

Abstract
We present a homology based model of the ligand binding domain (LBD) of the homopentameric alpha1 glycine receptor (GlyR). The model is based on multiple sequence alignment with other members of the nicotinicoid ligand gated ion channel superfamily and two homologous acetylcholine binding proteins (AChBP) from the freshwater (Lymnaea stagnalis) and saltwater (Aplysia californica) snails with known high resolution structure. Using two template proteins with known structure to model three dimensional structure of a target protein is especially advantageous for sequences with low homology as in the case presented in this paper. The final model was cross-validated by critical evaluation of experimental and published mutagenesis, functional and other biochemical studies. In addition, a complex structure with strychnine antagonist in the putative binding site is proposed based on docking simulation using Autodock program. Molecular dynamics (MD) simulations with simulated annealing protocol are reported on the proposed LBD of GlyR, which is stable in 5 ns simulation in water, as well as for a deformed LBD structure modeled on the corresponding domain determined in low-resolution cryomicroscopy structure of the alpha subunit of the full-length acetylcholine receptor (AChR). Our simulations demonstrate that the beta-sandwich central core of the protein monomer is fairly rigid in the simulations and resistant to deformations in water.
Keywords: glycine receptor, ligand-gated ion channel, ligand binding domain, nicotinicoid receptor, cys-loop receptor, protein structure modeling, protein structure prediction, ligand docking, alpha1 subunit
INTRODUCTION
Glycine receptors (GlyRs) are members of the nicotinicoid superfamily of ligand-gated ion channels that are also referred to as the Cys-loop receptors due to conserved disulfide-bonded cysteines in the ligand binding domain (LBD) of eukaryotic receptors that are separated by 13 amino acids. This family includes inhibitory GlyR and γ-aminobutyric acid receptors (GABAAR), and excitatory acetylcholine receptors (nAChR) and serotonin receptors (5HT3R).1 These receptors mediate signaling across synapses of the central and peripheral nervous systems. They are sensitive to many neuroactive drugs such as n-alcohols, volatile anesthetics and inhalants, neurotoxins, and neurosteroids.2–6 Dysfunctions of nicotinicoid receptors are associated with a number of nervous system diseases, underscoring their critical role in neuronal health and development.7–12 In mammals, the anion-selective GlyR channels are primarily located in the brain stem and spinal cord.13 The GlyRs, similar to all nicotinicoid receptors, are hetero- or homo-pentamers of homologous gene products arranged quasi-symmetrically around a central ion-conducting pore. To date, four types of α subunits and a single β subunit of GlyR have been identified.13 However, the α1 subunit is capable of forming homomeric functional receptors exhibiting the pharmacological properties of native receptors.14,15 Each α1 subunit is comprised of an extracellular LBD, a pore-forming domain composed of four transmembrane segments (TM1-4), and a variable (i.e., least well-conserved amongst family members) cytoplasmic domain formed primarily by the long intracellular loop connecting TM3 and 4. Binding of agonist ligands to the subunit–subunit LBD interfaces causes allosteric activation of ion channel and ion conduction. A successful recent construction of a functional chimeric proteins consisting of the LBD of α7 nAChR and the TM domain of the 5HT3 receptors16 or α7 nAChR-GlyR receptors17 strongly support the hypothesis that members of the nicotinicoid family share not only similar secondary folds and geometries but also a surprisingly similar, nearly identical mechanism of activation, i.e., a common signal transduction network coupling ligand binding in the LBD to channel gating in the transmembrane domain. Nicotinicoid receptor homologs have been identified in prokaryotes, and it appears that the coupling of the LBD with the pore domain and the channel-gating mechanisms of this receptor type have been remarkably well-conserved evolutionarily, and at least one aromatic residue is typically involved in ligand-binding, with cation–pi interactions implicated in binding interactions.17
Structurally, the best-characterized member of the nicotinicoid family is the nAChR.18 The nAChR isolated from electric ray has been extensively studied by cryomicroscopic studies,18–20 the most recent of which has reported the structure of the resting state of the receptor at 4 Å resolution.21 Recently, the three-dimensional structure of the soluble homopentameric acetylcholine binding protein (AChBP) from freshwater snail Lymnaea stagnalis with reported homology to the extracellular LBD of nicotinicoid receptors has been determined.22–24 While AChBP has low sequence similarity (15–25%) with the LBDs of nicotinicoid receptors, sequence–structure conservation analysis predicts that the secondary structure similarity between AChBP and the α7 nAChR subunit is as high as 80%.25,26 Lys scanning mutagenesis studies of the LBD of nAChR also has provided excellent experimental evidence that the LBD of nicotinicoid receptors shares a common fold with AChBP.27 Given that the sequence similarity between different subunits of nicotinicoid receptors is in range of 20–35% and several sequence alignment schemes were reported for series of LBDs28,29 as well as in comparison with AChBP protein,22 the crystal structures of AChBPs provides a good initial template for modeling of all the LBDs of nicotinicoid receptors. The sequence similarity between the α1 GlyR ligand-binding domain and AChBP is 25% (which is in fact higher than that of α7). Recently, several homology models of the LBD of GlyR30,31 as well as of LBDs of other members of the family, including the nAChR, 5HT3R, and GABAAR32–35 have been constructed.
