

Abstract
Background
Invasive fungal infections cause considerable morbidity and mortality in neutropenic patients. White blood cell transfusions are a promising treatment for such infections, but technical barriers have prevented their widespread use.
Methods
To recapitulate white blood cell transfusions, we are developing a cell-based immunotherapy using a phagocytic cell line, HL-60. We sought to stably transfect HL-60 cells with a suicide trap (herpes simplex virus thymidine kinase), to enable purging of the cells when desired, and a bioluminescence marker, to track the cells in vivo in mice.
Results
Transfection was stable despite 20 months of continuous culture or storage in liquid nitrogen. Activation of these transfected cells with retinoic acid and dimethyl sulfamethoxazole enhanced their microbicidal effects. Activated transfected killer (ATAK) cells were completely eliminated after exposure to ganciclovir, confirming function of the suicide trap. ATAK cells improved the survival of neutropenic mice with lethal disseminated candidiasis and inhalational aspergillosis. Bioluminescence and histopathologic analysis confirmed that the cells were purged from surviving mice after ganciclovir treatment. Comprehensive necropsy, histopathology, and metabolomic analysis revealed no toxicity of the cells.
Conclusions
These results lay the groundwork for continued translational development of this promising, novel technology for the treatment of refractory infections in neutropenic hosts.
Neutropenia is a major risk factor for invasive candidiasis [1–3] and aspergillosis [1, 4–7]. The primary predictor of survival of neutropenic patients with these infections is the duration of neutropenia [8–10], suggesting that exogenous replacement of phagocytes could be an effective treatment [11]. Although neutrophil transfusions have shown promising results [12, 13], daunting technical difficulties have prevented their general availability. For example, harvesting a sufficient number of neutrophils to mediate a protective effect (≥1 × 1011 neutrophils/day in infected patients) [14– 16] is difficult to achieve [11, 17]. Also, ex vivo neutrophils undergo rapid apoptosis, and they very quickly lose their ability to chemotax and kill microorganisms [17]. This loss of microbicidal activity is particularly severe for killing fungal pathogens, such as Candida organisms, compared with smaller bacterial organisms [18]. Therefore, despite the promising results of neutrophil transfusion at major transplant centers [19], they remain experimental.
To garner the therapeutic benefit of neutrophil transfusions but avoid the aforementioned technical obstacles, we are developing an immortal, phagocytic cell line to provide protection to neutropenic mice until their own phagocyte counts are restored. We have found that HL-60 cells, a human phagocytic cell line, can be activated by exposure to a combination of retinoic acid (RA) and dimethyl sulfamethoxazole (DMSO), which results in differentiation of the cells toward a neutrophil phenotype [20, 21]. This activation enhances the microbicidal capacity of the cells and diminishes their replication. These cells markedly improved the survival of neutropenic mice with disseminated candidiasis.
Continued preclinical development of this strategy required establishment of redundant protective mechanisms to ensure its safety as it moves toward clinical testing. To provide a critical layer of safety, we sought to stably transfect the cells with a suicide trap to enable the cells to be purged from infected neutropenic hosts when desired. Furthermore, we sought to use a luminescence marker to enable tracking of the cells in real time in vivo in infected, neutropenic mice. In the present study, we report that such activated transfected killer (ATAK) cells, with a stably integrated suicide trap and bioluminescence marker, protected neutropenic mice from lethal invasive yeast and mold infections. Furthermore, the cells were purged from surviving mice, and no evidence of toxicity was found by comprehensive toxicology protocols.
Materials and Methods
Cells and culture
HL-60 cells (American Type Culture Collection) were cultured and activated as described elsewhere [20, 21]. In brief, cells were activated for 3 days by incubation in the presence of 1.3% (vol/vol) DMSO and 2.5 μmol/L RA. For harvesting, cells were centrifuged at 250 g, washed in phosphate-buffered saline (PBS), and resuspended at the appropriate concentration.
Candida albicans SC5314 [21, 22], a well-characterized clinical isolate that is highly virulent in animal models, was serially passaged 3 times in yeast peptone dextrose broth (Difco) and washed twice with PBS. Inocula of Aspergillus fumigatus AF293 (a gift of P. Magee) were prepared by growth on Sabouraud dextrose agar plates for 2 weeks at 37°C Conidia were collected by flooding the plates with sterile PBS containing 0.2% (vol/ vol) Tween 80. Infectious inocula were prepared by counting in a hemacytometer.
Construction of lentiviral transfer plasmids
Lentiviral constructs were created using the backbone plasmid RRL-SIN (UCLA vector core; from Luigi Naldini). Based on this backbone plasmid, 2 plasmids were created for stable transfection of HL-60 cells: plenti-RRL-CMV-TK-SV40-NEO and plenti-RRL-hRluc-IRES-Pure. To generate the plenti-RRL-hRluc-IRES-Pure (8847-bp) construct, the 936-bp human luciferase reporter gene (hRluc) was cloned by polymerase chain reaction (PCR) with the phRL-CMV Vector (Promega) template. The hRluc gene was subcloned into the pIRES vector (Clontech) between NheI and XhoI. The puromycin resistance gene (600 bp) (Pure) was subcloned from pLKO.1 (Sigma) into phRluc-IRES between XbaI and SalI. hRluc-IRES-Pure was further subcloned into the SpeI and SalI of lentivirial vector pRRL.sin.cPPT.hCMV.MCS.
