

Abstract
A decline of norepinephrine transporter (NET) level is associated with several psychiatric and neurological disorders. Therefore positron emission tomography (PET) imaging agents are greatly desired to study the NET pathway. We have developed a C-fluoropropyl analog of nisoxetine: (R)-N-methyl-3-(3′-[18F]fluoropropyl)phenoxy)-3-phenylpropanamine (18F-MFP3) as a new potential PET radiotracer for NET with the advantage of the longer half-life of fluorine-18 (110 min compared with carbon-11 (20 min). Synthesis of (R)-N-methyl-3-(3′-fluoropropyl)phenoxy)-3-phenylpropanamine (MFP3) was achieved in five steps starting from (S)-N-methyl-3-ol-3-phenylpropanamine in approx. 3–5% overall yields. In vitro binding affinity of nisoxetine and MFP3 in rat brain homogenates labeled with 3H-nisoxetine gave Ki values of 8.02 nM and 23 nM, respectively. For radiosynthesis of 18F-MFP3, fluorine-18 was incorporated into a tosylate precursor, followed by the deprotection of the N-BOC-protected amine group with a 15% decay corrected yield in 2.5 h. Reverse-phase chromatographic purification provided 18F-MFP3 in specific activities of >2000 Ci/mmol. Fluorine-18 labeled 18F-MFP3 has been produced in modest radiochemical yields and in high specific activities. Evaluation of 18F-MFP3 in animal imaging studies is in progress in order to validate this new fluorine-18 radiotracer for PET imaging of NET.
Keywords: 18F-MFP3, PET, norepinephrine transporter, nisoxetine, autoradiography
Introduction
The norepinephrine transporter (NET) present in presynaptic adrenergic neurons is responsible for the reuptake of the neurotransmitter norepinephrine (NE), regulating the synaptic NE concentration.1 High densities of NET can be found in the hippocampus, thalamus, and especially the locus coeruleus (LC), from which the NET pathway is projected.2,3 Low densities are found in the cerebellum and the striatum, which receive sparse projections from the LC.2,4 Decreased NET concentration is associated with numerous psychiatric and neurological syndromes such as major depressive disorder and Alzheimer’s disease.5–7
Currently, there are several clinically used antidepressants that affect NE levels in the brain. Antidepressants, such as venlafaxine (or effexor) and duloxetine (or cymbalta), are serotonin–norepinephrine reuptake inhibitor (SNRI), and block presynaptic reuptake of both NE and serotonin (via serotonin reuptake transporter or SERT), thereby increasing the availability of both neurotransmitters in the synapse.8 These compounds increased the levels of NE and serotonin in rat prefrontal cortex.9,10 Clinical studies have shown these drugs to be effective antidepressants.11,12 More selective NET inhibitors (norepinephrine reuptake inhibitor (NRI)) such as reboxetine have been reported with relatively few side effects.13 Other compounds such as mirtazapine is an α-2 adrenoreceptor antagonist effective in major depression and has been identified as a noradrenergic and serotonergic antidepressant.14,15
Many positron emission tomography (PET) and single photon emission computed tomography (SPECT) tracers radiolabeled with carbon-11 (20.4 min half-life), fluorine-18 (109.8 min half-life), and iodine-123 (13 h half-life) have been reported (Figure 1, 1–9). Radiolabeled 11C-nisoxetine (Figure 1, 1) demonstrated modest specific NET binding, with findings of 60–75% nonspecific binding.16 The compound 11C-MeNER (Figure 1, 4, IC50 = 2.5 nM) is an O-methyl analog of reboxetine (IC50 = 8.2 nM) and showed a hypothalamus to striatum ratio of 2.5.17,18 Another methyl analog of reboxetine, 11C-(S,S)-MRB, showed a ratio of approximately 1.5–1.6 between thalamus and striatum.19 The fluorine-18 radiolabeled analog of reboxetine 18F-FRB (Figure 1, 6) displayed a thalamus to reference region ratio of 1.3–1.6.20 Talopram (1,3-dihydro-N-3,3-trimethyl-1-phenylbezo[c]furan-1-propanamine, 8) and talsupram (1,3-dihy-dro-N-3,3-trimethyl-1-phenylbezo[c]thiophene-1-propanamine, 9 ) exhibited high affinity and selectivity for the human NET in vitro. However, neither compound was able to enter the central nervous system in adequate amounts to be used in PET studies.21
Figure 1.

Radiolabeled imaging agents for NET. (a) Nisoxetine analogs; (b) reboxetine analogs; (c): other backbones.
Figure 4.

Specific Binding of 3H-Nisoxetine and MFP-3. Increase concentration of the drug, decrease binding affinity. MFP-3: IC50 = 2.30 × 10−8 M. 3H-Nisoxetine: IC50 = 8.02 × 10−9 M.