In this work sequences of the freshwater snail L. stagnalis AChBP22 and the saltwater snail A. californica AChBP36 proteins were aligned with the GlyR α1 subunit. The sequence similarity between L. stagnalis AChBP and A. californica AChBP is 33%. Structures of both AChBP proteins then served as templates for 3D homology modeling of the α1 GlyR LBD. Five nanoseconds molecular dynamics (MD) simulations of its pentameric assembly were then performed in explicit water solvent at room temperature to assess structural stability of monomers and a pentamer as a whole and refine positions of side-chains. Our simulations resulted in a stable structure in the course of the MD trajectory. Our proposed model is consistent with the earlier mutagenesis studies implicating variety of residues in ligand-binding function of the receptor. A putative ligand binding site is also proposed based on comparison of the three-dimensional structure and data found in the literature.
While it seems reasonable to model a GlyR LBD domain using AChBP proteins as templates, when associated with the transmembrane domain and/or in contact with lipid headgroups, the LBD conformation in the holoreceptor may be somewhat different. One indication on how an AChBP template conformation may deviate from the conformations of LBDs in nicotinicoid receptors is the relative conformational distortion that had been proposed for the LBD of an nAChR holoreceptor based on recent electron diffraction studies of N. Unwin.21 However, given that many residues of the LBD are not defined in the cryo-EM structure (PDB code, 2BG9), it is not a suitable template for a comparative modeling (e.g. with Modeller). Therefore, in this study we use a simulated annealing technique37 to “deform” our GlyR LBD model based on an AChBP template into a nAChR LBD-like conformation. Using a similar approach, a QBP protein template was used to model an unliganded open conformation structure of the GluR2 S1S2 protein38 (glutamate receptor LBD) starting from its ligand-bound (closed) structure which was known and forcing it to open to match a QBP template. Subsequent determination of the GluR2 S1S2 apo conformation X-ray structure proved the model to be very reasonable, and validated this approach. We have also further examined the stability of the GlyR LBD model [modeled using the nAChR LBD as a template]. A stable structure (during an extended simulated annealing MD simulation) was obtained and is described in the Discussion section. The model presented herein is unique in that it is the first model of the LBD of GlyR based on multiple alignments using various AChBP structures as templates whose stability in MD simulations has been rigorously tested.
MATERIALS AND METHODS
Homology Model
The sequences of GlyR α1 LBD, L. stagnalis AChBP, and A. californica AChBP were obtained from the data-bank in the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). The crystal structure of AChBP (PDB code, 1I9B) was downloaded from Protein Data Bank.39 The alignment between L. stagnalis AChBP, A. californica AChBP, and GlyR α1 LBD was obtained using web-server Clustal W.40 This alignment was then manually adjusted as described in the Results section. The program Modeller 7.741 was used to generate a tertiary structure model of the GlyR α1 LBD pentamer. All five subunits of the pentamer were modeled simultaneously. The positions of cysteines at positions 198 and 209 that form a disulfide bond in each subunit were constrained using the Modeller “patch” command. The Modeller's variable target function method (VTFM) and MD simulated annealing were used to generate 15 initially randomized models. Quality of these models was characterized in terms of Z-scores using the WHAT_IF program.42 Z-scores are standardized statistically-derived structure quality assessment scales that include packing quality, Ramachandran plot appearance, chi-1/chi-2 rotamer normality, and backbone conformation. The structure with the highest Z-scores was used for further simulations.
MD Simulations
Hydrogen atoms were added to the model structure using the Xleap module of AMBER 7 software package.43 The total charge in the computational box was neutralized by the addition of 20 Na+ ions to the simulation. The structure was then solvated in a box of TIP3P water molecules with dimensions 112 × 112 × 117 Å3 and periodic boundary conditions were applied. This solvated complex was subjected to the energy minimization followed by a 100 ps MD simulation with constant pressure at T = 10 K. At this stage the protein Cα atoms were restrained by a harmonic force of 100 kcal/(mol Å2). Next, all restraints were removed and the temperature was gradually increased to 300 K. The equilibrium simulation was performed for 5.3 ns at 300 K under NTV ensemble conditions. Long-range electrostatic interactions were calculated using particle mesh Ewald44 method with 12 Å cut-off. The time step for integration was 2 fs, and the coordinates of all atoms were saved every 10 ps. Bonds involving hydrogen atoms were constrained via the SHAKE algorithm.45 All MD simulations were performed using the PMEMD43,46 software package with the Cornell et al. force field.47
To develop a model of a “deformed” GlyR LBD (LBDdeformed) that corresponds to the nAChR LBD derived from the electron density profile21 we used a simulated annealing method.37 The nAChR LBD structure (PDB code, 2BG9) contains information only on a limited subset of atoms insufficient for comparative modeling (e.g. with Modeller). To deform the initial structure of GlyR LBD to match the structure of the nAChR α subunit template we used constrained MD simulations as follows. Using the sequence alignment of GlyR LBD and α-nAChR LBD from Ref. 29, each subunit of the GlyR LBD pentamer was overlapped with the α-subunit from the nAChR structure, and harmonic constraints of 100 kcal/mol were applied to a subset of the backbone atoms of the GlyR LBD as described in detail in the Results section. The centers of constraining potentials were placed at positions of the corresponding atoms of the nAChR template. Only the least mobile parts of the protein, namely β2, β5, β6, β7, β9, β10 strands (c.f. Figs. 1 and 2), were constrained (see Results and Discussion sections for discussion of the mobility of different parts of the protein derived from equilibrium unconstrained MD simulations). After initial equilibration simulation of this constrained pentamer in TIP3P water at room temperature, two cycles of the simulated annealing protocol were applied to optimize positions of the side chain and further relax the structure. Our simulated annealing protocol is similar to the standard protocols used in refinement of the NMR and X-ray derived protein structures. Maximum temperature in simulated annealing was 700 K. Following simulated annealing all constraints were removed and 500 ps MD simulation of the free pentamer in water was performed.