To generate the plenti-RRL-CMV-TK-SV40-Neo (9230-bp) plasmid, the 1131-bp herpes simplex virus (HSV)-thymidine kinase (TK) gene was cloned by PCR from the pAD-HSV-TK vector (Aldevron) and introduced into the XbaI and NotI site of pIRES to generate the pIRES-TK-SV-Neo (neomycin resistance gene) construct. TK-SV40-Neo was subcloned into the XbaI and BamH1 site of the lentivirial vector pRRL.sin.cPPT.hCMV.MCS. The Neo gene was driven by the SV40 enhancer/early promoter. All PCR products were confirmed by DNA sequencing.
Lentivirus production and stable transfection
Lentivirus was produced by transient cotransfection of 293T cells with lentiviral transfer plasmids and 3 lentiviral packaging plasmids (provided by Luigi Naldini): pMDLg/p (encoding gap-pol), pMD.G (encoding VSV-G pseudotype envelope), and pRSV-Rev (encoding rev).
Packaged virus was harvested from supernatant of the 293T transfected cells. HL-60 cells were incubated overnight at 37°C with packaged virus and Polybrene (Invitrogen) at a final concentration of 6 μg/mL. Cells were washed and incubated in fresh medium for 48 h with G418 (1 mg/mL) and/or puromycin (0.5 μg/mL), to select stable gene-transfected HL-60 cells.
In vitro killing
To quantify the candidacidal effect of HL-60 cells, our previously described killing assay was utilized [21]. In brief, 8 × 104 phagocytes were cocultured with 4 × 103 C. albicans (20:1 ratio) for 4 h at 37°C in RPMI + 10% pooled human serum (Sigma). At the end of the incubation, the cultures were sonicated, serially diluted, overlaid with yeast peptone dextrose agar, and incubated overnight at 37°C Colony-forming units were counted to assess the killing of C. albicans, compared with control cultures of the fungus alone.
HL-60 cell irradiation
HL-60 cells were irradiated with gamma rays by exposure to cesium chloride (Cs-137) (irradiator model 143-45; JL Shepherd & Associates) in the blood-bank at Harbor-University of California Los Angeles Medical Center. Radiation doses were confirmed using thermoluminescent dosimetry chips (Global Dosimetry Solutions). Careful dosimetry mapping confirmed a mean delivered dose (± standard deviation) of 2182 ± 241 rads.
In vitro suicide trap activity
HL-60 cells transfected with HSV TK or empty plasmid were exposed in vitro to varying concentrations of ganciclovir (GCV; Western Medical Supply). Media were changed every 2 days. Cell viability was determined by trypan blue exposure followed by counting on a hemacytometer.
In vitro luminescence and fluorescence
Fluorescence of HL-60 cells transfected with green fluorescent protein (GFP) was confirmed by flow cytometry performed using a FAC-SCalibur II, with gating on size and density to isolate viable cells. In vitro luminescence was confirmed using a luminometer (Berthold) and coelenterazine substrate (Goldbio).
For intracellular fluorescence staining of TK and hRluc expression, HL-60-tk-hRluc cells were fixed and permeabilized with Fix-Perm Buffer (BD Pharmingen) and stained with mouse anti-HSV TK monoclonal antibody (Bioworld Consulting laboratories) or isotype control antibody, followed by a fluorescein isothiocyanate goat anti-mouse immunoglobulin (Ig) G secondary antibody (Sigma-Aldrich), and with mouse anti-Renilla luciferase monoclonal antibody (Millipore) or isotype control antibody, followed by phycoerythrin (PE)-goat anti-mouse IgG secondary antibody (Sigma-Aldrich). Cells were quantified by flow cytometry, with gating by size and density on viable cells. For immunofluorescence confocal microscopy, antibodies were labeled with Alexa Fluor 488 in lieu of fluorescein isothiocyanate or Alexa Fluor 568 in lieu of PE, for superior confocal images (both from In-vitrogen). The samples were transferred to slides, sealed with nail polish, and observed under a confocal laser scanning microscope (Olympus).
In vivo experiments
Female BALB/c mice (body weight, 18-20 g) were obtained from the National Cancer Institute (Frederick, Maryland). Mice were made neutropenic by a single intraperitoneal injection of cyclophosphamide (230 mg/kg), resulting in 7 days of neutropenia, as described elsewhere [20, 21, 23]. For the aspergillosis model, a second dose of cyclophosphamide was administered on day 3 after infection. As well, for the aspergillosis model, cortisone acetate (250 mg/kg; Sigma-Aldrich) was given by subcutaneous injection with both doses of cyclophosphamide. For the aspergillosis model, mice were treated daily with 5 mg in 0.2 mL of ceftazidime sub-cutaneously to prevent bacterial superinfection.
