect crystals formed by competing processes of crystallization and phase separation, as was found in PCL-b-PEG-b-PCL triblock copolymers by An et al.34
In Table 3, incorporating PEG into the soft segment increased the melting enthalpy, which could be explained by flexible PEG blocks providing conformational freedom for neighboring PCL segments to relax and more readily crystallize.35,36 Increasing the PEG length increased PCL crystallinity. When the central PEG block was uniform, increasing the PCL block length decreased the glass transition temperature and increased the melting temperature and melting enthalpy. This can be explained by the longer PCL block in the soft segment not readily mixing with the hard segment, resulting in increased hard- and soft-segment separation. This would increase soft-segment flexibility and promote crystallization of the soft segment.
The synthesized PUs possessed mechanical properties that would be attractive for soft-tissue engineering, assuming that such a scaffold should possess mechanical parameters comparable to the native tissue prior to initiation of the degradation period.37 When comparing PUs with the same PCL length but different PEG length, tensile strength decreased after adding PEG to the soft segment as well as with an increase in PEG block length. This trend was contrary to the effect of PEG length on crystallinity where introducing and increasing PEG length increased crystallinity. A limitation of this study is that we did not quantitatively monitor the loss of mechanical properties during the degradation period. Future studies will need to examine the performance of these materials in the in vivo setting to address questions as to appropriate mechanical loss characteristics during degradation. This study demonstrates that mass loss during degradation can be tuned by polymer design.
Considering polymer hydrophilicity, the surface contact angle and bulk swelling data confirmed that adding PEG to PU soft segments increased hydrophilicity. By adjusting the PEG and PCL length, PU hydrophilicity could be manipulated in a predictable fashion, similar to what was found for polyurethanes synthesized with PCL-b-PEG-b-PCL as a soft segment but putrescine as chain extender.12 When comparing putrescine chain-extended polyurethanes with the current AAK chain-extended polyurethanes, AAK chain extension increased the hydrophilicity of the polymer. This was expected, given the higher relative hydrophilicity of AAK.
The 56-day mass loss in PBS for PEG-containing PUs was greater than for non-PEG-containing PUs when they had the same PCL length. This difference in mass loss between PUs with or without PEG blocks might be attributed to differences in both chemical structure and water absorption. The ester bond between the PCL block and the PEG block should be more labile than the ester bond in PCL. Perhaps more importantly, since hydrolysis requires water access to labile ester bonds, polymers capable of absorbing larger amounts of water generally will degrade more rapidly than those absorbing little water. Furthermore, a high degree of water absorption will generally result in a swollen polymer matrix with a greater free volume for mass transfer,38 leading to increased transport of soluble degradation products from the material bulk.
All of the AAK-containing PUs had significantly increased mass loss when incubated in an elastase solution relative to PBS. In Figure 7, the PUs with longer PEG length and shorter PCL length exhibited more extensive degradation. This effect could be attributed to increased hydrolysis, as the swelling ratio increased when PEG length increased and PCL length decreased, and possibly due to increased relative access of elastase to scissible AA bonds in the more swollen polymer systems. Even with a swollen PU system, it is still likely that most of the enzymatically facilitated degradation is occurring on the surface rather than in bulk.21,22,28 As bulk degradation proceeds, however, increased surface area should allow increasing elastase degradation.
The molecular weight changes of model conjugates mPEG-AAK-mPEG and mPEG-GGK-mPEG after enzymatic incubation confirmed that the AAK sequence, but not the structurally similar GGK, was sensitive to elastase and that AAK was not responsive in this assay to collagenase. It is worth noting that the use of only two alanines was sufficient for the demonstrated elastase sensitivity, whereas previous studies have utilized greater numbers of sequential alanines.30,32 An increase in the repeating alanine number could increase the elastase sensitivity and the number of potential attack sites. Gertler and Hofmann32 revealed that a substrate with three alanines was highly elastase specific. West et al.30 used a nine-alanine sequence as an elastase-sensitive peptide incorporated into a PEG-based hydrogel and showed a rapid degradation rate. Increasing the alanine number in this PU system could be expected to increase polymer hydrophilicity and cost, while the effect on mechanical properties would not be clear.
The test for cytotoxicity from the four-week degradation solutions of two of the AAK-containing PUs demonstrated a lack of a negative effect on endothelial cells, although this assay is certainly not exhaustive. Of note, the solution from the PU having a PCL soft segment actually fostered slightly higher cell numbers, similar to the effect seen with a structurally analogous PU chain-extended with putrescine.11 One potential explanation for this effect would be a positive influence of putrescine, which has been implicated as a mediator of cell responsiveness to growth and differentiation factors.39–41
Endothelial cell adhesion on PU/CE0C20AAK and PU/CE6C26AAK was higher than on corresponding polyurethanes using putrescine as a chain extender.11 Surface modification with RFGD under ammonia gas to introduce surface amine functionality, followed by attachment of the adhesion peptide RGDS, improved cell adhesion at 1 day and effectively reduced the time for HUVECs to reach confluence on the films. The improved adhesiveness of the modified surfaces thus did not have a detrimental effect on the speed of cell coverage. Given the slight effect of the structurally similar RGES, the improved cell adhesion and growth was attributed to specific adhesive interactions with endothelial cell integrins.