Fig. 1.

Sequence of the extracellular LBD of GlyR α1 subunit aligned with those of L. stagnalis AChBP and A. californica AChBP subunits. The numbering is given for GlyR. The asterisks indicate residues conserved in all three structures.
Fig. 2.
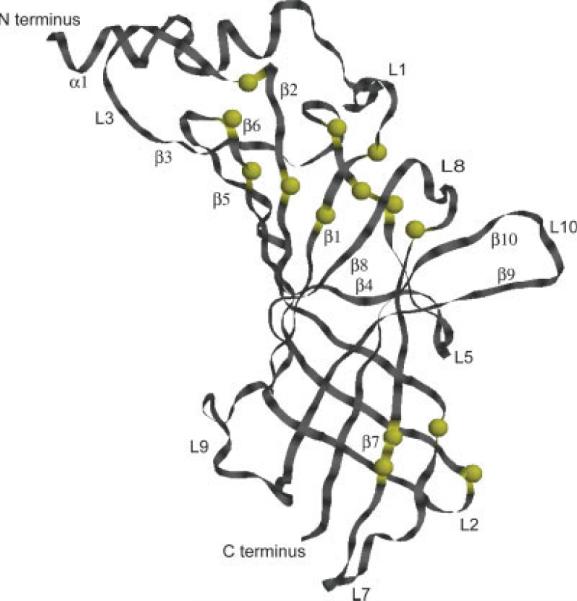
The three-dimensional structure of an α1 GlyR LBD monomer is shown as modeled using alignment in Figure 1 and MODELLER.38 Yellow spheres mark position of conserved residues. Strands and loops are labeled according to Ref. 22 nomenclature. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Ligand Docking
Strychnine antagonist docking to the putative binding pocket of the protein was performed using the Autodock 3.0.5 program.48 The protein coordinates were fixed during docking simulations, while the strychnine ligand was flexible and moved on the grid as implemented in Autodock. An initial population of 300 starting structures was used for energy optimization with a maximum number of energy evaluations set to 106. Grid spacing of 0.375 Å was used. All other parameters remained at their default values. Grid searching was performed using the Lamarckian genetic algorithm. Hundred different docking runs were performed to find the best conformation and orientation of ligand based on the binding energy.
RESULTS
Homology Modeling
The alignment between L. stagnalis AChBP, A. californica AChBP, and GlyR α1 LBD was obtained using Clustal W. The alignment was additionally manually adjusted to agree with the alignment reported by Nevin et al.31 for His in positions 107 and 109 (alignment C) as well as to satisfy all published biochemical data (collected in Ref. 29) and data obtained for nAChR through lysine scanning mutagenesis.27 Our final alignment (Fig. 1) is similar to alignments published by other groups22,31 except for the following differences. In the present paper Cys 198 is aligned with Val 183 in the L. stagnalis AChBP sequence. Also the alignment of the less conserved region 1–13 differs from previously published studies. In addition to the alignment with the sequence of L. stagnalis AChBP we have also aligned the GlyR α1 sequence to the A. californica AChBP. The alignment shows the additional conserved residues that do not exist in L. stagnalis AChBP sequence. For example, region 76–79 have conserved residues in GlyR α1 and A. californica AChBP sequences but is different in L. stagnalis AChBP sequence. Thus, we believe that using multiple homologues sequences we achieved more reliable alignment than those used previously.
Using the alignment of α1 GlyR LBD with the two AChBP proteins shown in Figure 1, a set of the 3D structural models was produced employing the Modeller's randomization algorithm of the initial structure. Predicted structures were analyzed in WHAT_IF program. However, the 3D superposition of several structures with different WHAT_IF scores showed only small differences in the β-sheet core of the protein. The major variations were observed in the geometry of the loops. In Figure 2 the proposed 3D structure of a monomer is shown with positions of conserved residues (corresponding to the alignment in Fig. 1) indicated as yellow spheres. In our alignment the conserved signature disulfide bridge (Cys 138–Cys 152 in α1 GlyR) is preserved. As expected, regions that are less well conserved include residues that define putative ligand binding pockets and subunit interfaces, as these regions would be expected to be specific to their respective receptor subfamily.
