

Abstract
Human tear lipocalin (TL) exhibits diverse functions, most of which are linked to ligand binding. To map the binding site of TL for some amphiphilic ligands, we capitalized on the hydrophobic and hydrophilic properties of 8-anilino-1-naphthalenesulfonic acid (ANS). In single Trp mutants, resonance energy transfer from Trp to ANS indicates that the naphthalene group of ANS is proximate to Leu105 in the cavity. Binding energies of TL to ANS and its analogues reveal contributions from electrostatic interactions. The sulfonate group of ANS interacts strongly with the nonconserved intracavitary residue Lys114 and less with neighboring residues His84 and Glu34. This trigonal cluster of residues may play a role in the ligand recognition site for some negatively charged ligands. Because many drugs possess sulfonate groups, the trigonal cluster–sulfonate interaction can also be exploited as a lipocalin-based drug delivery mechanism. The binding of lauric acid and its analogues shows that fatty acids assume heterogeneous orientations in the cavity of TL. Predominantly, the hydrocarbon tail is buried in the cavity of TL and the carboxyl group is oriented toward the mouth. However, TL can also interact, albeit relatively weakly, with fatty acids oriented in the opposite direction. As the major lipid binding protein of tears, the ability to accommodate fatty acids in two opposing orientations may have functional implications for TL. At the aqueous–lipid interface, fatty acids whose carboxyl groups are positioned toward the aqueous phase are available for interaction with TL that could augment stability of the tear film.
TL,1 also known as lipocalin-1, is secreted by the lacrimal glands and is the principal lipid binding protein in tears (1). However, TL is also found in a number of other secretory glands including lingual glands, nasal mucosal glands, secretory glands of the tracheobronchial tract, sweat glands, mammary glands, adrenal glands, the prostate, the thymus, the testis, and the pituitary gland (2-5). Putative functions for TL include scavenging lipid from the corneal surface to prevent the formation of lipid-induced dry spots (6, 7), solubilization of lipid in tears (6), antimicrobial activity (8), cysteine proteinase inhibition (9), transport of sapid molecules in saliva (10), transport of retinol in tears (11), scavenging potentially harmful lipid oxidation products (12), transport of antioxidants in tears (13), endonuclease activity (14), and stabilization of the lipid layer of the tear film (15). Most of these diverse functions are linked to binding various classes of ligands, but very little is known about the binding sites and specificity for individual classes of lipids.
The binding cavity of TL as with other members of the lipocalin family of proteins is formed by a continuously hydrogen-bonded β-barrel comprised of eight antiparallel β-strands (16). TL contains a single Trp17 and a single free Cys101, creating an ideal situation for conducting both SDTF and SDSL studies (17-19). The solution structure of TL was resolved by SDTF and revealed a capacious cavity that confers promiscuity in ligand binding (18). These findings were verified by crystallography of TL (20).
The lipocalin scaffold has been used to engineer anticalins and duocalins for novel drug delivery platforms (21-23). Targeted random mutagenesis of the lipocalin loops results in ligand binding proteins with high affinity for drugs. More detailed knowledge of the intracavitary binding sites of lipocalins could enhance the potential for this type of therapeutic intervention.
In tears, TL is found complexed with an assortment of endogenous ligands including fatty acids, alkyl alcohols, glycolipids, phospholipids, and cholesterol (1). The most studied ligands of TL are fatty acids. Fatty acids bind inside the cavity of TL with the carboxyl group oriented toward the mouth of the cavity and the alkyl chain extended internally (19, 24).
TL is believed to stabilize the lipid layer of the tear film (25, 26). Although the structure of the tear film is not certain, one proposal is that a complex lipid layer exists on the surface of the aqueous layer of tears that is composed of wax esters, triglycerides, and hydrocarbons, at the air–lipid interface, and polar lipids such as fatty acids reside at the aqueous–lipid interface. Some suggest that the polar lipid phase provides stability to the overall lipid layer via a monolayer with charged groups oriented toward the aqueous layer (27, 28). The rationale for interaction between tear lipocalin and the polar lipids at the tear film interface has not been explained in this model.
In the present study, ANS and its analogues serve as “reporter ligands” to map the internal binding site of TL and determine the binding specificity for a particular group of ligands. ANS, a fluorescent molecule that has both hydrophobic and hydrophilic properties, has been extensively utilized to probe protein binding sites. Previously, two ANS binding sites in TL (internal and external) have been identified by their distinct fluorescence lifetimes and dissociation constants (29, 30). The selective displacement of ANS from the ANS–apoTL complex by stearic acid has revealed the internal binding site, which is associated with a long fluorescence lifetime (30).
Here, steady-state and time-resolved RET and site-directed mutagenesis were employed to locate the intracavitary side chains in close proximity to bound ANS. The intracavitary sites for the experiments were chosen to encompass B, D, F, G, and H strands and the AB loop of TL (Figure 1). RET from Trp to ANS reveals that the naphthalene group of ANS is proximal to Leu105 in the cavity of TL. The sulfonate group of ANS interacts with residue Lys114 and neighboring residues His84 and Glu34 to confer unique functional features to TL. The trigonal cluster of charged residues permits TL to accommodate two orientations of fatty acids in the cavity. A position with the hydrocarbon tail buried in the cavity and the carboxyl group oriented toward the mouth (24) is energetically most favorable in an aqueous environment. However, a relatively weak interaction in the opposite direction may stabilize the lipid layer of the tear film.
Figure 1.

Positions of the residues that are considered for mapping the binding site of TL. Gray circles show locations of the Cα atoms of the residues. The side chains of all residues, but 35 and 115, are oriented inside the cavity. Single and double letters denote the identities of the β-strands and loop, respectively. The arrow shows the ligand entrance site of the cavity. The ribbon diagram of TL was generated from PDB 1XKI (20). The missing loop parts were built by DeepView/Swiss-PdbViewer v.3.7 (GlaxoSmithKline R&D) in accord with the structural data of tear lipocalin (18, 20).
MATERIALS AND METHODS
Materials
ANS, TNS, tetradecanedioic acid, 1,12-diaminododecane, dodecylamine, and lauric acid were purchased from Sigma (St. Louis, MO). 1NPN and DAUDA were purchased from Invitrogen (San Diego, CA).
Site-Directed Mutagenesis and Plasmid Construction
The TL cDNA in PCR II (Invitrogen), previously synthesized (31), was used as a template to clone the TL gene spanning bases 115–592 of the previously published sequence (11) into pET 20b (Novagen, Madison, WI). Flanking restriction sites for NdeI and BamHI were added to produce the native protein sequence as found in tears but with an initiating methionine (32). To construct mutant proteins with a single tryptophan, the previously well characterized TL mutant W17Y was prepared with oligonucleotides (Universal DNA Inc., Tigard, OR) by sequential PCR steps (33, 34). Using this mutant as a template, mutant cDNAs were constructed in which selected amino acids were additionally substituted with tryptophan or cysteine. Amino acid 1 corresponds to His, bases 115–118, according to Redl (11).
To map the binding site of tear lipocalin, single Cys and single Trp mutants that reside on both sheets of the barrel were considered (Figure 1). Because previous findings showed that the β-strand G was involved in the fatty acid binding site of TL (24), the current study was mainly focused on the central β-sheet (β-strands F, G, and H). However, to encompass the entire barrel of TL, a few sites from the peripheral β-sheet (β-strands B and D) and the loop AB were also included. Mutants with side chains oriented outside the cavity, sites 115 and 35, served as additional controls for the binding experiments with TL.
Single Trp mutants include W17Y/E34W (for simplicity denoted as E34W), W17Y/S35W (S35W), W17Y/M39W (M39W), W17Y/A66W (A66W), W17Y/H84W (H84W), W17Y/I88W (I88W), W17Y/F99W (F99W), W17Y/C101W (C101W), W17Y/L105W (L105W), W17Y/K114W (K114W), and W17Y/L115W (L115W).
Single Cys mutants include C101L/H84C (H84C) and C101L/K114C (K114C). The recombinant wild type of tear lipocalin is also denoted as TL.
The Trp-less mutant W17Y was used as a control in the RET experiments.
Expression and Purification of Mutant Proteins
The mutant plasmids were transformed in Escherichia coli, BL 21 (DE3) cells were cultured, and proteins were expressed according to the manufacturer’s protocol (Novagen). Following cell lysis (35), the supernatant was treated with methanol (40% final concentration) at 4 °C for 2½ h. Alternatively, mutant proteins expressed in inclusion bodies were dissolved in 8 M urea at room temperature for 2 h. In either case, the resulting suspension was centrifuged at 3000g for 30 min. The supernatant was dialyzed against 50 mM Tris–HCl, pH 8.4. The dialysate was treated with ammonium sulfate, 45–75% saturation. The resulting precipitate was dissolved in 50 mM Tris–HCl, pH 8.4, and applied to a Sephadex G-100 column (2.5 × 100 cm) equilibrated with 50 mM Tris–HCl, 100 mM NaCl, pH 8.4. The fraction containing the mutant protein was dialyzed against 50 mM Tris–HCl, pH 8.4, and applied to a DEAE Sephadex A-25 column. Bound protein was eluted with a 0–0.8 M NaCl gradient. Eluted fractions containing mutant proteins were centrifugally concentrated (Amicon, Centricon-10). The purity of mutant proteins was verified by SDS–tricine gel electrophoresis (1). To obtain lipid-depleted TL (denoted as apoTL), the delipidation procedure was carried out by chloroform/methanol extraction as described previously (1). The protein concentrations were determined by the biuret method (36).