Radioiodinated MIPP has high affinity to NET (Ki = 1.33 nM) compared with nisoxetine (Ki = 7.35 nM) but also has some affinity for SERT.22–24 The compound 125I-(S,S)-IPBM is a derivative of reboxetine that is iodinated at position 2 of the phenoxy ring with Ki value of 4.2 nM. Ex vivo experiments of 125I-IPBM showed high levels of radioactivity in the LC and the anteroventricular thalamic nucleus and regional distribution correlated well with reported 3H-nisoxetine binding.25 The same chemical structure 7, radiolabeled with iodine-123 referred as 123I-INER (Ki = 0.84 nM) was found to have comparable properties reported for MIPP.26 This group has recently reported additional carbon-11 and fluorine-18 derivatives of reboxetine, with promising properties for carbon-11 labeled analogs.27
Our goal was to develop a fluorine-18 labeled analog of nisoxetine for the following reasons: (1) Nisoxetine has a high affinity for NET and has been shown to bind to the transporter sites selectively4; (2) As the clearance of the reported agents, including 11C-nisoxetine, from nonspecific binding regions is slow, longer imaging times with fluorine-18 will be needed; (3). Concentration of NET in the brain is low, thus high specific activity of the radiotracer would be desired28; (4) Pharmacological challenge experiments may be carried out with greater ease with the longer half-life; (5) Fluorine-18 is suitable for high-resolution PET studies; (6) Fluorine-18 is clinically more amenable because centers without a cyclotron may be able to use these agents; and (7) As defluorination has been an issue with O-fluoroalkyl derivatives of reboxetine,20 a C-fluoroalkyl derivative may be more stable to defluorination. We report here synthesis of a fluorine-18 radiolabeled derivative (methoxy group in nisoxetine was replaced with a fluoropropyl group) to provide (R)-N-methyl-3-(3′-fluoropropyl)phenoxy)-3-phenylpro-panamine (Figure 2, 10a; MFP3) as a potential NET imaging agent for PET when radiolabeled to give (R)-N-methyl-3-(3′-[18F]fluoropropyl)phenoxy)-3-phenylpropanamine (Figure 2, 10b; 18F-MFP3).
Figure 2.
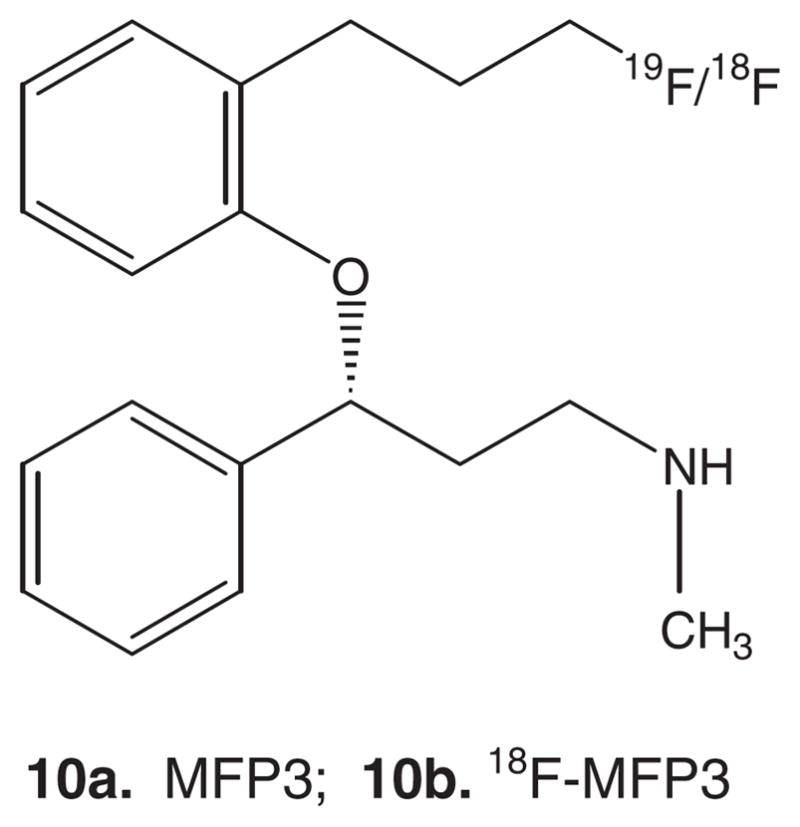
Chemical structure of (R)-N-Methyl-3-(3′-fluoropropyl)phenoxy)-3-phenyl-propanamine (MFP3, 10a) and (R)-N-Methyl-3-(18F-3′-fluoropropyl)phenoxy)-3-phenylpropanamine (18F-MFP3 10b).