Molecular Dynamics
The potential energy of the protein during the MD simulations is shown in Figure 3(a). An initial equilibration phase of the simulations with constrained atoms (as described in the precious section) was not included in Figure 3 and the following data analysis. The MD simulations of the model pentamer in explicit water solvent resulted in stabilization of the potential energy of the total system (of which the largest change occurred during the initial 500 ps relaxation phase). The additional 4.5 ns of MD simulation brought the potential energy to a relatively stable value. At the same time minor structural changes in the protein occurred as shown in Figure 3(b). The relative structural drift or stability of the pentameric structure is measured as the root mean square deviation (RMSD) of Cα atoms from the initial structure as a function of time (we define the initial structure as the structure produced using Modeller, which corresponded closely to the AChBP structure). The RMSD in Figure 3(b) was calculated for each MD snapshot after the coordinates of the pentamer were superimposed on the coordinates of the initial structure and averaged over all five subunits. As seen in Figure 3(b) the pentamer structure deviates rapidly from the initial structure within the first nanosecond of the MD simulation. This increase in the RMSD value is due to optimization of interactions within the protein structure, as well as with water solvent. After about 1 ns the total RMSD has stabilized at 3.0–3.3 Å. Taking into account that this simulated system is a homology model (thus, is not expected to be highly accurate), we propose that this is a satisfactory structural drift.
Fig. 3.

(a) The potential energy of the LBD of α1 GlyR during the MD simulations. (b) Average RMSD of all Cα atoms of the LBD homopentamer relative to the initial structure during the MD trajectory. The error bars are given to show the variability between subunits.
The difference between the final structure of the MD refined GlyR LBD pentamer and the initial structure modeled to closely correspond to an AChBP template is assessed by looking at the RMSD of individual residues over time as compared to the initial structure. The average structural shift of our final model from the initial model is illustrated in Figure 4. In Figure 4(a) some residues in the core of the protein (colored blue) deviated very little from the initial structure, indicating that core of the protein is well represented by the AChBP template and is stable. This observation is better illustrated in Figure 4(b) in which the superposition of structures is performed using only core residues (not including loop regions). As expected, the biggest deviations from the initial structure are found in peripheral loop regions.
Fig. 4.

The GlyR LBD structure is color coded according to the RMSD per residue averaged over the last nanosecond of the trajectory (only one subunit of the pentamer is shown). Color scheme is as follows blue is <1.0 Å, green ≤3.8 Å, and red >3.8 Å. (a) Calculated relative to the initial structure. (b) Fit to the initial structure using only core residues. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The relative mobility of residues within each subunit can be obtained through the analysis of individual residue movement with respect to the average structure of the equilibrated part of the trajectory, and the calculated mobility of Cα atoms of individual residues show different degrees of residue fluctuations. In Figure 5(b) the monomer structure is colored according to the RMSD values plotted in Figure 5(a). As described in the figure legend, the deep blue color indicates the least mobile residues, residues colored red are mobile residues, and green residues exhibited intermediate average mobility. Residues in the core of a protein subunit were the least mobile in our simulations with RMSD values within 1 Å. The most mobile parts of the protein are its loops that have fluctuations in the range 1.5–3 Å. The resulting refined structure of the GlyR LBD pentamer, which corresponds to the protein solvated in water at room temperature, is shown in Figure 6(a,b).
Fig. 5.

(a) RMSF of Ca atoms relative to the average structure. (b) Flexibility of the residues calculated relative to the average structure. Blue regions have fluctuations with values less than 1 Å and green regions are less than 1.2 Å. Maximum fluctuations, shown in red, do not exceed 3.0 Å. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 6.

GlyR α1 LBD pentamer model colored by chain (a) top view. (b) Side view (only three front chains are shown). The subunits are labeled by capital letters. (c) The interface between neighboring units of the pentamer is shown with strychnine (blue) docked in a putative binding site. The loop L10 is labeled. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Ligand Docking
Flexible docking of strychnine, a strongly-binding antagonist (with Kd in the nM range), was performed as described in the Methods section. Given that agonists and antagonists bind at the interface between subunits, structure of two neighboring subunits extracted from the LBD pentamer model [described above and shown in Fig. 6(a,b)] was used for ligand docking. Ligand orientation and position were restricted to the vicinity of subunit interface in all docking simulations. Structures with the minimum binding energy generated in multiple Autodock runs, which started with randomized initial protein–ligand configurations, deviated little from each other, indicating reliable positioning of the docked ligand. The resulting docked position of the strychnine molecule is shown in Figure 6(c).
DISCUSSION AND CONCLUSIONS
The stability of the pentameric model of the LBD of GlyR in the MD simulations supports a widely accepted hypothesis that the crystal structures of the snail AChBPs serve as appropriate templates to model the ligand binding domains of nicotinicoid superfamily of ligand-gated ion channels. In this study the pentamer structure was modeled using the program Modeller. The analysis of the several generated structures using the scores produced by WHAT_IF program did not show significant variations in the central, core, part of the protein. The water-soluble loops varied in structures generated by Modeller. We performed 5 ns of MD simulations to further optimize the structure with the best WHAT_IF score and to assess dynamics and stability of the pentameric protein in a simulation with explicit water solvent. The modeled structure of the pentamer is shown in Figure 6(a,b).