Circular Dichroism
CD spectra were recorded (Jasco 810 spectropolarimeter, 0.2 and 10 mm path lengths for far- and near-UV spectra, respectively) using protein concentrations of 1.2 mg/mL in 10 mM sodium phosphate at pH 7.3. Eight and sixteen scans from 190 to 260 nm and from 250 to 320 nm (scan speed 50 nm/min, bandwidth 1.0 nm, time constant 0.5 s) were averaged, respectively. The results were recorded in mdeg and converted to mean residue ellipticity in deg•cm2•dmol−1.
Steady-State Fluorescence
Steady-state fluorescence measurements were made on a Jobin Yvon-SPEX (Edison, NJ) Fluorolog Tau-3 spectrofluorometer; bandwidths for the excitation and emission monochromators were 2 nm. The excitation λ of 360 nm was used for ANS, TNS, and 1NPN fluorescence. All measurements were conducted in 10 mM sodium phosphate buffer (pH 7.3) at room temperature. The fluorescence spectra were corrected for light scattering from buffer. For quantum yield measurements, Trp spectra were additionally corrected for the instrument response using the appropriate correction curve. NATA in water, for which the quantum yield is 0.13 (37), was used as a fluorescence standard. The details of quantum yield calculation have been described elsewhere (38).
Excitation spectra of the mutant proteins with and without ANS were measured at an emission λ of the Trp emission maximum and 500 nm (for ANS), respectively. Bandwidths for the excitation and emission monochromators were 2 and 5 nm, respectively. Sixteen scans were averaged to increase the signal/noise ratio. The spectra were corrected for light scattering from solution and inner filter effects; the formula is shown below. The excitation spectra for the protein–ANS complexes at 500 nm were additionally corrected for direct Trp excitation using the protein without ANS.
To account for the wavelength dependency of excitation light and the excitation monochromator, the excitation spectra were corrected by means of the excitation correction curve supplied with the instrument. To resolve the tryptophan excitation spectra in the ANS–single tryptophan mutant complexes at the emission of ANS (500 nm), the excitation spectrum of tryptophan-less mutant complex W17Y–ANS was subtracted from the spectra of the tryptophan-containing mutant–ANS complexes. In the subtraction procedure the intensity of the excitation spectrum of W17Y–ANS was varied to match the excitation spectrum of the corresponding tryptophan residue in the mutant protein that was excited directly (L105W and C101W without ANS).
Fluorescence Binding Assays
The samples of apoTL (5 μM) in 10 mM sodium phosphate at pH 7.3 were titrated by addition of ANS, and the fluorescence was measured at emission λmax. The concentration of an ethanol stock solution of ANS was confirmed by absorbance at 350 nm, using an extinction coefficient ε350 = 6300 M−1 cm−1. Following each addition of ligand, the solution was mixed and allowed to equilibrate for 3 min. At the end of each titration experiment, the ethanol concentration did not exceed 2%, which does not affect the structure of TL. The ANS binding data were analyzed with the following formula for one binding site, which can be derived from the law of mass action (39):
where F is a fluorescence scaling factor, Kd is the apparent dissociation constant, P is the total protein concentration, Lt is the total ligand concentration, and n is the stoichiometry. The 1NPN and TNS binding experiments were performed similarly to that of ANS. The fluorescence (ANS, 1NPN, TNS) intensities were corrected for the inner filter effect, if necessary, by the formula
where Fcorr and Fobs are the corrected and observed fluorescence intensities, respectively, and Aex and Aem are the absorbance values at the excitation and emission wavelengths, respectively. For single Trp mutants, in addition to ANS fluorescence experiments, the dissociation constants were also obtained by quenching Trp fluorescence at various ANS concentrations. The dissociation constants were calculated by a formula that is essentially the same as the above shown formula for one binding site, except “y” was subtracted from Fmax (fluorescence intensity of Trp fluorescence without ANS). The fluorescence of NATA titrated with ANS was used to estimate the correction factors for each wavelength.
Competitive Displacement Assays To Assess the Ligand Charge Preference of the TL Cavity
To determine the ligand charge preference of the TL cavity, competitive displacement assays were performed with lauric acid and its analogues that carry double negative, double positive (one at each end of the hydrocarbon chain), and single positive charges. DAUDA exhibits little fluorescence in buffer but shows enhancement of fluorescence and a blue shift when bound to tear lipocalin (34). The displacement of DAUDA by ligands is accompanied by a decrease in fluorescence intensity. Samples of TL (4 μM) were incubated with DAUDA (2 μM) in 10 mM sodium phosphate, pH 7.3, and competitors were added in increasing concentration. The IC50 parameter (the concentration of a ligand required to achieve a 50% decrease in fluorescence intensity) for each ligand was extrapolated from the graph of the fluorescence intensity of bound DAUDA versus the competitor concentration using a model for one binding site with Microcal Origin software (Microcal Software, Inc.).
Fluorescence Lifetime Measurements
The fluorescence intensity decays were measured using a PTI TimeMaster fluorescence lifetime instrument, which consists of a nitrogen laser (GL-3300) linked to a dye laser (GL 302), a frequency doubler (GL 303), and a stroboscopic detector. Rhodamine 6G (Exciton, Inc., Dayton, OH) dye solution was used to obtain a wavelength of 295 nm. The 295 nm pulses (fwhm ≈ 1.5 ns) were used for the excitation of the single Trp mutants. The decay curves were analyzed at the wavelengths of the respective emission maxima. The emission monochromator slit was 3 nm. All measurements were conducted at room temperature. The IRF was determined by measuring scattered light from a solution of glycogen. A DPU-15 optical depolarizer (Optics for Research, Caldwell, NJ) was placed before the emission monochromator to eliminate the polarization dependence of the detection train. Each data point on a lifetime decay curve represents the average of nine laser flashes, and each decay represents 300 of these data points evenly spaced over the collection time interval.
The fluorescence intensity decay data were analyzed by the multiexponential decay law, using the software supplied with the PTI instrument:
where I is fluorescence intensity and αi and τi are the normalized preexponential factors and decay times, respectively. For the calculation of the efficiency of RET, the amplitude-averaged lifetime 〈τ〉 = Σαjτj was used.
RET by Steady-State and Time-Resolved Fluorescence
RET between the Trp residue (donor) and ANS (acceptor) was used to determine the efficiency of energy transfer, ET, which can be calculated from
where 〈τDA〉 and 〈τD〉 are the fluorescence lifetimes of the donor in the presence and absence of the acceptor, respectively, and IDA and ID are the fluorescence intensities of the donor in the presence and absence of the acceptor, respectively. The single Trp mutant proteins (5–6 μM) were used to calculate the donor lifetimes in the absence and presence of the acceptor (16.4 μM ANS). In these conditions, donor/acceptor ratios will vary because the dissociation constants for ANS binding differ among the tested mutants. Therefore, the efficiency of RET was extrapolated to the ratio donor/acceptor = 1 by using the corresponding dissociation constant of ANS binding for each mutant protein.
Modeling of the ANS–TL Complex
Docking ANS into the TL cavity was simulated by ArgusLab 4.0.1 software (Planaria Software LLC, Seattle, WA), which is freely available at http://www.arguslab.com.
X-ray coordinates of the crystal structure of TL (PDB 1XKI) were used for the docking program.
The ANS molecule was borrowed from the ANS–MurA (UDPGlcNAc enolpyruvyl transferase (EC)) complex (PDB 1EVN). The “high-precision” mode was employed for the docking run. The docking results were saved as a PDB file and visualized with DeepView/Swiss-PdbViewer v.3.7 (GlaxoSmithKline R&D).