Mice were infected via the tail vein with 0.2 mL of PBS containing blastoconidia of C. albicans ∼32 h after cyclophosphamide injection, as described elsewhere [22]. Our standard inhalational model of aspergillosis was used [24, 25]. In brief, mice in the aerosolization chamber were exposed for 1 h to A. fumigatus AF293 conidia aerosolized with a small-particle nebulizer (Hudson Micro Mist; Hudson RCI). Immediately after exposure, 3 mice were euthanized, and lungs were homogenized and quantitatively cultured to confirm the infectious inoculum.
After activation of HL-60 cells for 3 days in DMSO/RA, infected neutropenic mice were treated with 1.5 × 107 ATAK cells (∼7.5 × 108 cells/kg) or placebo in 0.25 mL of PBS administered intraperitoneally ∼1 h after infection on day 0 and again on days 2, 4, and 6 after infection. To confirm that ATAK cells expressed luciferase in vivo, whole-mouse imaging was performed using an in vitro imaging system (IVIS; Caliper Life Sciences). The day after treatment with ATAK cells or control, 3.5 mg/kg coelenterazine in 5% ethanol/PBS was administered intravenously via the tail vein. The mice were anesthetized using inhaled isofluorane, and luminescence was measured using IVIS. Some mice were also treated with GCV at a dose of 5 mg/kg administered intraperitoneally on days 16 and 18 after infection (during the second week after neutrophil recovery).
All procedures involving mice were approved by the institutional animal use and care committee, in accordance with guidelines for animal housing and care established by the National Institutes of Health.
Toxicology testing
Mice were euthanized 3 months after resolution of neutropenia. Complete necropsies with histopathological evaluation were conducted by Charles Rivers Laboratories Pathology Associates. The gross examination included evaluation of the carcass and musculoskeletal system; all external surfaces and orifices; the cranial cavity and external surfaces of the brain; and thoracic, abdominal, and pelvic cavities, with their associated organs and tissues. Brain, heart, lung, liver, spleen, kidney, sternum with bone marrow, and all gross lesions from all animals were fixed in formalin, embedded in paraffin, sectioned, stained with hematoxylin-eosin, and examined by a board-certified veterinary pathologist.
Serum samples from those same mice were collected at 2 time points (2 weeks after resolution of neutropenia and again 3 months after resolution of neutropenia). Comprehensive metabolomic analysis of the serum was conducted by Metabolon by use of liquid chromatography/gas chromatography mass spectroscopy and proprietary in silica technology, to perform global biochemical profiling, analyzing and identifying every metabolite present in serum and the biochemical pathways resulting in synthesis of those metabolites. Metabolite fingerprints were compared between treated and control mice, to identify any differences.
Statistics
Kaplan-Meier curves underwent pairwise comparison with the nonparametric log-rank test. Other statistical comparisons were made using the Mann-Whitney U test. In addition, t tests were used for metabolomics comparisons.
Results
Stable transfection of HL-60 cells with fluorescence and luminescence markers and a suicide trap
HL-60 cells were transfected with a myeloid-active lentivirus containing GFP as a reporter to determine transfection efficiency. After 48 h, GFP expression was confirmed by fluorescence microscopy (Figure 1). According to flow cytometry, 199% of cells were transfected (Figure 2A). To evaluate genetic stability, the transfected cells were continuously cultured for 20 months in the absence of selective pressure, after which time 199% of cells still expressed GFP (Figure 2B). Furthermore, cells frozen for 20 months in liquid nitrogen were thawed and evaluated for GFP expression, and 199% of such cells were fluorescent (Figure 2C).
Figure 1.
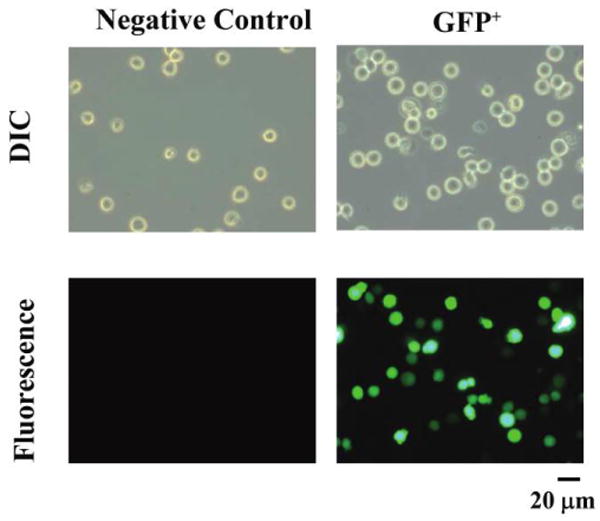
Fluorescence of HL-60 cells transfected with lentivirus containing green fluorescent protein (GFP) or not (negative control). A, Differential interference contrast (DIC) and fluorescence microscopy of individual, transfected cells. The same field of view is shown by DIC above and fluorescence below. Total original magnification, ×400. GFP+, GFP positive.