Energetic and structural analysis of the MD results showed that the system initially deviated slightly from the initial model and remained stable during nearly 4 ns of the simulations. This initial relaxation in the pentamer structure can be attributed to optimization of the positions of the side chain and backbone atoms in Cornell et al. force field,47 formation of better hydrogen bonds and salt bridges, and favorable interactions with water solvent. The overall structural drift did not exceed 3.5Å. This is an encouraging result since the AChBP protein template has low sequence similarity with our target sequence. The structural drift observed in our simulations is similar to the results obtained previously for the nAChR LBD.49 The most mobile residues were observed in the loop regions of the structure, while the core was relatively less mobile. The core also deviated the least from the initial model structure (with structural drift less then 1Å in some β-strands). The observed flexibility of the loops that are proposed to be membrane-proximal [i.e. bottom residues in Fig. 5(b)] is not unexpected, since we simulated the ligand binding domain solvated in water while in the native protein these loops are at the interface with the lipid bilayer interfacial region as well as with the surface loops and transmembrane domain of the GlyR receptor.20 These interactions are expected to stabilize the loop structure and reduce their flexibility.
One important test of validity of proposed model is to check whether amino acids that were previously implicated in ligand binding in biochemical and structural studies29 are located in a relative proximity to each other (in three dimensional space) and whether the location of these residues maps out a plausible binding site for agonists and antagonists. It seems logical to assume that if LBDs of all members of the ligand-gated receptor super-family share a common mechanism for allosteric regulation of gating, then location of agonist and antagonist binding sites should be similarly positioned with respect to the overall structure. Using this approach, Grudzinska and colleagues mutated residues in homo-oligomeric α1 GlyR that corresponded to residues putatively involved in ligand binding in nicotinicoid receptors.50 In Figure 7 the positions of all residues implicated in ligand binding are mapped onto our proposed structure of the pentamer. As expected, the putative binding pocket for agonist and antagonists is located at the interface between neighboring α1 subunits (shown in Fig. 7). This putative binding site is well defined by the pocket observed at the interface of the two subunits and correlates well with all published experimental data, strongly supporting our proposed model structure. We were able to dock the antagonist strychnine within this putative binding site [see Fig. 6(c)]. In this model the antagonist is positioned such that it directly interacts with the residues whose important role in the strychnine binding have been previously deduced from experimental observations, further supporting the validity of this model structure.
Fig. 7.
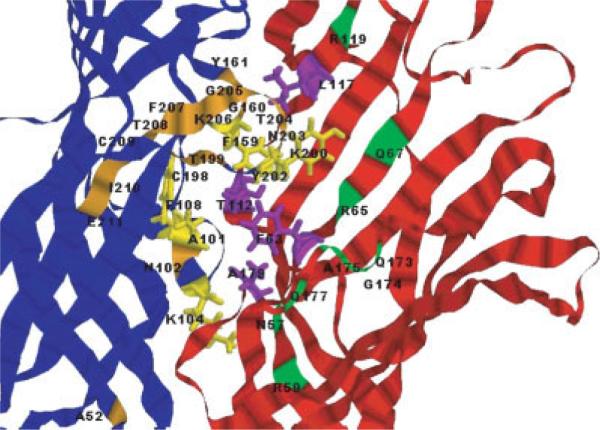
View of two neighboring subunits of the LBD of α1 GlyR. Residues implicated in ligand binding are shown in all-atom stick representations, labeled and colored according to the following scheme: in blue subunit, yellow residues are those that are closest to the putative binding site and their complements are purple residues of the red subunit. Orange and green residues that are further away from the binding site correspond to the blue and red subunits on Figure 6, respectively.
The overall stability and dynamics of the pentameric structure in simulations may be visualized by plotting the calculated distances between residues as a function of time in our simulations. Two sets of intersubunit Cα distances for Cys 41 or Lys 193 were calculated (Fig. 8). In both cases, the simulations showed that the pentameric structures are stable and do not fluctuate to any great degree. With respect to Cys 41, in our model this residue is located in the interface between two subunits, approximately in the middle of the protein height (i.e., the axis normal to the bilayer). The initial distance between the Cys 41 Cα atoms of the neighboring subunits is ~31 Å and between Cαs of the subunits positioned across the pentameric structure is ~50 Å. Average values of these distances over all five subunits varied by 3–4 Å during the MD simulations for both neighboring subunits and across the pentamer [Fig. 8(a)], which is in the range of the fluctuations in the central part of the protein. In preliminary ESR measurements, spin–spin distances between nitroxide labels presumed to be on Cys 41 of the full length wild-type GlyR (via covalent modification of its free thiol) were observed to be 31 Å in neighboring subunits and 50 Å to other subunits (Stone, Yang, Bonora, Cascio and Saxena, unpublished observation). Thus, our computational model is consistent with the experimentally-determined distances at this site.
Fig. 8.

(a) The distance between residues Cys 41 of neighboring (dashed) and across the pentamer subunits (solid). (b) Distances between Lys 193 of neighbor (dashed) subunits and across (solid) subunits.