Results and discussion
Our goal has been to develop a fluorine-18 labeled derivative for imaging NET in the living brain. Previous efforts have focused on carbon-11 and fluorine-18 derivatives of nisoxetine and reboxetine. These derivatives were radiolabeled by either N-methylation or O-methylation using 11C-methyl iodide or by O-fluoroalkylation using 18F-fluoroalkyl groups. Our approach in this work was to develop a C-fluoropropyl derivative, since our previous work on dopamine and nicotine receptor imaging agents has demonstrated their in vivo stability, particularly towards defluorination.29,30 For this purpose, we undertook the synthesis of MFP3, Figure 2, 10a, which is an analog of nisoxetine (where the methoxy group in nisoxetine, Figure 1, 1 is replaced by the fluoropropyl group) or MIPP (where the iodine in MIPP, Figure 1, 2 is replaced by the fluoropropyl group). As nisoxetine appears to be tolerant to the replacement of the methoxy group (as in tomoxetine, Figure 1, 3) and reboxetine appears to retain affinity upon incorporation of two carbon chain at the position (Figure 1, 5, 6), we envisaged that MFP3 would maintain binding characteristics to NET sites.
Figure 3.

Chemical synthesis of MFP3: (a) Di-tert-butyl dicarbonate, THF, 0–5°C, 90 min; (b) 13, Ph3P, DIAD, THF, 0–5°C → 22°C, 24 h; (c) (i) BH3-THF, 0–5°C → 22°C, 24 h, (ii) H2O2, NaOH, 22°C, 12 h; (d) p-TsCl, Et3N, CH2Cl2, 22°C, 24 h; (e) TBAF, THF, 68 °C, 1.5 h.
Figure 5.

(a) Radiosynthesis of 18F-MFP3 (b) HPLC chromatogram of 18F-MFP3 purification (Reverse phase C18, CH3CN:H2O 0.25%Et3N, 68%:32%; flow rate = 2.5 mL/min). The retention time of 18F-MFP3 was 18 min.
The synthesis of MFP3 and the tosylate 16 were carried out using general procedures for this class of compounds (Figure 3). As the R-isomer of nisoxetine is more pharmacologically active, we started out with (S)-N-methyl-3-ol-3-phenylpropanamine 11 reported previously.31 Using previously synthesized (S)-N-methyl-3-ol-3-phenylpropanamine31 the secondary amine was protected using the tert-butyloxycarbonyl group in modest yields.32 This N-BOC-protected secondary alcohol was coupled with 2-allylphenol using the Mitsunobu reaction. The yields were in the range of approximately 40% for this reaction which was carried out in the presence of diisopropyl azodicarboxylate (DIAD). Similar yields were observed with other phenol–alcohol coupling reactions.33 The reaction provided (R)-N-tert-butyloxycarbonyl-N-methyl-3-(2′-allyl)phenoxy)-3-phenylpropanamine 14, since an inversion of configuration occurs in the Mitsunobu reaction.34 Hydroboration of the allyl group followed by peroxide treatment under basic conditions provided the propanol derivative 15 in modest yields. The alcohol was treated with p-toluenesulfonyl chloride in the presence of triethylamine, using previously described methods.30 Yields of this reaction were surprisingly poor. It is unclear whether steric effects contributed to the low yields of the tosylate. Nucleophilic substitution of the tosylate with TBAF followed by deprotection of the N-BOC protecting group with trifluoroacetic acid (TFA) provided MFP3, 10a in modest yields.
We performed in vitro experiments on rat brain homogenates in order to measure the binding affinity of MFP3. Competition studies were performed using different concentrations (between 1 nM and 50 μM) of the NET-selective agent nisoxetine and the cold compound MFP3 (Figure 4). The reported Ki value for nisoxetine is 1–1.5 nM25,35 and is lower compared with our findings of 8.02 nM for nisoxetine. The Ki of 23 nM for MFP3 suggests that MFP3 is about three times weaker in binding compared with nisoxetine. Incorporation of the 3-carbon long propyl group could potentially result in some steric effects in the binding site resulting in a lower affinity. Similar lowering of affinity was reported in fluoroalkyl derivatives of reboxetine.27
Radiosynthesis of 18F-MFP3 involved two steps as described in Figure 5(a). The first step involves incorporation of fluorine-18 into (R)-N-tert-butyloxycarbonyl-N-methyl-3-(3′-tosyloxypropyl)-phenoxy)-3-phenylpropanamine 16, and the second step involved deprotection of the N-BOC-protected amine group. The radiolabeling step by 18F-fluorine was carried out using the nucleophilic substitution reaction in the presence of Kyptofix and K2CO3 in acetonitrile at 95°C for 30 min. This reaction provided high, reproducible radiochemical yields as previously observed in the case of propyl tosylates.29,30 After radiolabeling, product was taken directly to the second step of deprotection. Deprotection of the N-BOC group was accomplished with a strong acid, TFA, similar to the procedures we have used for the nicotinic receptor radioligands.33 After the reaction, the neutralized mixture was separated by reverse-phase high-performance liquid chromatography (HPLC) purification. It must be noted that at a radioactive peak at approximately 7 min (Figure 4(b)) there was a major volatile side-product. This may be a result of the breakdown of the ether linkage under the strong acidic conditions of deprotection. To minimize this, milder conditions will have to be explored. The retention time of 18F-MFP3 was 18 min (Figure 5(b)). The total synthesis time was 2.5 h with radiochemical yields of 15–20%, decay corrected. The specific activities of 18F-MFP3 were 1500–2000 Ci/mmol.