Figure 2.
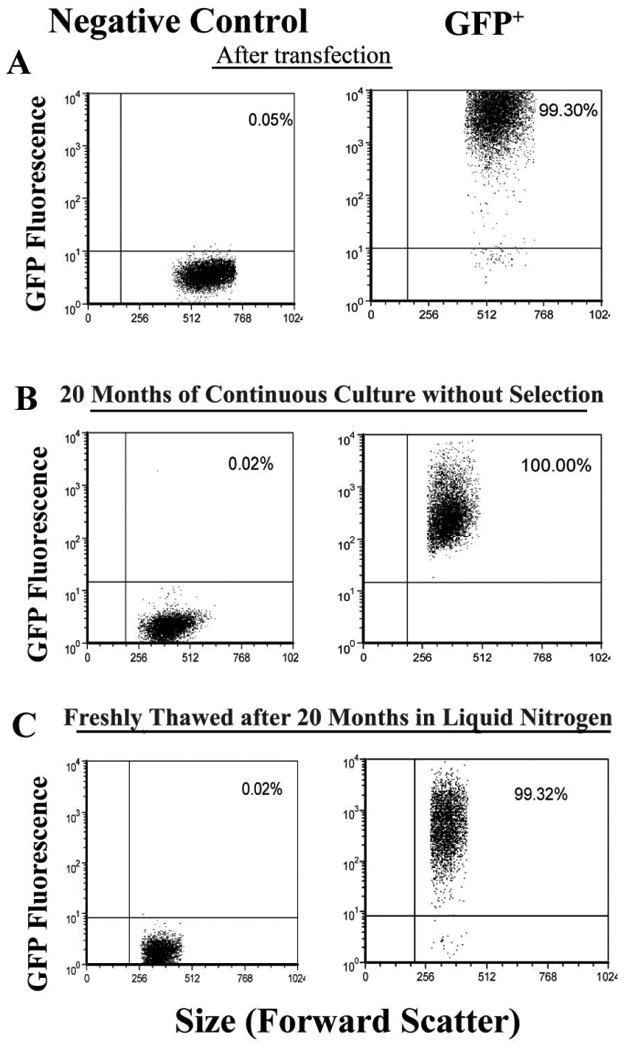
Fluorescence of HL-60 cells sequentially transfected with 2 different lentiviral constructs. A, Flow cytometry performed after transfection of green fluorescent protein (GFP)–transfected or control HL-60 cells, with the percentage of fluorescent cells shown in the right upper quadrant. Transfection efficiency was >98%. B, Flow cytometry performed >20 months after continuous culture without selection of cells from panel B, showing stable integration as evidenced by maintenance of fluorescence in >99% of cells. C, Flow cytometry performed after rapid thawing of transfected HL-60 cells that had been in liquid nitrogen storage for >20 months, showing minimal loss of fluorescence activity after thawing. GFP+, GFP positive.
After confirming successful transfection of HL-60 cells with the GFP-lentiviral construct, we performed sequential transfection with TK and luciferase by the same lentiviral vector. Reverse-transcriptase (RT)–PCR was used to confirm TK and luciferase (hRluc) gene messenger RNA expression from the transfected cells (Figure 3A). Expression at the protein level of both genes was confirmed by flow cytometry (Figure 3B) and by dual-color confocal microscopy (Figure 3C) after intracellularly staining the cells with anti-TK and antiluciferase antibodies.
Figure 3.
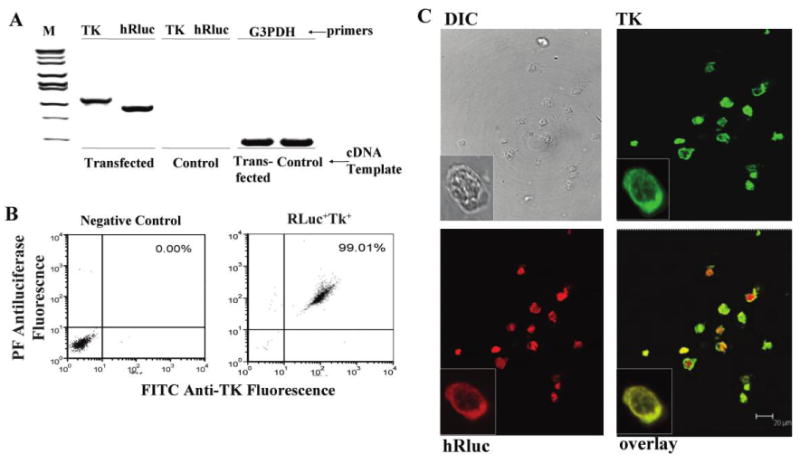
High level of expression of both the luciferase bioluminescence marker and herpes simplex virus (HSV) thymidine kinase (TK) suicide trap in dually transfected cells. A, Expression by reverse-transcriptase polymerase chain reaction (RT-PCR) of luciferase and HSV TK in dually transfected HL-60 cells or control HL-60 cells. B, Flow cytometry of dually transfected or control HL-60 cells stained intracellularly for both luciferase and TK protein expression. Flow was conducted after 2 months of growth in no selection media. C, Confocal microscopy of dually transfected or control cells stained for expression of luciferase and TK, or visualized by differential interference contrast optics. cDNA, complementary DNA; DIC, differential interference contrast; G3PDH, RT-PCR using primers for this as a constitutive control; hRluc, RT-PCR using primers specific for luciferase; M, molecular weight markers; PE, phycoerythrin; TK, RT-PCR using primers specific for HSV TK.