It is clear that while overall fold and the quaternary structure of the AChBP proteins is a good low resolution template for the ligand binding domains of all ligand-gated receptors in the superfamily, the conformational state of a given LBD in a native receptor may differ significantly from the solution structure of either AChBP protein or a water-soluble construct of an LBD. One indication of how the overall structure of a pentameric LBD may differ from the AChBP protein can be inferred from a recent study by Unwin,21 in which the core structure of each subunit monomer of the AChBP pentamer template has been deformed in order to fit the low resolution cryo electron microscopy derived electron density profile of the LBD of nAChR. With respect to the AChBP template, the proposed structure of the LBD in the nAChR holoreceptor21 was deformed only at positions of central core residues, which form the β-sheet sandwich in each subunit (the corresponding strands β2, β5, β6, β7, β9, β10 in GlyR LBD shown in Fig. 2). Assuming that the general conformation and configuration of a LBD should be very similar for all members of the superfamily and that the main differences are expected to be in the loops and other peripheral regions of the proteins, we attempted to model a GlyR LBD structure in an α-subunit nAChR LBD-like conformation and assess its stability in the simulations. Presumably, such a configuration more closely represents the corresponding domain in the whole receptor rather than to the water soluble form of the AChBP proteins. We performed simulated annealing MD simulations of the GlyR LBD pentamer with its β-sandwich core residues constrained in the conformation of the α subunit of the nAChR LBD (refer to the Materials and Methods section for the details of the system set up and simulated annealing MD). The starting structure of our simulations was the stable pentamer GlyR LBD structure shown in Figure 6(a,b) and described earlier (also shown in Fig. 9 in blue). The final (after the simulated annealing) nAChR LBD like structure termed LBDdeformed is shown in Fig. 9 in red. After simulated annealing further unconstrained MD simulation was performed for 500 ps and the resulting conformation of a subunit deviated slightly from the LBDdeformed template structure. The final equilibrated “whole receptor” structure is also shown in Figure 9 with its β-sandwich core colored green (only backbone trace of the protein is shown in the figure). We termed this modeled structure LBDsa. Note that while three structures of a subunit shown in Figure 9 deviate slightly from each other, the overall structure of the pentamer did not significantly change in the simulations.
Fig. 9.
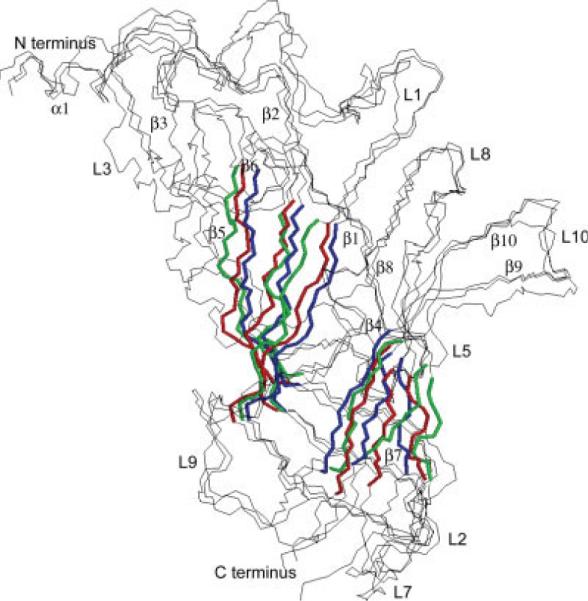
Shown are the superimposed backbone traces of a single subunit from each of the three simulated pentameric GlyR α1 systems. The view is towards the central axis from outside the pentamer. The inner and outer parts of the β-sandwich core of each model protein are colored blue, red, and green. The three structures are as follows: a model produced using the AChBP proteins as templates and equilibrated in further MD simulations (the same protein as in Fig. 6) is colored blue, a model (LBDdeformed) produced by the deformation of the first model using the nAChR α subunit backbone of the β-sandwich core residues as a template is colored red, and a resulting equilibrium configuration (LBDsa) of the second structure after simulated annealing in the absence of any constraints is colored green.
The stable LBDsa model (Fig. 9, green) differs from both templates derived from the AChBP and the α subunit of nAChR. RMS deviation between LBDsa and either template structures is 1.4 Å calculated using only β-sheet core Cα atoms. We also measured the distance between the centers of mass of the outer and inner β-sheets (notation is taken from Ref. 51). The difference between these distances for the LBDsa structure and AChBP derived structure is 2.4 Å, while for the LBDsa structure and the LBDdeformed structure it is 1.2 Å, indicating that the LBDsa structure is fairly similar to the LBDdeformed template. However, it seems that the β-sandwich core of a LBD subunit is very stable and resistant to deformation, supporting our earlier conclusion that the core of this protein is relatively inflexible.
While the deformed structure deviated from its LBD nAChR α template, a new stable configuration which resulted from the simulated annealing refinement has the same main features as its both model templates (e.g., the same content of the secondary structure elements). The results of this simulation should be considered with a certain degree of skepticism. First of all, our equilibration simulations could have been insufficiently slow to let the structure to converge into a more deformed template-like structure. However, the protocol used for these equilibration simulations has been successfully used by us to model other structures that needed to be deformed into a template configuration, such as in examination of a dimer formed from identical monomers by bringing them together in a constrained simulation (Speranskiy and Kurnikova, unpublished data). Therefore, while we can not rule out a possibility that longer equilibration under constrains will produce a better initial structure for the deformed protein, we are confident that the structure proposed in Ref. 21 is not a good template for the GlyR LBD solvated in water. However, it should be noted that in native receptors the membrane-proximal surface of the LBD is closely associated with the lipid and protein from the TM domain and connecting loops of the receptor. Thus the presence of the protein/lipid and protein/protein interface may play important role in further stabilizing the structure of the LBD, giving rise to an overall conformation slightly different from our model. Regardless, the model presented in this study appears to be valid as an initial model for the LBD of GlyR in that it is stable throughout the 5 ns of our simulation, and its structural details are in agreement with all published biochemical data.