RESULTS AND DISCUSSION
Characterization of Mutant Proteins
To reveal any possible structural perturbations produced from point mutations, far-UV CD was performed on mutant proteins. The far-UV circular dichroic spectra of mutant proteins used in this study are shown in Figure 2. The CD spectra of S35W and A66W have been described previously (18). In some cases, the variation of peak-to-peak intensities can be attributed to the errors in estimation of the protein concentration, which do not exceed 5%. It has been shown that CD intensities can vary as much as 10% for two different preparations of the same mutant (40). Therefore, retention of the overall shape of the CD spectrum, particularly the negative lobe of the spectrum, is more indicative of unperturbed secondary structure than the signal intensity. Among mutant proteins, S35W shows the most noticeable perturbation, a shifted minimum at about 207 nm. This modification in the CD spectrum may be attributed to changes in the packing of secondary structural elements (40). However, the result is surprising because the side chain of Ser35 is located outside the cavity of TL (Figure 1) and does not interact with other side chains that may have a role in stabilization of the protein (20). It should be emphasized that the fluorescence λmax value of every single Trp mutant of TL was consistent with its site-specific location in the crystal structure (18, 20). Therefore, the mutant proteins have a native fold, and each site-directed mutant reports the features of the native structure. Compared with the wild type of TL or single Cys mutants (K114C and H84C), some single Trp mutants show an expected loss of the positive contribution at 230 nm (Figure 2) that is attributable to the absence of Trp17 rather than the structural changes (34). The negative deviations of the CD spectra around 230 nm vary among the mutant proteins (e.g., A66W and H84W) relative to that of wild-type TL. The mutants K114W and S35W can be considered to have positive contributions to the far-UV CD that are similar to that of native Trp17. It has been shown that contributions of Trp side chains to the far-UV CD of proteins result from interactions between Trp side chains and the nearest neighbor peptide group. The net sign (positive or negative) and intensity of the contribution depend on the backbone conformation as well as the distribution of the side chain rotamers (41). There are no other significant differences in the far-UV CD spectra of these mutants compared with the spectrum of the W17Y mutant or that of wild-type TL.
Figure 2.
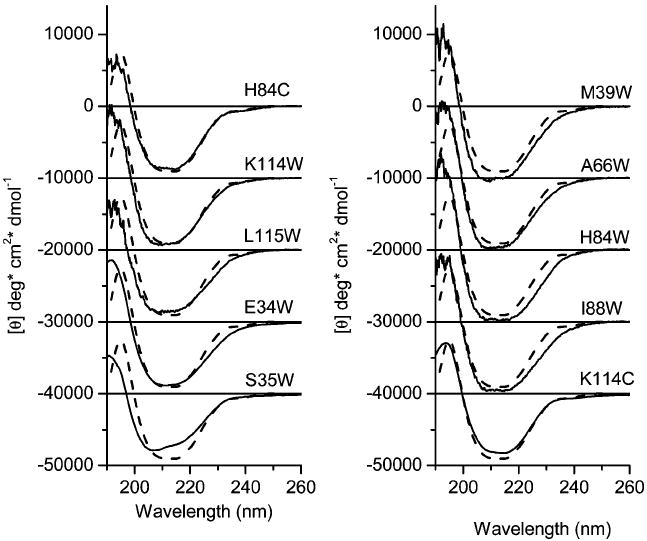
Far-UV CD spectra of TL mutants. CD spectra were recorded using protein concentrations of 1.2 mg/mL in 10 mM sodium phosphate at pH 7.3. To reduce overlap, the spectra were shifted vertically. For comparison, the spectrum of wild-type TL (dashed line) was added to each spectrum of the mutant protein.
The contributions of aromatic side chains to the near-UV CD spectra of proteins depend on their conformations, environments, and nearest (<10 Å) spatial arrangement of neighboring residues (42). Therefore, the near-UV CD spectra of proteins are unique for the individual proteins and reflect the tertiary structure. For TL, the near-UV CD spectrum is mainly influenced by the contribution of the sole Trp17 and five Tyr residues (34). Two single Cys mutant proteins, H84C and K114C, contain aromatic amino acids identical to those of wild-type TL. The near-UV CD spectra of H84C and K114C are very similar to that of wild-type TL (Figure 3); the conformation of TL appears unaffected by these point mutations. In protein, to recover the desolvation penalties, charged side chains in a hydrophobic cavity are expected to form a salt bridge with neighboring oppositely charged side chains. The stabilization effect from the salt bridge formations vary greatly in proteins. In TL, the side chain of Lys114 is buried and has close proximity to that of Glu34; the distance between NZ (Lys114) and OE1 (Glu34) is 4.38 Å (20). However, the distance and geometry of these amino acid residues, Lys114 and Glu34, do not fulfill the criteria for salt bridge formation (43). Therefore, the relation between Lys114 and Glu34 can be considered a weak electrostatic interaction. Indeed, the point mutations (E34W, K114W, and K114C) at position 34 or 114 do not significantly change the CD spectra (Figures 2 and 3).
Figure 3.

Near-UV CD spectra of TL mutants. The experimental conditions were the same as in Figure 2.
Binding Experiments with ANS, 1NPN, and TNS: Electrostatic Contribution to Binding Energy
It has been shown that TL has two binding sites for ANS (29, 30). At pH 7.3, the dissociation constants for the internal (intracavitary) and external binding sites are 0.58 and 5.7 μM, respectively. The 16.99 and 2.76 ns lifetime species were attributed to the internal and external binding sites, respectively. The fractional contribution of intracavitary ANS to the overall steady-state fluorescence intensity of the ANS–apoTL complex is over 90%. Therefore, the steady-state fluorescence was employed to study the intracavitary ANS–apoTL binding. To elucidate the electrostatic contributions to the free energy of binding for ANS–apoTL, titration experiments were performed with ANS and 1NPN (Figure 4). 1NPN, the analogue of ANS that lacks the sulfonate group, binds to apoTL with significantly less affinity than ANS. The dissociation constants for ANS– and 1NPN–apoTL complexes are 0.48 and 9.1 μM, respectively. Consequently, the contribution of the sulfonate group to the free energy of ANS–apoTL binding is −1.74 kcal/mol (ΔΔG = ΔGANS − ΔG1PN = −8.61 kcal/mol + 6.87 kcal/mol). This energy can be attributed to the effect of electrostatic attraction between the sulfonate group of ANS and an intracavitary positively charged side chain, for which Lys114 is the best contender.
Figure 4.
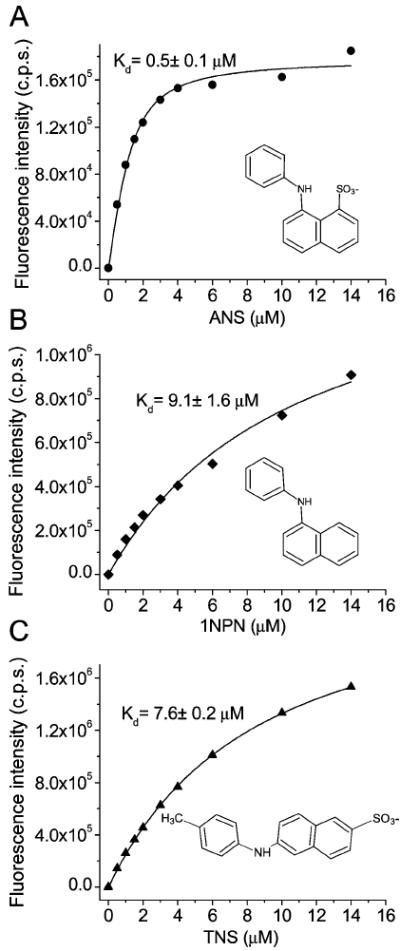
Binding curves of the fluorescence probes ANS (A), 1NPN (B), and TNS (C) to apoTL (5 μM). Emission λ values for ANS, 1NPN, and TNS binding were 465, 416, and 432 nm, respectively.
Previously, it has been shown that the electrostatic interaction alone is not sufficient to provide high binding affinity for ANS (Kd < 1 mM) (44). For internal ANS binding, in addition to the electrostatic interaction, hydrophobic and van der Waals interaction should be considered. The fluorescence probe TNS is similar to ANS, but has a different configuration. If the binding affinity of ANS to TL were primarily determined by the electrostatic interaction and/or hydrophobic effect, one would expect similar dissociation constants for ANS and TNS. It is evident that TNS has significantly decreased affinity for TL (Figure 4C). Therefore, van der Waals interactions between ANS and intracavitary side chains are greater for ANS than for TNS. Obviously, van der Waals interactions depend on the cavity shape, the arrangement of the side chains inside the cavity, and the relative position of ANS, all of which influence the maximum number of contacts between ANS and the side chain atoms.
Evidence for Ion Pair Formation with Lys114
Previously, it has been shown that the side chain of the residue Lys114 is oriented inside the cavity (18, 20). To uncover a possible electrostatic interaction between Lys114 and ANS, binding studies were performed on mutants K114C and K114W (Figure 5). The side chain of Leu115 is oriented outside the cavity of TL (18, 20). Accordingly, the mutant L115W is included in the ANS binding study as a control (Figure 5). Indeed, compared to the wild-type TL, this mutation does not significantly alter ANS binding affinity. However, it is evident that the substitution of Lys with Cys at position 114 results in a decrease of ANS binding affinity by about 1 order of magnitude. The associated change in free energy of ANS binding is 1.34 kcal/mol (ΔΔG = ΔGK114C − ΔGL115W = −6.90 kcal/mol + 8.24 kcal/mol), comparable with the difference in free energies of ANS and 1NPN binding. The Cys mutation at position 114, K114C, of TL does not induce any changes in secondary or tertiary structure (Figure 2 and 3). Therefore, the decreased affinity observed for the K114C mutant can be attributed to the lack of interaction between the side chain of Lys114 and ANS, i.e., electrostatic interaction.
Figure 5.