The development of the elastase-sensitive poly(ether ester urethane)ureas reported here extends the array of biodegradable elastomers available for soft-tissue engineering and other biomedical material applications. These PUs are noteworthy for their combination of attractive mechanical properties and thermoplastic structure, with tunable sensitivity to both hydrolytic and elastase degradation. Earlier work with biodegradable polyurethanes has similarly incorporated soft-segment hydrolytic lability,20–22 and most notably, Woodhouse and colleagues have developed polyurethanes with a chain extender based on a phenylalanine diester together with a polyester soft segment.20,21 In that work, sensitivity to chymotrypsin and, to a lesser extent, trypsin was shown to be related to the introduction of a segment with two phenylalanine residues linked by a cyclohexane dimethanol. Following up on this report, Ciardelli and colleagues recently described a similar group of polymers where increased hydrophilicity was incorporated in the soft segment by using PCL-PEG-PCL triblocks in addition to PCL alone.22 In their work, more substantial sensitivity was shown to chymotrypsin when incorporating the more hydrophilic soft segment. Our focus on elastase sensitivity was a result of an interest in enzymatic activity characteristic of the physiologic wound healing response. Macrophages at the site of material implantation would be expected to secrete this enzyme when activated. It would also be of interest in future work to examine peptide sequences sensitive for the matrix metalloproteinases that facilitate extracellular matrix remodeling in vivo.
This report also extends earlier work wherein PEG-based hydrogel systems have been designed to exhibit a variety of enzymatic sensitivities relevant for native tissue remodeling processes.27–31 The PU system provides this type of activity in a thermoplastic elastomer system that is amenable to more rigorous mechanical demands that may be required in some physiologic settings. In previous work with similar hydrolytic polyurethanes, we have been able to take advantage of processing techniques to form this type of polymer into highly porous scaffolds using thermally induced phase-separation methodology26 and into submicron-scale fibrous scaffolds in a blend with collagen.25
In summary, we have synthesized and characterized a class of elastase-sensitive PU elastomers where introduction of the AAK sequence into the polymer backbone provided enzymatic lability and where manipulating the polyester/polyether ratio in the soft segment allowed manipulation of the polymer degradation rate in buffer, with and without elastase present. The synthesized polymers were shown to have high tensile strength and distensibility and exhibited elastomeric behavior. Furthermore, we demonstrated that the degradation products from these polymers did not appear to be cytotoxic, that endothelial cell adhesion and growth were supported, and that by surface modification with the cell adhesion peptide Arg-Gly-Asp-Ser (RGDS) endothelial cell adhesion could be augmented. The synthesized PUs thus combine chemical, mechanical, and bioresponsive properties that might be employed in future soft-tissue engineering applications as well as other biomedical materials applications.
Acknowledgments
This work was supported by the National Institutes of Health (HL069368).
References and Notes
- 1.Niklason LE, Gao J, Abbott WM, Hirschi KK, Houser S, Marini R, Langer R. Science. 1999;284:489–493. doi: 10.1126/science.284.5413.489. [DOI] [PubMed] [Google Scholar]
- 2.Hoerstrup SP, Zund G, Sodian R, Schnell AM, Grunenfelder J, Turina MI. Eur J Cardiothorac Surg. 2001;20:164–169. doi: 10.1016/s1010-7940(01)00706-0. [DOI] [PubMed] [Google Scholar]
- 3.Tiwari A, Salacinski HJ, Punshon G, Hamilton G, Seifalian AM. FASEB J. 2002;16:791–796. doi: 10.1096/fj.01-0826com. [DOI] [PubMed] [Google Scholar]
- 4.Opitz F, Schenke-Layland K, Richter W, Martin DP, Degenkolbe I, Wahlers T, Stock UA. Ann Biomed Eng. 2004;32:212–222. doi: 10.1023/b:abme.0000012741.85600.f1. [DOI] [PubMed] [Google Scholar]
- 5.Albertsson AC, Varma IK. Adv Polym Sci. 2002;157:1–40. [Google Scholar]
- 6.Hiki S, Miyamoto M, Kimura Y. Polymer. 2000;41:7369–7379. [Google Scholar]