In conclusion, the stability of the homopentameric LBD of GlyR under the rigorous conditions presented herein, coupled with the validation of the model with respect to published experimental data, provides confidence that this unique model is an excellent template for the ligand binding domain of GlyR. This model provides us with a template for developing and assessing structural studies of the receptor aimed at elucidating our understanding of ligand binding and nicotinicoid receptor allostery. Future computational studies may help further refine our model by including a membrane interface as well as with interacting surface loops and the transmembrane domain of the receptor. In addition, future studies will also analyze the dynamics of the receptor in the presence and absence of ligands, as these dynamics are responsible for gating of the channel.
ACKNOWLEDGMENTS
MD simulations reported in this work have been performed on computers at Pittsburgh Supercomputer Center sponsored by NSF under the PACI grant.
REFERENCES
- 1.Betz H. Ligand-gated ion channels in the brain: the amino acid receptor superfamily. Neuron. 1990;5:383–392. doi: 10.1016/0896-6273(90)90077-s. [DOI] [PubMed] [Google Scholar]
- 2.Yamakura T, Bertaccini E, Trudell JR, Harris RA. Anesthetics and ion channels: molecular models and sites of action. Annu Rev Pharmacol Toxicol. 2001;41:23–51. doi: 10.1146/annurev.pharmtox.41.1.23. [DOI] [PubMed] [Google Scholar]
- 3.Hemmings HC, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Beckstead MJ, Phelan R, Trudell JR, Bianchini MJ, Mihic SJ. Anesthetic and ethanol effects on spontaneously opening glycine receptor channels. J Neurochem. 2002;82:1343–1351. doi: 10.1046/j.1471-4159.2002.01086.x. [DOI] [PubMed] [Google Scholar]
- 5.Eser D, Romeo E, Baghai T, di Michele F, Schule C, Pasini A, Zwanzger P, Padberg F, Rupprecht R. Neuroactive steroids as modulators of depression and anxiety. Neuroscience. 2006;138:1041–1048. doi: 10.1016/j.neuroscience.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Belelli D, Herd M, Mitchell E, Peden D, Vardy A, Gentet L, Lambert J. Neuroactive steroids and inhibitory neurotransmission: mechanisms of action and physiological relevance. Neuro-science. 2006;138:821–829. doi: 10.1016/j.neuroscience.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 7.Li M, Lester HA. Ion channel diseases of the central nervous system. CNS Drug Rev. 2001;7:214–240. doi: 10.1111/j.1527-3458.2001.tb00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gundlach AL. Disorder of the inhibitory glycine receptor-inherited myoclonus in Poll Hereford calves. FASEB J. 1990;4:2761–2766. doi: 10.1096/fasebj.4.10.2165010. [DOI] [PubMed] [Google Scholar]
- 9.Becker CM, Schmieden V, Tarroni P, Strasser U, Betz H. Isoform-selective deficit of glycine receptors in the mouse mutant spastic. Neuron. 1992;8:283–289. doi: 10.1016/0896-6273(92)90295-o. [DOI] [PubMed] [Google Scholar]
- 10.Shiang R, Ryan SG, Zhu YZ, Hahn AF, Oconnell P, Wasmuth JJ. Mutations in the α-1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- 11.Jones-Davis DM, Macdonald RL. GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Curr Opin Pharmacol. 2003;3:12–18. doi: 10.1016/s1471-4892(02)00015-2. [DOI] [PubMed] [Google Scholar]
- 12.Wong CGT, Bottiglieri T, Snead OC. GABA, γ-hydroxybutyric acid, and neurological disease. Ann Neurol. 2003;54:S3–S12. doi: 10.1002/ana.10696. [DOI] [PubMed] [Google Scholar]
- 13.Legendre P. The glycinergic inhibitory synapse. Cell Mol Life Sci. 2001;58(5/6):760–793. doi: 10.1007/PL00000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grenningloh G, Schmieden V, Schofield PR, Seeburg PH, Siddique T, Mohandas TK, Becker CM, Betz H. α-Subunit variants of the human glycine receptor—primary structures, functional expression and chromosomal localization of the corresponding genes. EMBO J. 1990;9:771–776. doi: 10.1002/j.1460-2075.1990.tb08172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cascio M, Schoppa NE, Grodzicki RL, Sigworth FJ, Fox RO. Functional expression and purification of a homomeric human-α-1 glycine receptor in baculovirus-infected insect cells. J Biol Chem. 1993;268:22135–22142. [PubMed] [Google Scholar]
- 16.Eisele JL, Bertrand S, Galzi JL, Devillersthiery A, Changeux JP, Bertrand D. Chimeric nicotinic serotonergic receptor combines distinct ligand-binding and channel specificities. Nature. 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- 17.Grutter T, de Carvalho LP, Dufresne V, Taly A, Edelstein SJ, Changeux JP. Molecular tuning of fast gating in pentameric ligand-gated ion channels. Proc Natl Acad Sci USA. 2005;102:18207–18212. doi: 10.1073/pnas.0509024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unwin N. Nicotinic acetylcholine-receptor at 9-Angstrom resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 19.Unwin N. Acetylcholine-receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 20.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 21.