In vitro and in vivo binding studies are currently underway with 18F-MFP3 in order to evaluate its ability to bind to NET and if this binding can be displaced by nisoxetine.
Materials and methods
General
All chemicals and solvents were of analytical or HPLC grade from Aldrich Chemical Co. and Fisher Scientific. Nisoxetine was purchased from Sigma Biochemicals. Electrospray mass spectra (MS) were obtained on a Model 7250 mass spectrometer (Micromass LCT) and proton NMR spectra were recorded on a Bruker OMEGA 500 MHz spectrometer. Analytical thin layer chromatography (TLC) was carried out on silica-coated plates (Baker-Flex, Phillipsburg, NJ). Chromatographic separations were carried out on preparative TLC (silica gel GF 20 × 20 cm 2000 μm thick; Alltech Assoc. Inc., Deerfield, IL) or silica gel flash column or semi-preparative reverse-phase columns using the Gilson HPLC systems. High specific activity aqueous 18F-fluoride was produced in the MC-17 cyclotron or the CTI RDS-112 cyclotron using the 18O (p,n)18F reaction. The high specific activity 18F-fluoride was used in subsequent reactions which were carried out in automated radiosynthesis units (either a chemistry processing control unit (CPCU) or a nuclear interface fluorine-18 module). Fluorine-18 radioactivity was counted in a Capintec dose calibrator and radioactive thin layer chromatographs were obtained by scanning in a Bioscan system 200 Imaging scanner (Bioscan, Inc., Washington, DC). Tritium was assayed by using a Packard Tri-Carb Liquid scintillation counter with 65% efficiency. All animal studies were approved by the Institutional Animal Care and Use Committee of University of California-Irvine.
Chemistry
(S)-N-tert-butyloxycarbonyl-N-methyl-3-ol-3-phenylpropanamine (12)
The synthesis of (S)-N-methyl-3-ol-3-phenylpropanamine (11) was previously described.31 The secondary amine 11 (0.62 g; 3.74 mmol) was dissolved in tetrahydrofuran (8 mL) and cooled to 0–5°C. Dissolved di-tert-butyl dicarbonate (1.33 g; 6.1 mmol) in tetrahydrofuran (8 mL) was further added to this cold (0–5°C) mixture and allowed to react for 90 min. The reaction was then permitted to reach ambient temperature and allowed to react overnight. Tetrahydrofuran was removed and 20 mL of dichloromethane was added and washed with saturated NaHCO3 (2 × 20 mL) and water (1 × 20 mL). The organic layer was dried (magnesium sulfate) and filtered while the crude product was purified on a silica column (30% ethyl acetate in hexane) to provide 0.56 g of pure (S)-N-BOC-N-methyl-3-ol-3-phenylpropanamine, 12 (57% yield) comparable to previous findings.32 MS, m/z, 266 (9%, [M+H]+), 288 (100%, [M+Na]+). NMR (500 MHz; CDCl3; δ ppm): 7.30–7.36 (m, 5H, Ar); 4.59 (br, 1H, CH); 2.87 (s, 3H, NCH3); 2.81 (m, 2H, CH2N); 1.68 (m, 2H, CH2 β to N); 1.42 (s, 9H, t-butyl).