In vitro functions of transfected ATAK cells
To confirm that transfection of the cells did not alter their microbicidal capacity, we conducted in vitro C. albicans kill assays with unactivated or activated HL-60 cells (ATAK cells) dually transfected with HSV TK and luciferase. ATAK cells demonstrated a 3-fold increase in candidacidal capacity versus transfected but unactivated HL-60 cells (Figure 4A), as we have previously seen with untransfected, activated HL-60 cells [20, 21]. Hence, dual transfection did not abrogate the candidacidal capacity of the cells after activation.
Figure 4.
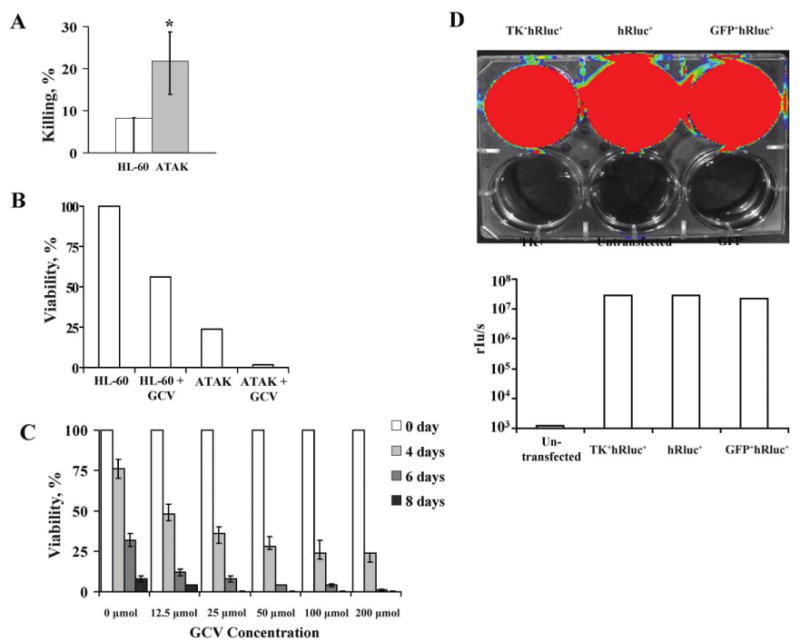
In vitro effects of activated transfected killer (ATAK) cells. A, Unactivated transfected HL-60 cells were compared with ATAK cells for their ability to kill Candida albicans in vitro. Results are from 3 experiments, each done in triplicate. Medians and interquartile ranges are shown. *P< .01 vs. control. B, Viability of unactivated (HL-60) or activated (ATAK) cells transfected with herpes simplex virus (HSV) thymidine kinase (TK) and cultured for 4 days in normal media or media plus 25 μmol/L ganciclovir (GCV). C, Viability of ATAK cells after irradiation with a mean dose (± standard deviation) of 2182 ± 241 rads cultured for 8 days in the presence or absence of varying concentrations of GCV. Medians and interquartile ranges are shown. Results are from 3 experiments combined. D, HL-60 cells that were untransfected; transfected with green fluorescent protein (GFP), thymidine kinase (TK), or hRluc alone; or transfected dually with GFP-hRluc or TK-hRluc. Cells were activated with dimethyl sulfamethoxazole and retinoic acid, and luminescence was imaged in tissue culture plate using an in vitro imaging system (Caliper Life Sciences) and quantified by a luminometer.
The function of the suicide trap was determined by exposure of 105 transfected, unactivated HL-60 cells or ATAK cells grown in media with or without 25 μmol/L of GCV (which is below the ∼30-mmol/L and ∼60-mmol/L serum concentrations achievable in transplant patients treated with oral valganciclovir and intravenous ganciclovir, respectively [26, 27]). After 4 days of exposure, GCV considerably decreased the viability of transfected, unactivated HL-60 cells (Figure 4B). As previously described, activation (which results in differentiation toward an end-differentiated neutrophil phenotype) also reduced viability of cells even in the absence of GCV. The combination of activation and exposure to GCV resulted in a >95% reduction in cell viability by 4 days.