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 22.Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 23.Celie PHN, Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 24.Bourne Y, Talley TT, Hansen SB, Taylor P, Marchot P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake a-neurotoxins and nicotinic receptors. EMBO J. 2005:1512–1522. doi: 10.1038/sj.emboj.7600620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Novere N, Grutter T, Changeux JP. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc Natl Acad Sci USA. 2002;99:3210–3215. doi: 10.1073/pnas.042699699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell RB, Saqi MAS, Sayle RA, Bates PA, Sternberg MJE. Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J Mol Biol. 1997;269:423–439. doi: 10.1006/jmbi.1997.1019. [DOI] [PubMed] [Google Scholar]
- 27.Sine SM, Wang HL, Bren N. Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J Biol Chem. 2002;277:29210–29223. doi: 10.1074/jbc.M203396200. [DOI] [PubMed] [Google Scholar]
- 28.Menziani MC, De Rienzo F, Cappelli A, Anzini M, De Benedetti PG. A computational model of the 5-HT3 receptor extracellular domain: search for ligand binding sites. Thermochim Acta. 2001;106(1/2):98–104. [Google Scholar]
- 29.Leite JF, Cascio M. Structure of ligand-gated ion channels: critical assessment of biochemical data supports novel topology. Mol Cell Neurosci. 2001;17:777–792. doi: 10.1006/mcne.2001.0984. [DOI] [PubMed] [Google Scholar]
- 30.Absalom NL, Lewis TM, Kaplan W, Pierce KD, Schofield PR. Role of charged residues in coupling ligand binding and channel activation in the extracellular domain of the glycine receptor. J Biol Chem. 2003;278:50151–50157. doi: 10.1074/jbc.M305357200. [DOI] [PubMed] [Google Scholar]
- 31.Nevin ST, Cromer BA, Haddrill JL, Morton CJ, Parker MW, Lynch JW. Insights into the structural basis for zinc inhibition of the glycine receptor. J Biol Chem. 2003;278:28985–28992. doi: 10.1074/jbc.M300097200. [DOI] [PubMed] [Google Scholar]
- 32.Reeves DC, Lummis SCR. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel. Mol Membr Biol. 2002;19:11–26. doi: 10.1080/09687680110110048. [DOI] [PubMed] [Google Scholar]
- 33.Henchman RH, Wang HL, Sine SM, Taylor P, McCammon JA. Asymmetric structural motions of the homomeric α7 nicotinic receptor ligand binding domain revealed by molecular dynamics simulation. Biophys J. 2003;85:3007–3018. doi: 10.1016/S0006-3495(03)74720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cromer BA, Morton CJ, Parker MW. Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–287. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- 35.Amiri S, Tai K, Beckstein O, Biggin PC, Sansom MSP. The α7 nicotinic acetylcholine receptor: molecular modelling, electrostatics, and energetics. Mol Membr Biol. 2005;22:151–162. doi: 10.1080/09687860500063340. [DOI] [PubMed] [Google Scholar]
- 36.Hansen SB, Talley TT, Radic Z, Taylor P. Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J Biol Chem. 2004;279:24197–24202. doi: 10.1074/jbc.M402452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkpatrick S, Gelatt CD, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 38.Mendieta J, Ramirez G, Gago F. Molecular dynamics simulations of the conformational changes of the glutamate receptor ligand-binding core in the presence of glutamate and kainate. Proteins: Struct Funct Genet. 2001;44:460–469. doi: 10.1002/prot.1111. [DOI] [PubMed] [Google Scholar]
- 39.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. Clustal-W—improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 42.Vriend G. What If—a molecular modeling and drug design program. J Mol Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 43.Case DA, Pearlman DA, Caldwell JW, Cheatham Iii TE, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER 7. University of California; San Francisco: 2002. [Google Scholar]
- 44.Darden T, York D, Pedersen L. Particle Mesh Ewald—An N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 45.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 46.Duke ER, Pedersen LG. PMEMD 3. University of North Carolina; Chapel Hill: 2003. [PubMed] [Google Scholar]
- 47.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 48.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 49.Henchman R, Hai-Long W, Sin SM, Taylor P, McCammon JA. Ligand-induced conformational change in the α7 nicotinic receptor. Biophys J. 2005;88:2564–2576. doi: 10.1529/biophysj.104.053934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H, Laube B. The β subunit determines the ligand binding properties of synaptic glycine receptors. Neuron. 2005;45:727–739. doi: 10.1016/j.neuron.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 51.Unwin N, Miyazawa A, Li J, Fujiyoshi Y. Activation of the nicotinic acetylcholine receptor involves a switch in conformation of the a subunits. J Mol Biol. 2002;319:1165–1176. doi: 10.1016/S0022-2836(02)00381-9. [DOI] [PubMed] [Google Scholar]