(R)-N-tert-butyloxycarbonyl-N-methyl-3-(2′-allyl)phenoxy)-3-phenylpropanamine(14)
To a solution of (S)-N-BOC-N-methyl-3-ol-3-phenylpropanamine 12 (0.25 g, 0.94 mmol), 2-allylphenol 13 (0.14 g, 1 mmol) and triphenylphosphine (0.27 g, 1 mmol) in THF (10 mL), stirred under nitrogen and cooled at 0°C, DIAD (0.22 mL, 1.1 mmol) in THF (2 mL) was added. The reaction mixture was stirred at room temperature for 24 h and evaporated until dried. The residue was purified by preparative TLC (30% EtOAc in hexane) to give pure product (R)-N-BOC-N-methyl-3-(2′-allyl)phenoxy)-3-phenylpropanamine 14 0.15 g (42% yield). MS, m/z, 382 (9%, [M+H]+), 404 (100%, [M+Na]+). NMR (500 MHz; CDCl3; δ ppm): 7.65–7.69 (m, 3H, Ar); 7.56–7.53 (m, 1H, Ar); 7.48–7.45 (m, 3H, Ar); 7.12–7.07 (m, 1H); 6.86–6.83 (m, 1H); 5.12 (m, 1H), 4.98 (m, 2H), 4.12 (m, 1H, CH); 2.83 (s, 3H, NCH3); 2.81 (m, 2H, CH2N); 1.74 (m, 2H, CH2 β to N); 1.26 (s, 9H, t-butyl).
(R)-N-tert-butyloxycarbonyl-N-methyl-3-(3′-propanol)phenoxy)-3-phenylpropanamine (15)
The olefin, (R)-N-BOC-N-methyl-3-(2′-allyl)phenoxy)-3-phenylpropanamine 14 (0.11 g, 0.28 mmol) in THF (5 mL) was treated with borane-tetrahydrofuran complex (0.58 mL, 0.3 mmol) drop wise at 0°C stirred under nitrogen atmosphere. The reaction mixture was stirred at room temperature over night and then was treated with NaOH (3N, 5 mL) followed by the addition of 30% H2O2 (200 μL) at 0°C. This mixture was stirred at room temperature for 12 h and dried. The residue was poured into saturated ammonium chloride solution and extracted with ethyl acetate. The organic layer was dried over MgSO4 and the solvent evaporated. The residue was purified by silica column chromatography (30% ethyl acetate in hexane) to furnish alcohol, (R)-N-BOC-N-methyl-3-(3′-propanol)phenoxy)-3-phenylpropanamine 15 (55 mg, 49% yield). MS, m/z 400 (8%, [M+H]+), 422 (100%, [M+Na]+). NMR (500 MHz; CDCl3; δ ppm) 7.71–6.75 (m, 9H, Ar); 3.40 (m, 2H, CH2–OH); 2.83 (s, 3H, NCH3); 2.35 (m, 2H, CH2N); 1.66 (m, 2H, CH2 β to N); 1.30 (s, 9H, t-butyl).
(R)-N-tert-butyloxycarbonyl-N-methyl-3-(3′-tosyloxypropyl)phenoxy)-3-phenylpropanamine (16)
The alcohol, (R)-N-BOC-N-methyl-3-(3′-propanol)phenoxy)-3-phenylpropanamine 15 (50 mg, 0.13 mmol) was reacted with p-toluenesulfonyl chloride (25 mg, 0.13 mmol) in the presence of Et3N (60 μL) in CH2Cl2 for 24 h at room temperature. Solvent was removed by rotary evaporation. Dichloromethane was added and washed with saturated NaHCO3 and water. The organic layer was removed, dried with MgSO4, and filtered to give (R)-N-BOC-N-methyl-3-(3′-tosyloxypropyl)phenoxy)-3-phenylpropanamine, 16 which was purified by silica column chromatography using 5–30% ethyl acetate in hexane to provide 14 mg of pure product (19% yield). MS, m/z, 554 (58%. [M+H]+), 576 (100%. [M+ Na]+). NMR (500 MHz; CDCl3; δ ppm) 7.81–7.78 (m, 3H, Ar); 7.75–7.70 (m, 1H, Ar); 7.36–7.31 (dd, 4H, tosyl ArH-2,3,5,6); 7.30–7.28 (m, 3H, Ar); 6.96–6.94 (m, 1H); 6.74–6.71 (m, 1H); 4.10 (m, 2H, CH2–OSO2); 3.95 (m, 1H, CH) 2.81 (s, 3H, NCH3); 2.47 (m, 2H, CH2N); 2.44 (s, 3H, tosyl CH3); 1.65 (m, 2H, CH2 β to N); 1.32 (s, 9H, t-butyl).
(R)-N-methyl-3-(3′-fluoropropyl)phenoxy)-3-phenylpropanamine (10a)
The tosylate, (R)-N-BOC-N-methyl-3-(3′-tosyloxypropyl)phenoxy)-3-phenylpropanamine 16 (20 mg, 36.1 μmol) in THF (0.5 mL) was treated with a solution of tetrabutylammonium fluoride TBAF. THF (1.0 M, 54 μL). The solution was heated at 68°C for 1.5 h. The reaction mixture was cooled to room temperature and dried. The residue was titrated with ether (3 × 5 mL) three times until none of the product showed in the TLC. The ether solution was concentrated to dryness. The residue was purified by preparative TLC (1:1 hexane and ethyl acetate) to provide (R)-N-BOC-N-methyl-3-(3′-fluoropropyl)phenoxy)-3-phenylpropanamine (7.2 mg, 49% yield).