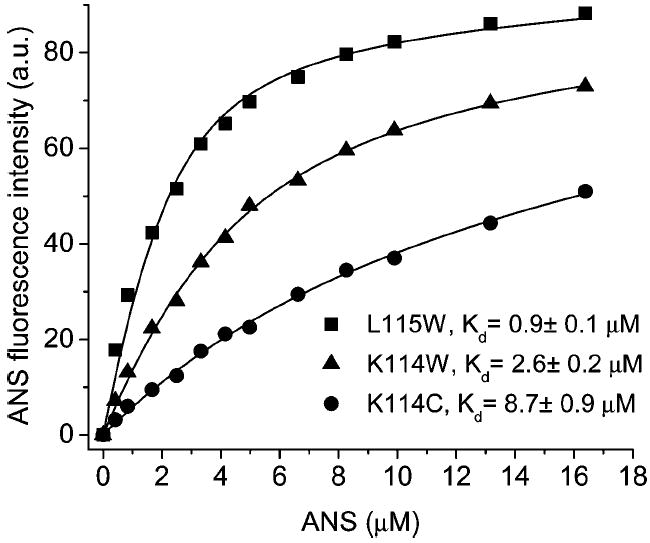
ANS binding to the mutants L115W, K114W, and K114C. Emission λ values in ANS binding experiments with L115W, K114W, and K114C were 462, 475, and 475 nm, respectively. Concentrations of the proteins were in the range of 5–6 μM.
The decreased ANS binding that results from the substitution of the Lys residue with Trp at position 114 is not as dramatic as that of the Cys substitution. Presumably, the interaction between the aromatic ring of Trp and ANS partially compensates for eradicated electrostatic influences. In summary, the data shown in Figures 4 and 5 corroborate the electrostatic basis for intracavitary binding between Lys114 of TL and ANS.
RET from Trp to ANS in Single Trp Mutants by Steady-State and Time-Resolved Fluorescence
To assess the physical proximity of bound ANS to the intracavitary amino side chains, steady-state and time-resolved fluorescence RET experiments were performed with selected single Trp mutant proteins. For RET measurements Trp and ANS are a good donor–acceptor pair for which the Förster distance is about 23 Å (45). The mutation sites, which encompass strands B, D, F, and G as well as loop AB of TL, are shown in Figure 1.
ANS quenches the fluorescence of Trp34 in a dose-dependent manner (Figure 6). A decrease in the Trp fluorescence intensity accompanies an increase of ANS fluorescence. The fluorescence quenching data for some single Trp mutants titrated with ANS are shown in Figure 7. The fraction of fluorescence quenched by ANS, and therefore the efficiency of RET, varies with the position of the Trp residue in TL. Obviously, the distance between bound ANS and Trp residues is the major factor for efficiency of RET. RET between Trp residues (in single Trp mutants) and ANS can be culled from the means of time-resolved fluorescence. In accord with steady-state fluorescence, time-resolved fluorescence shows that the efficiency of RET is much greater for the L105W–ANS complex than for E34W–ANS (Figures 7 and 8).
Figure 6.
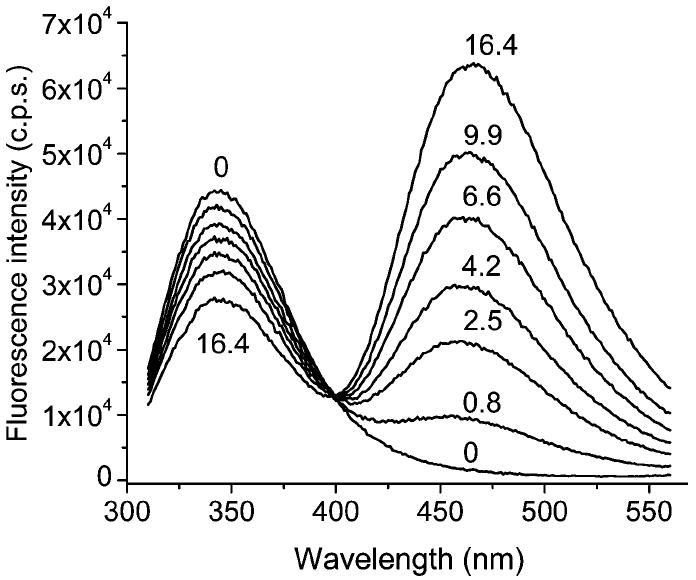
Fluorescence spectra of E34W at various ANS concentrations. The numbers in the figure represent the ANS concentration (μM). The fluorescence spectra have been corrected for the inner filter effect.
Figure 7.
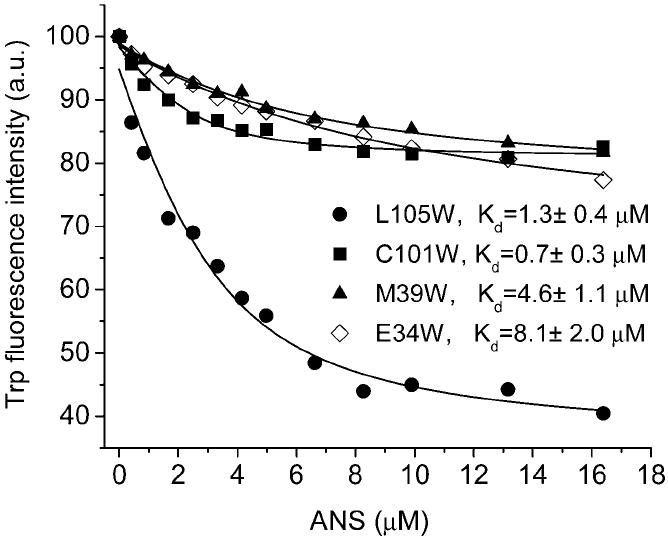
Quenching the steady-state fluorescence of single Trp mutants by ANS. The fluorescence intensities were measured at the wavelengths of the respective emission maxima. Emission λ values in the experiments with L105W, C101W, M39W, and E34W were 348, 337, 332, and 344 nm, respectively. The excitation λ was 295 nm. The solid curves are the best fit for one binding site model.
Figure 8.
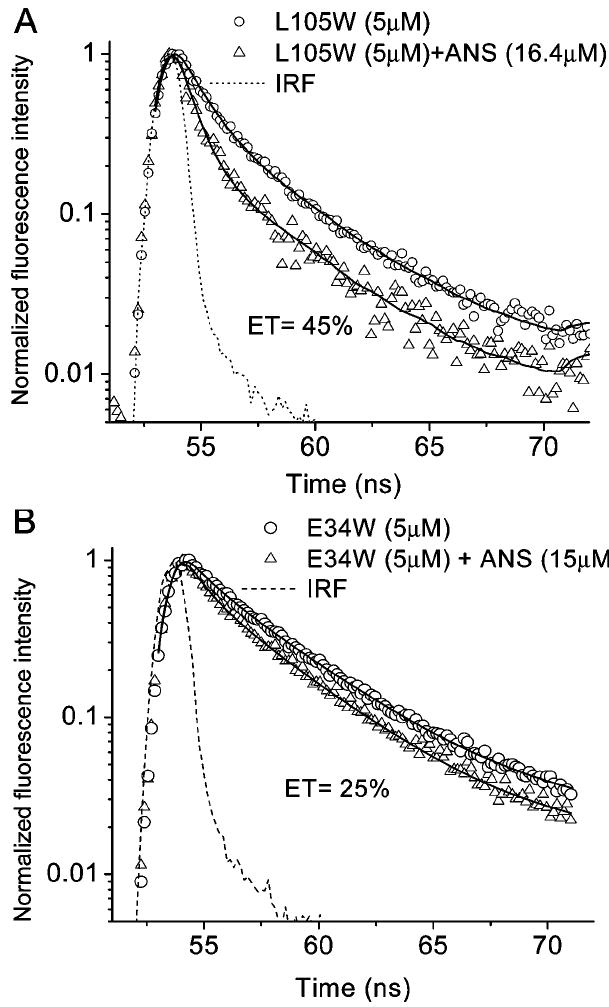
Time-resolved fluorescence decay of single Trp mutants (A) L105W and (B) E34W with and without ANS. Solid curves are the best fit for a double exponential decay. Emission λ values in the experiments with L105W and E34W were 348 and 344 nm, respectively. The excitation λ was 295 nm.