- 7.Kwon IK, Park KD, Choi SW, Lee SH, Lee EB, Na JS, Kim SH, Kim YH. J Biomater Sci Polym Ed. 2001;12:1147–1160. doi: 10.1163/15685620152691904. [DOI] [PubMed] [Google Scholar]
- 8.Wang YD, Ameer GA, Sheppard BJ, Langer R. Nat Biotechnol. 2002;20:602–606. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 9.Webb AR, Yang J, Ameer GA. Expert Opin Biol Ther. 2004;4:801–812. doi: 10.1517/14712598.4.6.801. [DOI] [PubMed] [Google Scholar]
- 10.Storey RF, Herring KR, Hoffman DC. J Polym Sci Polym Chem Ed. 1991;29:1759–1777. [Google Scholar]
- 11.Guan J, Sacks MS, Beckman EJ, Wagner WR. J Biomed Mater Res. 2002;61:493–503. doi: 10.1002/jbm.10204. [DOI] [PubMed] [Google Scholar]
- 12.Guan J, Sacks MS, Beckman EJ, Wagner WR. Biomaterials. 2004;25:85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- 13.Cohn D, Stern T, Gonzalez MF, Epstein J. J Biomed Mater Res. 2002;59:273–281. doi: 10.1002/jbm.1242. [DOI] [PubMed] [Google Scholar]
- 14.Lendlein A, Neuenschwander P, Suter UW. Macromol Chem Phys. 1998;199:2785–2796. [Google Scholar]
- 15.Lendlein A, Colussi M, Neuenschwander P, Suter UW. Macromol Chem Phys. 2001;202:2702–2711. [Google Scholar]
- 16.Saad B, Hirt TD, Welti M, Uhlschmid GK, Neuenschwander P, Suter UW. J Biomed Mater Res. 1997;36:65–74. doi: 10.1002/(sici)1097-4636(199707)36:1<65::aid-jbm8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 17.Spaans CJ, de Groot JH, Dekens FG, Pennings AJ. Polym Bull. 1998;41:131–138. [Google Scholar]
- 18.Kylma J, Seppala JV. Macromolecules. 1997;30:2876–2882. [Google Scholar]
- 19.Gorna K, Polowinski S, Gogolewski S. J Polym Sci Part A: Polym Chem. 2002;40:156–170. [Google Scholar]
- 20.Skarja GA, Woodhouse KA. J Biomater Sci Polym Ed. 1998;9:271–295. doi: 10.1163/156856298x00659. [DOI] [PubMed] [Google Scholar]
- 21.Skarja GA, Woodhouse KA. J Biomater Sci Polym Ed. 2001;12:851–873. doi: 10.1163/156856201753113060. [DOI] [PubMed] [Google Scholar]
- 22.Ciardelli G, Rechichi A, Cerrai P, Tricoli M, Barbani N, Giusti P. Macromol Symp. 2004;218:261–271. [Google Scholar]
- 23.Storey RF, Hickey TP. Polymer. 1994;35:830–838. [Google Scholar]
- 24.Storey RF, Wiggins JS, Puckett AD. J Polym Sci Polym Chem. 1994;32:2345–2363. [Google Scholar]
- 25.Stankus JJ, Guan J, Wagner WR. J Biomed Mater Res. 2004;70A:603–614. doi: 10.1002/jbm.a.30122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan J, Fujimoto KL, Sacks MS, Wagner WR. Biomaterials. 2005;26:3961–3971. doi: 10.1016/j.biomaterials.2004.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci USA. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. J Biomed Mater Res. 2004;25:85–96. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 29.West JL, Hubbell JA. Macromolecules. 1999;32:241–244. [Google Scholar]
- 30.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Biomaterials. 2001;22:3045–3051. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 31.Kim S, Healy KE. Biomacromolecules. 2003;4:1214–1223. doi: 10.1021/bm0340467. [DOI] [PubMed] [Google Scholar]
- 32.Gertler A, Hofmann T. Can J Biochem. 1970;48:384–386. doi: 10.1139/o70-061. [DOI] [PubMed] [Google Scholar]
- 33.Lelah MD, Cooper SL. Polyurethanes in medicine. CRC Press; Boca Raton, FL: 1986. pp. 5–20. [Google Scholar]
- 34.An JH, Kim HS, Chung DJ, Lee DS. J Mater Sci. 1999;32:726–731. [Google Scholar]
- 35.D'Angelo S, Galletti P, Maglio G, Malinconico M, Morelli P, Palumbo R, Vignola MC. Polymer. 2001;42:3383–3392. [Google Scholar]
- 36.Bezemer JM, Oude Weme P, Grijpma DW, Dijkstra PJ, van Blitterswijk CA, Feijen J. J Biomed Mater Res. 2000;52:8–17. doi: 10.1002/1097-4636(200010)52:1<8::aid-jbm2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 37.Park JB, Lakes RS. Biomaterials: an introduction. 2nd. Plenum; New York: 1992. [Google Scholar]
- 38.Borkenhagen M, Stoll RC, Neuenschwander P, Suter UW, Aebischer P. Biomaterials. 1998;19:2155–2165. doi: 10.1016/s0142-9612(98)00122-7. [DOI] [PubMed] [Google Scholar]
- 39.Endean E, Toursarkissian B, Buckmaster M, Aziz S, Gellin G, Hill B. Growth Factors. 1996;13:229–242. doi: 10.3109/08977199609003224. [DOI] [PubMed] [Google Scholar]
- 40.Wang JY, Viar MJ, Li J, Shi HJ, McCormack SA, Johnson LR. Am J Physiol. 1997;272:G713–G720. doi: 10.1152/ajpgi.1997.272.4.G713. [DOI] [PubMed] [Google Scholar]
- 41.Milovic V, Turhanowa L, Fares FA, Lerner A, Caspary WF, Stein J. Z Gastroenterol. 1998;36:947–954. [PubMed] [Google Scholar]