Deprotection of (R)-N-BOC-N-methyl-3-(3′-fluoropropyl)phenoxy)-3-phenylpropanamine was carried out by treatment with TFA. The N-BOC derivative (7 mg, 17.9 μmol) was dissolved in dichloromethane (1 mL) and TFA (0.1 mL) was added at ambient temperatures. The reaction solution was stirred at ambient temperatures for 24 h and then dried. The resulting residue was neutralized with saturated NaHCO3 and its product was extracted into ether. The ether layer was dried (MgSO4), filtered, and the crude product was purified on preparative TLC to provide (R)-N-methyl-3-(3′-fluoropropyl)phenoxy)-3-phenylpropanamine 10a (1.5 mg, 28% yield). MS, m/z, 302 (100%. [M+H]+). NMR (500 MHz; CDCl3; δ ppm) 7.32–6.65 (m, 9H); 4.52 (dt, 2H, CH2F); 4.2 (m, 1H, CH); 2.88 (s, 3H, NCH3); 2.6 (br, 2H, CH2N); 2.2 (br, 2H, benzylic CH2); 1.59 (br, 2H, CH2 β to N); 1.35 (br, 2H, CH2).
In vitro binding affinity
Using prior published methods,25 rat cerebral cortex from male Sprague-Dawley rats was dissected and homogenized using Tekmar tissumizer (15 s, half maxima speed) in buffer A (containing 50 mM Tris-HCL, 120 nM NaCl, and 5 nM KCl at a pH of 7.4) using 1:100 (wt:vol). This homogenized mixture was then centrifuged at 28,000g for 10 min at 4°C. The procedure was repeated twice with the final pellet suspended in buffer B (containing 50 mM Tris-HCl, 300 mM NaCl, and 5 mM KCl at a pH of 7.4).
Using 3H-nisoxetine (nisoxetine hydrochloride N-methyl 3H, 85 Ci/mmol, Perkin-Elmer, Boston, MA) competition assays were performed using 100 μL of tissue, 50 μL of 3H-nisoxetine (1.0 nM) and 25 μL of various drug concentrations in buffer B, with the final volume made up to 500 μL per tube using buffer B. Incubation was carried out at 4°C for 4 h. The incubation was terminated by rapid vacuum filtration through Whatman GF/C filter paper (presoaked in 0.1% polyethyleneimine for 60 min) using Brandel tissue harvester. The filters were rapidly washed three times (4 mL each) with cold buffer B and the filters were suspended in 10 mL of Bio-Safe II scintillation fluid and counted for 10 min in the scintillation counter. The experiments were done in duplicates. Data was analyzed using following procedure: (1) the nonspecific binding of 3H-nisoxetine (measured using 10 μM nisoxetine) was subtracted for all samples; (2) the specific binding was normalized to 100% (no competitive ligand); and (3) the binding isotherms were fit to the Hill equation (KELL BioSoft software (v 6), Cambridge, UK) to provide inhibitor concentration (IC50), which is the inflection point of the isotherm where 50% of the 3H-nisoxetine binding (Figure 4). The Ki was calculated by the Cheng-Prussof equation and using the value of KD as 1 nM for 3H-nisoxetine.35,36
Radiosynthesis
The radiosynthesis of 18F-MFP3 was carried out using an automated synthesis procedure using a tosylate precursor which employs a CPCU (Figure 5(a)). Fluorine-18 in from a MC-17 cyclotron was passed through a QMA-light sep-pak (Waters Corp. Milford, MA), preconditioned with 3 mL of K2CO3, 140 mg/mL, followed by 3 mL of anhydrous acetonitrile. The fluorine-18 trapped in the QMA-light sep-pak was then eluted (using nitrogen gas) with 1 mL Kryptofix222/K2CO3 (360 mg/75 mg in 1 mL of water and 24 mL of acetonitrile) and transferred to the CPCU reaction vessel. The ‘SYNTH1’ program in the CPCU was used for the synthesis. This involved initial drying of the 18 F-fluoride, Kryptofix, and K2CO3 mixture at 120°C for 10 min. Subsequently, acetonitrile (2 mL) from CPCU reagent vial 2 was added and evaporated at 120°C for 7 min to ensure dryness of the 18F-fluoride mixture. Following this, the precursor, (R)-N-tert-butyloxycarbonyl-N-methyl-3-(3′-tosyloxypropyl)phenoxy)-3-phenylpropanamine, 16 (1–2 mg in 0.5 mL of anhydrous acetonitrile continued in CPCU reagent vial 3) was added and the reaction went for 15–30 min at 96°C. Subsequent to the reaction, CH2Cl2 (7 mL contained in CPCU reagent vial 4) was added to the mixture and the CH2Cl2 contents were passed through a neutral alumina sep-pak (prewashed with CH2Cl2 in order to remove any unreacted 18F-fluoride). The collected CH2Cl2 solution coming out of the CPCU now contained 18F-N-BOC-MFP3. The solvent was removed and the residue was taken in dichloromethane (1 mL) and trifluroacetic acid (0.2 mL). The solution was heated at 98°C for 30 min and evaporated until dried. The residue was neutralized with 10% NaHCO3 solution until pH of 7.0 was obtained. Semipreparative HPLC purification was performed using an Alltech C18 column (10 μm, 250 × 10 mm) and UV detector (254 nm), mobile phase 60% acetonitrile: 40% water containing 0.1% triethylamine with a flow rate of 2.5 mL/min. The retention time of 18F-MFP3 was found to be 18 min (Figure 5(b), HPLC chromatogram). The 18F-MFP3 fraction was collected into a flask and the solvent was removed in vacuo using a rotary evaporator. The radiosynthesis was accomplished in 2.5 h with an overall radiochemical yield of approx. 15% decay corrected. Specific activity was measured to be >2000 Ci/mmol.