Irradiation of 105 cells resulted in prolonged viability in the presence of GCV (Figure 4C), likely by preventing cellular replication, thereby protecting the cells from the effect of the GCV. For example, nonirradiated ATAK cells not exposed to GCV had 25% viability by day 4 of culture (Figure 4B), versus the 75% viability of irradiated ATAK cells not exposed to GCV by day 4 of culture (Figure 4C). Furthermore, irradiated cells exposed to 25 μmol/L GCV for 4 days had a viability of ∼40%, compared with the previously discovered viability of !5% after 4 days of exposure of unirradiated cells to the same concentration of GCV (compare Figure 4B with 4C). Nevertheless, by 8 days, viability of irradiated cells exposed to ≥=25 μmol/L GCV was 0%. The effect of the suicide trap was confirmed because activated, irradiated cells not exposed to GCV had 10% viability by day 8 versus 0% viability in the presence of GCV (Figure 4C).
Finally, as an added measure of precaution, we sought to ensure that the suicide trap was functional with a cell dose that was in excess of the treatment dose for mice. Irradiation plus 25 μmol/L GCV also resulted in 100% cell death by 11 days of culture, when the starting inoculum of cells was increased 4000-fold to 4 × 108 cells (∼30-fold greater than the dose administered to infected, neutropenic mice).
Luciferase activity in dually transfected ATAK cells was confirmed after exposure of the cells to coelentrazine substrate. Compared with untransfected control HL-60 cells, ATAK cells transfected with luciferase alone, luciferase and GFP, or luciferase and TK generated markedly increased luminescence, as detected by IVIS and by a quantitative luminometer (2.9 × 107, 2.3 × 107, and 2.9 × 107 rlu/s vs. 1199 rlu/s for untransfected negative control cells, respectively) (Figure 4D).
In vivo functions of ATAK cells
Neutropenic mice were infected intravenously with a sublethal inoculum of C. albicans (103 blastospores) and treated with irradiated ATAK cells. Mice were injected with the luciferase enzyme substrate coelentrazine at 24 h after the first dose of ATAK cells (day 1 after infection) and at 24 h after the last dose (on day 7 after infection, which is the day after neutrophil recovery begins [20]), and they were imaged with IVIS. GCV was administered intraperitoneally to mice on days 16 and 18 after infection, and mice were imaged with IVIS again 3 months after infection.
After administration of coelentrazine, mice were sedated with inhaled isofluorane and imaged for 5 min in the IVIS system. Control mice received substrate but had not been injected with ATAK cells. After the first dose of treatment, mice injected with ATAK cells had intense, diffuse luminescence signal, compared with control mice (Figure 5A). After recovery from neutropenia began, the signal intensity of ATAK cells began to decrease (Figure 5B), consistent with the cells being rejected by the reconstituting immune system. At 3 months of follow-up, luminescence signal in the treated mice was not greater than background in the control mice (Figure 5C).
Figure 5.
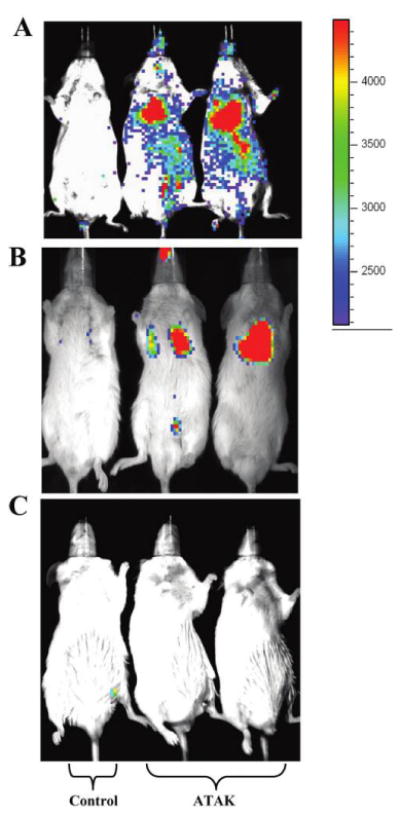
Activated transfected killer (ATAK) cells are bioluminescent in vitro and in vivo. Mice were made neutropenic and infected with a sublethal inoculum of Candida albicans. Mice were treated with irradiated ATAK cells or saline control starting 3 h after infection and repeated on days 2, 4, and 6 after infection. Mice were imaged with the in vitro imaging system (IVIS; Caliper Life Sciences) 24 h after the first treatment (A); 24 h after the last treatment, which was 1 day after neutrophil recovery (B); or 2 months after neutrophil recovery (C). The mice imaged at 2 months after neutrophil recovery had been treated with ganciclovir (5 mg/kg given intraperitoneally on days 16 and 18 after infection, which was 1 week after neutrophil recovery). One saline control mouse (left) and 2 ATAK-treated mice (middle and right) are shown in each panel and are representative of 5 mice per group. The photon intensity scale shown is as follows: red→yellow→green→blue.