Although RET is the most likely mechanism for the decreased fluorescence intensities and lifetimes in the single Trp mutants complexed with ANS, additional data substantiate this statement. Observation of the excitation spectrum of the Trp residue at the emission λ of ANS in single Trp mutant–ANS complexes could be considered direct evidence for RET. However, ANS has a dominant absorption band below 320 nm that completely envelops the relatively minor absorption band of Trp. The excitation spectrum of ANS in ethanol is shown in Figure 9A. The W17Y–ANS complex, which does not contain a Trp residue, was used to obtain the excitation spectra of bound ANS without the absorption band of Trp. Such an excitation spectrum may also contain any contribution of Tyr residues if there is RET between them and ANS (Figures 9A and 10A). In single Trp mutants complexed with ANS, relative contributions of Trp residues in the excitation spectra (at an emission λ of 500 nm) will depend on several factors, such as quantum yields and absorption of Trp and ANS as well as ET efficiency from Trp to ANS. Therefore, two mutants, L105W and C101W, which have the highest ET (~58%) and quantum yield (0.135), respectively, were chosen to confirm the presence of RET. The quantum yield of Trp105 is 0.059. Normalized excitation spectra (at an emission λ of 500 nm) of these mutants complexed with ANS show small but distinct differences compared to that of W17Y (Figures 9A and 10A). Difference excitation spectra of L105W and C101W complexed with ANS are very similar to the excitation spectra (at emission λ values of 348 and 337 nm, respectively) of directly excited Trp residues in these mutants (Figures 9B and 10B). Vibronic bands 0–0 (289 nm) and 0+850 (283 nm) of 1Lb electronic absorption transition are evident for both Trp105 and Trp101. Broad and featureless deviation of difference excitation spectra from that of corresponding Trp residues can be attributed to small differences between excitation spectra of ANS bound to W17Y and L105W (or C101W). Thus, it is evident that there is RET from both Trp105 and Trp101 to bound ANS in related single Trp mutant–ANS complexes.
Figure 9.

Evidence for RET between Trp105 and bound ANS in the L105W–ANS complex. (A) Key: dotted line, corrected excitation spectrum (emission at 500 nm) of ANS (1.5 μM) in ethanol; dashed line, corrected excitation spectrum (emission at 500 nm) of the W17Y (5.0 μM)–ANS (2.5μM) complex; solid line, corrected excitation spectrum (emission at 500 nm) of the L105W (4.4 μM)–ANS (2.5 μM) complex. (B) Key: dashed line, excitation spectrum (emission at 348 nm) of L105W (4.4 μM) without ANS; solid line, difference excitation spectrum at an emission of 500 nm [spectrum of L105W–ANS – 0.67(spectrum of the W17Y–ANS complex shown in (A))]. The arrow indicates the position of the 0–0 band of the 1Lb electronic absorption transition of Trp.
Figure 10.

Evidence for RET between Trp101 and bound ANS in the C101W–ANS complex. (A) Key: dashed line, corrected excitation spectrum (emission at 500 nm) of the W17Y (5.0 μM)–ANS (2.5μM) complex; solid line, corrected excitation spectrum (emission at 500 nm) of the C101W (5.0 μM)–ANS (2.5 μM) complex. (B) Key: dashed line, excitation spectrum (emission at 337 nm) of C101W (5.0 μM) without ANS; solid line, difference excitation spectrum at an emission of 500 nm [spectrum of C101W–ANS – 0.63(spectrum of the W17Y–ANS complex shown in (A))].
The fluorescence parameters for single Trp mutants, both alone and complexed with ANS, are shown in Table 1. Position 105 shows the most efficient RET and implies that Trp105 is closest to bound ANS. However, the ratio [bound ANS]/[TL mutant] may vary among the tested mutant proteins. To account for these possible differences, ANS binding experiments were performed for all TL mutants for which dissociation constants are shown in Table 2. The dissociation constants obtained by Trp fluorescence quenching experiments match those of ANS fluorescence, but have much higher errors. In some cases, the higher error values can be attributed to both the low quenching efficiency by ANS and higher inner filter correction factors. The dissociation constants for ANS binding (Table 2) have been used to extrapolate ET values to the ratio [bound ANS]/[TL mutant] = 1. This correction does not change the relative order of the RET efficiencies among the tested mutants (Table 1).
Table 1.
Fluorescence Decay Parameters for the Single Trp TL Mutants with and without ANSa
| TL mutant | location | α1 | α2 | τ1 (ns) | τ2 (ns) | 〈τ〉 (ns) | ETτ (%) | ETτ* (%) | ETss* (%) | χ2 |
|---|---|---|---|---|---|---|---|---|---|---|
| E34W | loop AB | 0.34 | 0.66 | 0.96 | 3.85 | 2.86 | 1.0 | |||
| E34WANS | 0.51 | 0.49 | 0.77 | 3.61 | 2.15 | 25 | 35 | 29 | 0.8 | |
| M39W | strand B | 0.52 | 045 | 2.94 | 5.24 | 4.05 | 0.9 | |||
| M39WANS | 0.52 | 045 | 2.94 | 5.24 | 4.05 | 0 | 0 | 21 | 0.9 | |
| A66W | strand D | 0.67 | 0.33 | 2.19 | 5.35 | 3.23 | 0.8 | |||
| A66WANS | 0.60 | 0.40 | 1.88 | 5.01 | 3.13 | 3 | 4 | 6 | 0.9 | |
| H84W | strand F | 0.78 | 0.22 | 1.37 | 4.11 | 1.97 | 1.2 | |||
| H84WANS | 0.79 | 0.21 | 1.47 | 4.43 | 2.09 | 0 | 0 | 3 | 1.2 | |
| I88W | strand F | 0.50 | 0.50 | 1.33 | 4.13 | 2.72 | 1.0 | |||
| I88WANS | 0.57 | 0.43 | 1.22 | 4.04 | 2.43 | 11 | 12 | <0 | 1.1 | |
| F99W | strand G | 0.79 | 0.21 | 1.77 | 5.22 | 2.49 | 1.0 | |||
| F99WANS | 0.71 | 0.29 | 1.84 | 4.73 | 2.68 | 0 | 0 | 8 | 1.1 | |
| C101W | strand G | 0.54 | 0.46 | 1.84 | 4.93 | 3.27 | 1.0 | |||
| C101WANS | 0.65 | 0.35 | 1.80 | 6.32 | 3.04 | 7 | 7 | 18 | 0.9 | |
| L105W | strand G | 0.73 | 0.27 | 1.04 | 3.81 | 1.77 | 1.2 | |||
| L105WANS | 0.89 | 0.11 | 0.64 | 3.68 | 0.97 | 46 | 52 | 64 | 1.0 |
Estimated errors for calculating ET efficiencies with steady-state and time-resolved fluorescence do not exceed 6%. An asterisk indicates the value was extrapolated to the ratio donor/acceptor = 1. ETτ values were calculated from the time-resolved fluorescence measurements. ETss values were calculated from the steady-state fluorescence measurements.
Table 2.
Dissociation Constant for ANS Binding to the TL Mutant
| TL mutant | Kda (μM) | Kdb (μM) | TL mutant | Kda (μM) | Kdb (μM) |
|---|---|---|---|---|---|
| E34W | 5.3 ± 0.5 | 8.1 ± 2.0 | H84C | 7.1 ± 0.8 | |
| S35W | 1.3 ± 0.4 | 5.4 ± 3.5 | I88W | 1.2 ± 0.2 | |
| M39W | 3.1 ± 0.2 | 4.6 ± 1.1 | F99W | 1.2 ± 0.1 | 2.7 ± 1.1 |
| A66W | 6.0 ± 0.6 | C101W | 0.7 ± 0.1 | 0.7 ± 0.3 | |
| H84W | 5.0 ± 0.4 | L105W | 1.7 ± 0.2 | 1.3 ± 0.4 |
Calculated from the increase of ANS fluorescence.
Calculated from the quenching of Trp fluorescence.
ET data obtained from the steady-state fluorescence are also shown in Table 1. Unlike the steady-state fluorescence, the time-resolved fluorescence experiments with ANS–TL complexes do not require the corrections for the inner filter effect. Therefore, the ET data from the time-resolved experiments can be considered more accurate. However, in some cases, e.g., I88W and M39W, the differences in ET values are out of the error range. This situation is plausible when bound ANS alters the Trp conformation that, in turn, changes the partial static quenching of Trp fluorescence by nearby groups. Therefore, discrepancies in ET data observed for the I88W and M39W steady-state and time-resolved fluorescence may be accounted for by a local conformational change in TL near the corresponding Trp side chain upon ANS binding. The static quenching decreases the quantum yield of Trp but does not alter the fluorescence lifetime. Therefore, ET data are more reliable when the values obtained by two methods, steady-state and time-resolved fluorescence, match closely. Because of the uncertainty in the relative orientation, no attempt has been made to determine the distance between bound ANS and the particular Trp residue; the proximity was judged qualitatively by ET values. It should be noted that the κ2 factor, which describes the relative orientation of the transition dipoles of the donor (Trp residues in the mutants) and the acceptor (bound ANS), can greatly affect corresponding the R0 values. This, in turn, will affect distance estimations between Trp residues and ANS. The widely used 2/3 value for κ2, which describes dynamic random averaging of the donor and acceptor rotating independently and rapidly in space within the lifetime of the donor, is not applicable for ANS–TL complexes. However, variation of κ2 between the broad limits of 1 and 4 would result in only a 26% change in the estimated distances (46). Because at least two fluorescence lifetimes were detected for tryptophan mutants, which are linked to different side chain rotamers, a 0 value for κ2 is very unlikely. Therefore, ET values for single Trp mutant–ANS complexes are an appropriate parameter to judge the donor–acceptor proximities. RET experiments show that position 105 is most proximate to the bound ANS. This result is very consistent with that obtained from the fatty acid binding study of TL (24). The residue Leu105 is not resolved in the crystal structure of TL (20). However, the solution structure obtained by SDTF and site-directed spin labeling studies reveal that the side chain of residue 105, located at the tip of strand G, is oriented inside the cavity (18, 19, 47). In another study (24), it has been shown that the nitroxide spin label of the lauric acid analogue C12SL interacts most with the tryptophan at 105, but also weakly with tryptophans at position 101 of the G strand. No such interaction was observed for the deeply buried position 99. The ET values for ANS–TL mutant complexes show exactly the same pattern for the G strand (Table 1).