Conclusion
The new NET radioligand, (R)-N-methyl-3-(18F-3′-fluoropropyl)-phenoxy)-3-phenylpropanamine (18F-MFP3) was prepared successfully. The compound showed moderate affinity for the NET pathway in rat brain homogenates. Potential use of 18F-MFP3 in the study of the NET pathway as well as in evaluating SNRI/NRI antidepressants remains to be determined.
Acknowledgments
This work was supported by NIH RO1 EB006110 from the National Institute of Biomedical Imaging and Bioengineering.
References
- 1.Blakely RD, De Felice LJ, Hartzell HC. J Exp Biol. 1994;196:263. doi: 10.1242/jeb.196.1.263. [DOI] [PubMed] [Google Scholar]
- 2.Charnay Y, Leger L, Vallet PG, Hof PR, Jovet M, Bouras C. Neuroscience. 1995;23:259. doi: 10.1016/0306-4522(95)00257-j. [DOI] [PubMed] [Google Scholar]
- 3.Ordway GA, Stockmeier CA, Cason GW, Klimek V. Neuroscience. 1997;17:1710. doi: 10.1523/JNEUROSCI.17-05-01710.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tejani-Butt SM. J Pharmacol Exp Ther. 1992;260:427. [PubMed] [Google Scholar]
- 5.Klimek V, Stockmeier C, Overholser J, Meltzer HY, Kalka S, Dilley G, Ordway GA. J Neurosci. 1997;17:8451. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marien MR, Colpaert FC, Rosenquist AC. Brain Res Brain Res Rev. 2004;45:38. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Tejani-Butt SM, Yang J, Zaffar H. Brain Res. 1993;63:147. doi: 10.1016/0006-8993(93)91201-3. [DOI] [PubMed] [Google Scholar]
- 8.Holliday SM, Benfield P. Drugs. 1995;49:280. doi: 10.2165/00003495-199549020-00010. [DOI] [PubMed] [Google Scholar]
- 9.Bymaster FP, Beedle EE, Findlay J, Gallagher PT, Krushinski JH, Mitchell S, Robertson DW, Thompson DC, Wallace L, Wong DT. Bioorg Med Chem Lett. 2003;13:4477. doi: 10.1016/j.bmcl.2003.08.079. [DOI] [PubMed] [Google Scholar]
- 10.Wong DT, Bymaster FP, Mayle DA, Reid LR, Krushinski JH, Robertson DW. Neuropsychopharmacology. 1993;8:23. doi: 10.1038/npp.1993.4. [DOI] [PubMed] [Google Scholar]
- 11.Morton WA, Sonne SC, Verga MA. Ann Pharmacother. 1995;29:387. doi: 10.1177/106002809502900410. [DOI] [PubMed] [Google Scholar]
- 12.Detke MJ, Lu Y, Goldstein DJ, Hayes JR, Demitrack MA. J Clin Psychiatry. 2002;63:308. doi: 10.4088/jcp.v63n0407. [DOI] [PubMed] [Google Scholar]
- 13.Page ME. CNS Drug Rev. 2003;9:327. doi: 10.1111/j.1527-3458.2003.tb00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bengtsson JH, Kele J, Johansson JM, Hjorth S. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:406. doi: 10.1007/s002100000294. [DOI] [PubMed] [Google Scholar]
- 15.Haddjeri N, Blier P, De Montigny C. J Pharmacol Exp Ther. 1996;277:861. [PubMed] [Google Scholar]
- 16.Haka MS, Kilbourn MR. Nucl Med Biol. 1989;16:771. doi: 10.1016/0883-2897(89)90160-8. [DOI] [PubMed] [Google Scholar]
- 17.Schou M, Halldin C, Sovago J, Pike VW, Gulyas B, Mozley PD, Johnson DP, Hall H, Innis RB, Farde L. Nucl Med Biol. 2003;30:707. doi: 10.1016/s0969-8051(03)00079-9. [DOI] [PubMed] [Google Scholar]