We tested the efficacy of the transfected ATAK cells as treatment for disseminated candidiasis and inhalational aspergillosis in our standard neutropenic mouse model. Neutropenic mice were infected intravenously via the tail vein with a lethal inoculum of C. albicans or infected in an inhalation chamber with A. fumigatus and treated with irradiated ATAK cells. Negative control mice received saline injection. ATAK cells significantly improved the survival of neutropenic mice infected with either organism (Figure 6).
Figure 6.
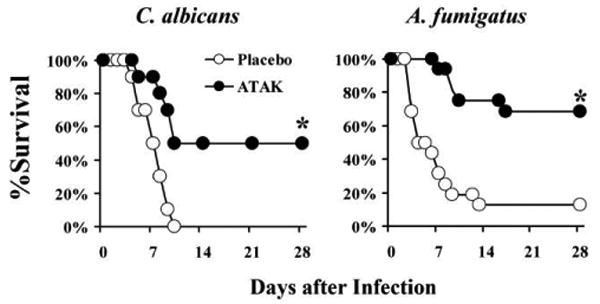
Activated transfected killer (ATAK) cells effectively treat neutropenic mice with disseminated candidiasis or inhalational aspergillosis. Neutropenic mice were infected intravenously via the tail vein with Candida albicans (n = 10 mice per group) or in an inhalational chamber with Aspergillus fumigatus (n = 16 mice per group from 2 experiments). Mice were treated ∼1 h after infection and again on days 2, 4, and 6 after infection with ATAK cells (1.5 × 107 cells) or saline placebo. *P< .01 versus control by log-rank test.
In vivo toxicology evaluation of the ATAK cells
The mice described above, which were made neutropenic and infected with a sublethal inoculum of C. albicans, treated with ATAK cells or saline control, serially imaged, and treated with GCV, were followed for 3 months. No deaths occurred during follow-up; there was no difference in gross appearance, behavior, or body weight between the mice (data not shown); and no tumors were evident on gross inspection.
Serum was obtained at 2 weeks and at 3 months after resolution of neutropenia, and the mice were then shipped to Charles Rivers Laboratories Pathology Associates for formal toxicology evaluation. At necropsy, there were no gross or microscopic findings attributed to the administration of ATAK cells, and no leukemic cells were found in the bone marrow or in any organ. In addition, serum metabolomic profiles were compared between ATAK- and placebo-treated mice at 2 weeks and 3 months after infection. The technology used to generate these profiles establishes a metabolic “fingerprint” of toxicity profiles by identifying metabolic pathways known to be activated in specific organs during organ toxicity. The technology is more sensitive than histopathological evaluation of tissue, because it can detect biochemical evidence of tissue injury significantly (ie, days) before the onset of tissue injury visible in a histopathological section [28–30]. Furthermore, the technology is extremely broad, being able to identify toxicity in every organ in the body.
Metabolomic analysis of serum from the ATAK- or placebo-treated mice found 323 compounds, 220 named and 103 unnamed, of which only 13 were present in differing amounts between ATAK-treated and placebo-treated mice (Table 1). Hierarchical cluster algorithms were unable to separate the groups into discrete pathways differing between ATAK-treated and placebo-treated mice (Table 1); this finding would be expected if the metabolites were generated by toxic metabolic pathways. The small differences in these metabolites between groups, the fact that some metabolites were higher in the placebo group and some were higher in the ATAK group, and the fact that the metabolites were randomly distributed from among diverse metabolic pathways rather than being clustered within individual pathways indicate that they do not reflect organ-system toxicity.
Table 1.
Metabolites Differing between Activated Transfected Killer (ATAK)– and Placebo-Treated Mice
| Superpathway | Pathway | Biochemical | Ratioa | P |
|---|---|---|---|---|
| Amino acid | Phenylalanine and tyrosine metabolism | Phenol sulfate | 1.45 | .04 |
| Valine, leucine, and isoleucine metabolism | 3-methyl-2-oxovalerate | 0.72 | .03 | |
| Lipid | Bile acid metabolism | β-muricholate | 0.50 | .05 |
| Deoxycholate | 0.70 | .01 | ||
| Peptide | Fibrinogen cleavage peptide | TDTEDKGEFLSEGGGV | 0.70 | .01 |
| Unidentified | Unidentified | Y-11835 | 2.07 | .003 |
| Y-11538 | 0.52 | .02 | ||
| Y-12138 | 0.52 | .02 | ||
| Y-11327 | 0.44 | .02 | ||
| Y-14603 | 2.65 | .03 | ||
| Y-14938 | 1.26 | .04 | ||
| Y-14588 | 0.80 | .05 | ||
| Y-12645 | 1.71 | .05 |
a Ratio of metabolite present in ATAK samples/placebo samples. Numbers >1 denote excess metabolite in ATAK samples, and numbers <1 denote diminished metabolite in ATAK samples.