Along with RET data, dissociation constants for ANS–TL mutant complexes are informative for the details of ANS binding. The substitution of the His with Cys at residue 84 significantly decreases ANS binding (Table 2). Trp substitution, compared to Cys substitution at position 84, results in slightly increased ANS binding affinity. This is much smaller compared to the difference observed between Trp and Cys substitution of Lys114 (Figure 5). Perhaps, the proximity of ANS is better suited for van der Waals interactions with Trp114 (K114W) than Trp84 (H84W). The decreased binding affinities of ANS for TL mutants H84C and H84W imply an electrostatic interaction with the histidine residue. Trp substitutions, particularly at position 84, could at least partially contribute to hydrophobic effects and van der Waals interactions. However, the effect of other interactions in addition to the electrostatic interaction in these mutations could not be excluded. The substitution of Glu34 also decreases ANS binding. The spatial arrangement of Lys114, His84, and Glu34 presents a trigone, which permits interaction of the sulfonate group of ANS with all three residues (Figure 11).
Figure 11.
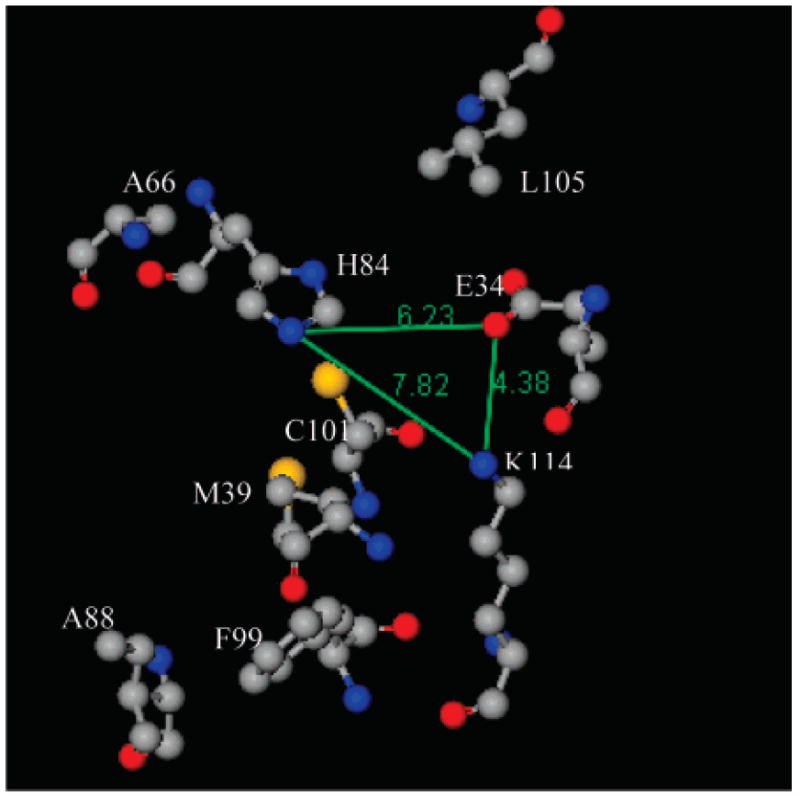
Arrangement of amino acid residues, considered for ANS binding, in the crystal structure of TL (PDB 1XKI (18)). Residue Leu105 is modeled from the solution structure of TL by SDTF (20). Gray, blue, red, and yellow balls represent carbon, nitrogen, oxygen, and sulfur atoms, respectively. The numbers represent distances between the atoms (Å). The image was generated by ViewerLite 5.0 (Accelrys Inc.).
It is well-established that the pKa values of the side chains are sensitive to the surrounding residues, particularly charged residues. A nearby positive charge always decreases pKa values; a negative charge results in an increase. The pKa values for the side chains of His and Glu residues are about 6.0 and 4.3, respectively, but vary considerably in buried sites. Because the sulfonate group of ANS has a very low pKa value, <2.0, it can be considered to have a negative charge that would not be affected by surrounding residues at pH 7.3. However, upon binding, the sulfonate group of ANS can induce protonation of adjacent His84 and Glu34 residues by increasing their corresponding pKa values. The protonation of His84 and Glu34 residues will increase ANS binding interactions via electrostatic interaction and hydrogen bonding (probably not true hydrogen bonding because of the geometry of the sulfonate group and the side chain of Glu34).
The Trp substitution at position 66 reduces the binding affinity for ANS (Table 2). This mutation is known to decrease the capacity of the cavity of TL with about a 3-fold decrease in the binding affinity of 16-doxylstearic acid (18). Presumably, the mutation also influences ANS binding affinity by restriction of the open entrance of the cavity.
A docking study was performed to test whether interaction between the sulfonate group of ANS and the side chains of Lys114, His84, and Glu34 is feasible. Similar to β-lactoglobulin, multiple solutions were generated for the ANS–TL complex with energy values ranging from −11.9 to −10.5 kcal/mol. In accord with multiple solutions, the decay-associated spectrum (for 17 ns) of the ANS–TL complex has been shown to be heterogeneous and could not be fit to a single log–normal component (29). However, β-lactoglobulin shows a much lower interaction energy with ANS, −6.2 kcal/mol (48). Despite 23% sequence identity and superimposable calyxes of these proteins (18, 20), the binding affinity to ANS for β-lactoglobulin is much lower (about 3.8 mM (49)) compared to that of TL (0.5 μM). In addition, the fluorescence lifetime of ANS bound into the cavity of β-lactoglobulin is shorter, about 14 ns.
The docking solution that best concurs with experimental data is shown in Figure 12. The sulfonate group of ANS is positioned favorably to interact with the side chains of Lys114, His84, and Glu34. In addition, the naphthalene group of ANS is in close proximity to Leu105 of strand G. Comparison of ANS binding to TL versus β-lactoglobulin is very insightful. In β-lactoglobulin, which does not have a positively charged residue corresponding to position Lys114 of TL, the sulfonate group of ANS has electrostatic interactions with Lys69 and Lys60. These positively charged residues are located at shallow sites of the calyx compared to Lys114 of TL. Previously, it has been shown that the cavity of TL is more capacious compared to that of β-lactoglobulin (18). Therefore, it is not surprising that the ANS binding site is located at a deeper position in the calyx of TL.
Figure 12.

Docking solution for interaction of ANS with TL. The backbone of TL (PDB 1XKI) is shown in cyan in a ribbon representation. Gray, blue, red, and yellow sticks represent carbon, nitrogen, oxygen, and sulfur atoms, respectively. The position of ANS (energy level –11.0 kcal/mol) is that in best agreement with the experimental data. Distances, indicated with green dotted arrows, between the sulfonate group of ANS and the side chains of Glu34, His84, and Lys114 are 2.5, 2.4, and 3.0 Å, respectively. The image was generated by DeepView/Swiss-PdbViewer v.3.7 (GlaxoSmithKline R&D).
In summary, the sulfonate group of ANS interacts mainly with Lys114 and to a lesser extent with neighboring residues His84 and Glu34. In addition, RET data show that the naphthalene group of ANS is in close proximity to Leu105. It is interesting to speculate that the cluster of residues Lys114, His84, and Glu34 comprise the ligand recognition site for some negatively charged ligands. Interaction of the sulfonate group with the trigonal cluster has implications for drug delivery using duocalins and anticalins. Sulfonation is widely used to make molecules more soluble, e.g., estradiol sulfate. Sulfotransferases, e.g., estrogen sulfotransferase, can be used for enzymatic sulfonation (50). Modified drugs can be activated by sulfonate removal by endogenous sulfatases (50). Accordingly, sulfonated (deactivated) and desulfonated (activated) forms of drugs can be controlled by these two antagonistic enzymes. Human tear lipocalin would require little modification, if any, to bind to some sulfonated drugs with high affinity. The trigonal cluster could improve the drug-carrying capacity for extremely hydrophobic drugs and could be selectively added to anticalins and duocalins (21-23). Similar charged residues exist in the cavity of other lipocalins. Human odorant binding protein, hOBP-2A, exhibits 45.2% sequence identity to TL. Lys112 of the H strand, analogous to Lys114 in TL, confers binding specificity of hOBP-2A to aldehydes and small carboxylic acids (51).