- 18.Wilson AA, Johnson DP, Mozley D, Hussey D, Ginovart N, Nobrega J, Garcia A, Meyer J, Houle S. Nucl Med Biol. 2003;30:85. doi: 10.1016/s0969-8051(02)00420-1. [DOI] [PubMed] [Google Scholar]
- 19.Ding YS, Lin KS, Garza V, Carter P, Alexoff D, Logan J, Shea C, Xu Y, King P. Synapse. 2003;50:345. doi: 10.1002/syn.10281. [DOI] [PubMed] [Google Scholar]
- 20.Ding YS, Lin KS, Logan J, Benveniste H, Carter P. J Neurochem. 2005;94:337. doi: 10.1111/j.1471-4159.2005.03202.x. [DOI] [PubMed] [Google Scholar]
- 21.McConathy J, Owens MJ, Kilts CD, Malveaux EJ, Camp VM, Votaw JR, Nemeroff CB, Goodman MM. Nucl Med Biol. 2003;31:705. doi: 10.1016/j.nucmedbio.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Kiyono Y, Kanegawa N, Kawashima H, Fujiwara H, Iida Y, Nishimura H, Saji H. Nucl Med Biol. 2003;30:697. doi: 10.1016/s0969-8051(03)00085-4. [DOI] [PubMed] [Google Scholar]
- 23.Kiyono Y, Kanegawa N, Kawashima H, Kitamura Y, Iida Y, Saji H. Nucl Med Biol. 2004;53:57. doi: 10.1016/j.nucmedbio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Kung MP, Choi SR, Hou C, Zhuang ZP, Foulon C, Kung HF. Nucl Med Biol. 2004;31:533. doi: 10.1016/j.nucmedbio.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Kanegawa N, Kiyono Y, Kimura H, Sugita T, Kajiyama S, Kawashima H, Ueda M, Kuge Y, Saji H. Eur J Nucl Med Mol Imaging. 2006;33:639. doi: 10.1007/s00259-005-0017-y. [DOI] [PubMed] [Google Scholar]
- 26.Tamagnan GD, Brenner E, Alagille D, Haile C, Cosgrove K, Koren A, Early M, Baldwin RM, Tarazi F, Baldessarini RJ, Jarkas N, Goodman MM, Staley JK, Seibyl JP. Bioorg Med Chem Lett. 2007;17:533. doi: 10.1016/j.bmcl.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng F, Mun J, Jarkas N, Stehouwer JS, Voll RJ, Tamagnan GD, Howell L, Votaw JR, Kilts CD, Nemeroff CB, Goodman MM. J Med Chem. 2009;52:62. doi: 10.1021/jm800817h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eckelman WC, Kilbourn MR, Mathis CA. Nucl Med Biol. 2006;33:449. doi: 10.1016/j.nucmedbio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Chattopadhyay S, Xue B, Pichika R, Collins D, Bagnera R, Leslie FM, Christian BT, Shi B, Narayanan TK, Potkin SG, Mukherjee J. J Nucl Med. 2005;46:130. [PubMed] [Google Scholar]
- 30.Mukherjee J, Yang ZY, Das MK, Brown T. Nucl Med Biol. 1995;22:283. doi: 10.1016/0969-8051(94)00117-3. [DOI] [PubMed] [Google Scholar]
- 31.Das MK, Mukherjee J. Appl Radiat Isot. 1993;44:835. [Google Scholar]
- 32.Kumar A, Ner DH, Dike SY. Tetrahedron Lett. 1991;32:1901. [Google Scholar]
- 33.Pichika R, Easwaramoorthy B, Collins D, Christian BT, Shi B, Narayanan TK, Potkin SG, Mukherjee J. Nucl Med Biol. 2006;33:295. doi: 10.1016/j.nucmedbio.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Hughes DL. Organic Reactions. Vol. 42. Wiley; New York: 1992. p. 335. [Google Scholar]
- 35.Cheetham SC, Viggers JA, Bulter SA, Prow MR, Heal DJ. Neuropharmocology. 1996;35:63. doi: 10.1016/0028-3908(95)00134-4. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Y, Prusoff WH. Biochem Pharmocol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]