Transposition of Fatty Acid in the Cavity of TL: Possible Role of Lys114
The orientation of fatty acids, which bind in the cavity of TL, has been previously elucidated by using a spin-labeled analogue of lauric acid (19, 24). In TL, fatty acids are oriented with the hydrocarbon tail buried in the cavity and the carboxyl group oriented toward the mouth. The side chains of Lys114, His84, and Glu34 are oriented inside an otherwise predominantly hydrophobic cavity. These side chains are in close proximity and appear well situated for interaction with the sulfonate group of ANS. This fact prompted us to test the hypothesis that fatty acids can bind within the cavity of TL but in the opposite direction, i.e., the carboxyl group oriented toward the cavity. To elucidate the possibility of an alternative orientation for lipids, the DAUDA displacement experiments were performed with analogues of lauric acid that contain either positive or negative charges at both ends (Figure 13). It is not surprising that the ligand that contains a positive charge at one end has the greatest affinity (therefore, lowest IC50 value), since the TL cavity has two negatively charged amino acids (Glu34 and Asp80) at the open end. The greater binding affinity of the singly charged analogues confirms that the preferred orientation casts the alkyl chain of the fatty acid into the cavity of TL (19, 24). The differences in binding affinity between the doubly charged compounds and their singly charged analogues reflect the impact of forcing either a positive or negative charge into the cavity. The fatty acid analogue, which contains positive charges at both ends, reduces the binding affinity by an order of magnitude. This finding is explained by an intracavitary position of the positively charged Lys114. However, the decrease in binding affinity for the doubly negatively charged analogue is not as dramatic as with the doubly positively charged analogue. This is consistent with dominant interaction between the carboxyl group of the fatty acid and the side chain of Lys114.
Figure 13.
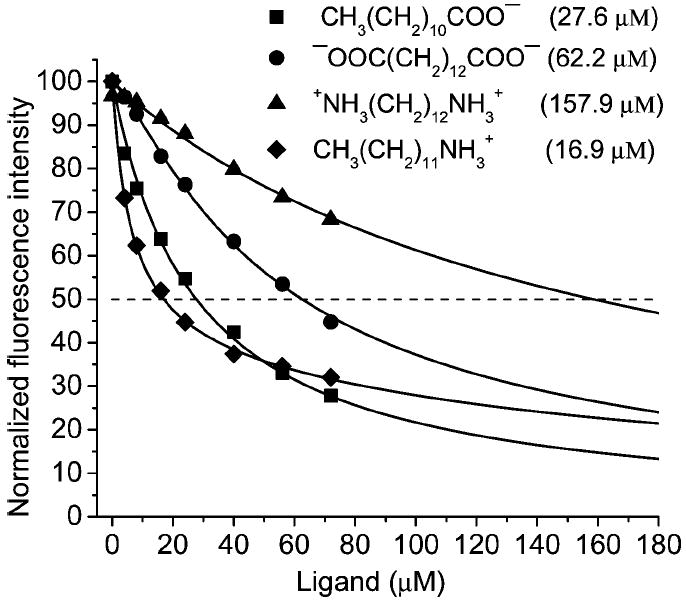
Displacement of DAUDA from the DAUDA(2 μM)–TL(4 μM) complex by various ligands at pH 7.3. Excitation and emission wavelengths were 345 and 498 nm, respectively. IC50 values for each ligand are shown in parentheses. Solid curves are generated by fitting of the experimental data to one binding site model.
Implications for TL Functions
TL is a multifunctional protein (6, 7). At the ocular surface, at least three functions related to ligand binding have been assigned to TL, scavenging lipid from the corneal surface, solubilization of lipid in tears (6, 52), stabilization of the tear film via interaction with the lipid layer (34). In tears, TL binds most strongly to the least soluble fatty acids (53). For interaction with the lipid layer of the tears, a pH-driven mechanism has been proposed for TL (20). This mechanism, obviously, has several steps: recognition, interaction, molten globule state transition, penetration, etc. The findings of a potential ligand recognition site composed of a charged trigonal cluster of residues Lys114, His84, and Glu34 suggest a plausible stabilizing mechanism between TL and negatively charged lipids at the lipid–tear interface. This motif permits TL to interact with fatty acids oriented so that the negative charge is oriented toward the cavity and the hydrocarbon tail is embedded in the lipid layer. In the bulk, this binding mode exhibits less affinity compared with the alkyl chain positioned in the cavity of TL. However, in the unique environment provided at the tear–lipid interface, this configuration might be the only one available for fatty acids. A weak interaction between TL and the carboxyl group of the fatty acid may help stabilize the lipid layer of the tear film.
Footnotes
Supported by U.S. Public Health Service Grants NIH EY11224 and EY00331 as well as the Edith and Lew Wasserman Endowed Professorship in Ophthalmology.
Abbreviations: ANS, 8-anilino-1-naphthalenesulfonic acid; CD, circular dichroism; cps, counts per second; DAUDA, 11-((5-dimethylaminonaphthalene-1-sulfonyl)amino)undecanoic acid; IRF, instrument response function; NATA, N-acetyl-l-tryptophanamide; 1NPN, N-phenyl-1-naphthylamine; RET, resonance energy transfer; SDTF, site-directed tryptophan fluorescence; SDSL, site-directed spin labeling, TL, human tear lipocalin; TNS, 6-(p-toluidino)-2-naphthalenesulfonic acid.
References
- 1.Glasgow BJ, Abduragimov AR, Farahbakhsh ZT, Faull KF, Hubbell WL. Tear lipocalins bind a broad array of lipid ligands. Curr Eye Res. 1995;14:363–372. doi: 10.3109/02713689508999934. [DOI] [PubMed] [Google Scholar]
- 2.Scalfari F, Castagna M, Fattori B, Andreini I, Maremmani C, Pelosi P. Expression of a lipocalin in human nasal mucosa. Comp Biochem Physiol. 1997;118B:819–824. [Google Scholar]
- 3.Lacazette E, Gachon AM, Pitiot G. A novel human odorant-binding protein gene family resulting from genomic duplicons at 9q34: differential expression in the oral and genital spheres. Hum Mol Genet. 2000;9:289–301. doi: 10.1093/hmg/9.2.289. [DOI] [PubMed] [Google Scholar]
- 4.Redl B. Human tear lipocalin. Biochim Biophys Acta. 2000;1482:241–248. doi: 10.1016/s0167-4838(00)00142-4. [DOI] [PubMed] [Google Scholar]
- 5.Wojnar P, Dirnhofer S, Ladurner P, Berger P, Redl B. Human lipocalin-1, a physiological scavenger of lipophilic compounds, is produced by corticotrophs of the pituitary gland. J Histochem Cytochem. 2002;50:433–435. doi: 10.1177/002215540205000314. [DOI] [PubMed] [Google Scholar]
- 6.Glasgow BJ, Marshall G, Gasymov OK, Abduragimov AR, Yusifov TN, Knobler CM. Tear lipocalins: potential lipid scavengers for the corneal surface. Invest Ophthalmol Visual Sci. 1999;40:3100–3107. [PubMed] [Google Scholar]
- 7.Gasymov OK, Abduragimov AR, Prasher P, Yusifov TN, Glasgow BJ. Tear lipocalin: evidence for a scavenging function to remove lipids from the human corneal surface. Invest Ophthalmol Visual Sci. 2005;46:3589–3596. doi: 10.1167/iovs.05-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selsted ME, Martinez RJ. Isolation and purification of bactericides from human tears. Exp Eye Res. 1982;34:305–318. doi: 10.1016/0014-4835(82)90079-3. [DOI] [PubMed] [Google Scholar]
- 9.van’t Hof W, Blankenvoorde MF, Veerman EC, Amerongen AV. The salivary lipocalin von Ebner’s gland protein is a cysteine proteinase inhibitor. J Biol Chem. 1997;272:1837–1841. doi: 10.1074/jbc.272.3.1837. [DOI] [PubMed] [Google Scholar]
- 10.Blaker M, Kock K, Ahlers C, Buck F, Schmale H. Molecular cloning of human von Ebner’s gland protein, a member of the lipocalin superfamily highly expressed in lingual salivary glands. Biochim Biophys Acta. 1993;1172:131–137. doi: 10.1016/0167-4781(93)90279-m. [DOI] [PubMed] [Google Scholar]
- 11.Redl B, Holzfeind P, Lottspeich F. cDNA cloning and sequencing reveals human tear prealbumin to be a member of the lipophilic-ligand carrier protein superfamily. J Biol Chem. 1992;267:20282–20287. [PubMed] [Google Scholar]
- 12.Lechner M, Wojnar P, Redl B. Human tear lipocalin acts as an oxidative-stress-induced scavenger of potentially harmful lipid peroxidation products in a cell culture system. Biochem J. 2001;356:129–135. doi: 10.1042/0264-6021:3560129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glasgow BJ, Abduragimov AR, Gassymov OK, Yusifov TN, Ruth EC, Faull KF. Vitamin E associated with the lipocalin fraction of human tears. AdV Exp Med Biol. 2002;506:567–572. doi: 10.1007/978-1-4615-0717-8_79. [DOI] [PubMed] [Google Scholar]
- 14.Yusifov TN, Abduragimov AR, Gasymov OK, Glasgow BJ. Endonuclease activity in lipocalins. Biochem J. 2000;347(Pt 3):815–819. [PMC free article] [PubMed] [Google Scholar]
- 15.Gouveia SM, Tiffany JM. Human tear viscosity: an interactive role for proteins and lipids. Biochim Biophys Acta. 2005;1753:155–163. doi: 10.1016/j.bbapap.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 16.Flower DR. The lipocalin protein family: structure and function. Biochem J. 1996;318(Pt 1):1–14. doi: 10.1042/bj3180001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glasgow BJ, Abduragimov AR, Yusifov TN, Gasymov OK, Horwitz J, Hubbell WL, Faull KF. A conserved disulfide motif in human tear lipocalins influences ligand binding. Biochemistry. 1998;37:2215–2225. doi: 10.1021/bi9720888. [DOI] [PubMed] [Google Scholar]
- 18.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Site-directed tryptophan fluorescence reveals the solution structure of tear lipocalin: evidence for features that confer promiscuity in ligand binding. Biochemistry. 2001;40:14754–14762. doi: 10.1021/bi0110342. [DOI] [PubMed] [Google Scholar]
- 19.Glasgow BJ, Gasymov OK, Abduragimov AR, Yusifov TN, Altenbach C, Hubbell WL. Side chain mobility and ligand interactions of the G strand of tear lipocalins by site-directed spin labeling. Biochemistry. 1999;38:13707–13716. doi: 10.1021/bi9913449. [DOI] [PubMed] [Google Scholar]
- 20.Breustedt DA, Korndorfer IP, Redl B, Skerra A. The 1.8-A crystal structure of human tear lipocalin reveals an extended branched cavity with capacity for multiple ligands. J Biol Chem. 2005;280:484–493. doi: 10.1074/jbc.M410466200. [DOI] [PubMed] [Google Scholar]
- 21.Skerra A. Anticalins as alternative binding proteins for therapeutic use. Curr Opin Mol Ther. 2007;9:336–344. [PubMed] [Google Scholar]
- 22.Skerra A. Lipocalins as a scaffold. Biochim Biophys Acta. 2000;1482:337–350. doi: 10.1016/s0167-4838(00)00145-x. [DOI] [PubMed] [Google Scholar]
- 23.Schlehuber S, Skerra A. Duocalins: engineered ligand-binding proteins with dual specificity derived from the lipocalin fold. Biol Chem. 2001;382:1335–1342. doi: 10.1515/BC.2001.166. [DOI] [PubMed] [Google Scholar]
- 24.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Resolution of ligand positions by site-directed tryptophan fluorescence in tear lipocalin. Protein Sci. 2000;9:325–331. doi: 10.1110/ps.9.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bron AJ, Tiffany JM, Gouveia SM, Yokoi N, Voon LW. Functional aspects of the tear film lipid layer. Exp Eye Res. 2004;78:347–360. doi: 10.1016/j.exer.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 26.Miano F, Calcara M, Millar TJ, Enea V. Insertion of tear proteins into a meibomian lipids film. Colloids Surf B: Biointerfaces. 2005;44:49–55. doi: 10.1016/j.colsurfb.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 27.McCulley JP, Shine W. A compositional based model for the tear film lipid layer. Trans Am Ophthalmol Soc. 1997;95:79–88. discussion 88–93. [PMC free article] [PubMed] [Google Scholar]
- 28.McCulley JP, Shine WE. The lipid layer of tears: dependent on meibomian gland function. Exp Eye Res. 2004;78:361–365. doi: 10.1016/s0014-4835(03)00203-3. [DOI] [PubMed] [Google Scholar]
- 29.Gasymov OK, Abduragimov AR, Glasgow BJ. Characterization of fluorescence of ANS-TL complex: Evidence for multiple species. Photochem Photobiol. 2007;83:1405–1414. doi: 10.1111/j.1751-1097.2007.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasymov OK, Abduragimov AR, Glasgow BJ. Evidence for internal and external binding sites on human tear lipocalin. Arch Biochem Biophys. 2007;468:15–21. doi: 10.1016/j.abb.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glasgow BJ, Heinzmann C, Kojis T, Sparkes RS, Mohandas T, Bateman JB. Assignment of tear lipocalin gene to human chromosome 9q34-9qter. Curr Eye Res. 1993;12:1019–1023. doi: 10.3109/02713689309029229. [DOI] [PubMed] [Google Scholar]
- 32.Glasgow BJ. Tissue expression of lipocalins in human lacrimal and von Ebner’s glands: colocalization with lysozyme. Graefes Arch Clin Exp Ophthalmol. 1995;233:513–522. doi: 10.1007/BF00183433. [DOI] [PubMed] [Google Scholar]
- 33.Cormack B. Current Protocol in Molecular Biology. Greene Publishing Associates and Wiley-Interscience; New York: 1987. pp. 158.51–58.59. [Google Scholar]
- 34.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Binding studies of tear lipocalin: the role of the conserved tryptophan in maintaining structure, stability and ligand affinity. Biochim Biophys Acta. 1999;1433:307–320. doi: 10.1016/s0167-4838(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 35.Marston FAO. In: DNA Cloning: a practical approach. Glover DM, editor. IRL Press; Oxford, England: 1987. pp. 59–88. [Google Scholar]
- 36.Bozimowski D, Artiss JD, Zak B. The variable reagent blank: protein determination as a model. J Clin Chem Clin Biochem. 1985;23:683–689. doi: 10.1515/cclm.1985.23.10.683. [DOI] [PubMed] [Google Scholar]
- 37.Lehrer SS. Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 38.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Interstrand loops CD and EF act as pH-dependent gates to regulate fatty acid ligand binding in tear lipocalin. Biochemistry. 2004;43:12894–12904. doi: 10.1021/bi049076o. [DOI] [PubMed] [Google Scholar]
- 39.Cogan U, Kopelman M, Mokady S, Shinitzky M. Binding affinities of retinol and related compounds to retinol binding proteins. Eur J Biochem. 1976;65:71–78. doi: 10.1111/j.1432-1033.1976.tb10390.x. [DOI] [PubMed] [Google Scholar]
- 40.Berengian AR, Bova MP, McHaourab HS. Structure and function of the conserved domain in alphaA-crystallin. Site-directed spin labeling identifies a beta-strand located near a subunit interface. Biochemistry. 1997;36:9951–9957. doi: 10.1021/bi9712347. [DOI] [PubMed] [Google Scholar]
- 41.Woody RW. Contributions of tryptophan side chains to the far-ultraviolet circular dichroism of proteins. Eur Biophys J. 1994;23:253–262. doi: 10.1007/BF00213575. [DOI] [PubMed] [Google Scholar]
- 42.Strickland EH. Aromatic contributions to circular dichroism spectra of proteins. CRC Crit Rev Biochem. 1974;2:113–175. doi: 10.3109/10409237409105445. [DOI] [PubMed] [Google Scholar]
- 43.Kumar S, Nussinov R. Salt bridge stability in monomeric proteins. J Mol Biol. 1999;293:1241–1255. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- 44.Gasymov OK, Glasgow BJ. ANS fluorescence: Potential to augment the identification of the external binding sites of proteins. BBA–Proteins and Proteomics. 2007;1774:403–411. doi: 10.1016/j.bbapap.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 46.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; New York: 2006. p. 954. [Google Scholar]
- 47.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Solution structure by site directed tryptophan fluorescence in tear lipocalin. Biochem Biophys Res Commun. 1997;239:191–196. doi: 10.1006/bbrc.1997.7451. [DOI] [PubMed] [Google Scholar]
- 48.Collini M, D’Alfonso L, Molinari H, Ragona L, Catalano M, Baldini G. Competitive binding of fatty acids and the fluorescent probe 1–8-anilinonaphthalene sulfonate to bovine beta-lactoglobulin. Protein Sci. 2003;12:1596–1603. doi: 10.1110/ps.0304403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D’Alfonso L, Collini M, Baldini G. Evidence of heterogeneous 1-anilinonaphthalene-8-sulfonate binding to beta-lactoglobulin from fluorescence spectroscopy. Biochim Biophys Acta. 1999;1432:194–202. doi: 10.1016/s0167-4838(99)00105-3. [DOI] [PubMed] [Google Scholar]
- 50.Goodsell DS. The molecular perspective: estrogen sulfotransferase. Oncologist. 2006;11:418–419. doi: 10.1634/theoncologist.11-4-418. [DOI] [PubMed] [Google Scholar]
- 51.Tcatchoff L, Nespoulous C, Pernollet JC, Briand L. A single lysyl residue defines the binding specificity of a human odorant-binding protein for aldehydes. FEBS Lett. 2006;580:2102–2108. doi: 10.1016/j.febslet.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 52.Tiffany JM, Nagyova B. The role of lipocalin in determining the physical properties of tears. Adv Exp Med Biol. 2002;506:581–585. doi: 10.1007/978-1-4615-0717-8_81. [DOI] [PubMed] [Google Scholar]
- 53.Gasymov OK, Abduragimov AR, Yusifov TN, Glasgow BJ. Structural changes in human tear lipocalins associated with lipid binding. Biochim Biophys Acta 1. 1998;386:145–156. doi: 10.1016/s0167-4838(98)00092-2. [DOI] [PubMed] [Google Scholar]