Discussion
To garner the therapeutic benefit of neutrophil transfusions but avoid the technical obstacles to their widespread adoption, we have developed a strategy that uses a single, immortal, phagocytic cell line to provide protection to neutropenic hosts until their own phagocyte counts are restored. To ensure the safety of our cell line strategy, it was necessary to integrate multiple, redundant protective mechanisms to prevent leukemic engraftment. The first protective strategy is activation with DMSO and RA, which diminishes cellular replication [20]. Second, irradiation with ∼2000 rads further inhibits replication of cells [20]. Such a dose is used for lymphocyte-contaminated blood products before infusion in neutropenic patients to prevent graft-versus-host disease [31]. A lower dose of radiation (1000 rads) has already been used in human patients receiving allogeneic immortal natural killer cells in a clinical trial [32]. Finally, we have achieved a critical, additional layer of protection: stable integration of an inducible suicide trap into the cells. We used as our suicide trap the HSV TK gene (tk). GCV-induced suicide of malignant cells transformed with tk has been shown to eliminate >99% of tumor mass in animal models [33], and tk-mediated gene therapy for malignancies has already been used in several clinical trials [33–37], underscoring the practicality and clinical relevance of this technique.
Because the tk suicide trap works by blocking DNA replication in cells that are dividing, the trap spares nonreplicating cells. Instead, the trap serves as a final safety layer that can be used to purge any rare cells that escape activation and irradiation and persistently replicate, thereby preventing engraftment in susceptible hosts by rare escape variants. We confirmed the function of the suicide trap in vitro, and we confirmed that mice treated in vivo with GCV had total purging of the cells. It was difficult to compare purging by GCV in vivo, because even mice not treated with GCV had no residual luminescence signal at long-term follow-up (data not shown).
The purging of cells from mice not treated with GCV was likely the result of the murine immune response to the cells after marrow reconstitution. In this context, the host immune response to the cells also serves as an additional safety layer, helping to ensure that the cells are not able to engraft.
In summary, we have developed a platform technology of immortal cells transfected with an inducible suicide trap that can be activated to provide protection against otherwise lethal invasive fungal infections in neutropenic mice. Of greatest importance was the confirmation of considerable efficacy of ATAK cells in treating both disseminated candidiasis and inhalational aspergillosis in neutropenic mice. Furthermore, no toxicity was observed during 3 months of follow-up, either by cageside evaluations, necropsy, extensive histopathological evaluation, or a global metabolomics evaluation. These results lay the groundwork for continued translational development of this promising, novel technology for the treatment of refractory infections in neutropenic hosts.
Acknowledgments
Financial support: Public Health Service (grant R01 AI081719 to B.S.).
Footnotes
Potential conflicts of interest: none reported.
References
- 1.Segal BH, Almyroudis NG, Battiwalla M, et al. Prevention and early treatment of invasive fungal infection in patients with cancer and neutropenia and in stem cell transplant recipients in the era of newer broad-spectrum antifungal agents and diagnostic adjuncts. Clin Infect Dis. 2007;44:402–9. doi: 10.1086/510677. [DOI] [PubMed] [Google Scholar]
- 2.Perlroth J, Choi B, Spellberg B. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med Mycol. 2007;45:321–46. doi: 10.1080/13693780701218689. [DOI] [PubMed] [Google Scholar]
- 3.Pappas PG, Kauffman CA, Andes D, et al. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:503–35. doi: 10.1086/596757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh TJ, Anaissie EJ, Denning DW, et al. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:327–60. doi: 10.1086/525258. [DOI] [PubMed] [Google Scholar]
- 5.Marr KA, Carter RA, Crippa F, Wald A, Corey L. Epidemiology and outcome of mould infections in hematopoietic stem cell transplant recipients. Clin Infect Dis. 2002;34:909–17. doi: 10.1086/339202. [DOI] [PubMed] [Google Scholar]
- 6.Marr KA. Primary antifungal prophylaxis in hematopoietic stem cell transplant recipients: clinical implications of recent studies. Curr Opin Infect Dis. 2008;21:409–14. doi: 10.1097/QCO.0b013e328307c7d9. [DOI] [PubMed] [Google Scholar]
- 7.Chamilos G, Luna M, Lewis RE, et al. Invasive fungal infections in patients with hematologic malignancies in a tertiary care cancer center: an autopsy study over a 15-year period (1989–2003) Haematologica. 2006;91:986–9. [PubMed] [Google Scholar]
- 8.Malik IA, Moid I, Aziz Z, Khan S, Suleman M. A randomized comparison of fluconazole with amphotericin B as empiric anti-fungal agents in cancer patients with prolonged fever and neutropenia. Am J Med. 1998;105:478–83. doi: 10.1016/s0002-9343(98)00326-x. [DOI] [PubMed] [Google Scholar]
- 9.Nucci M, Pulcheri W, Spector N, et al. Fungal infections in neutropenic patients: an 8-year prospective study. Rev Inst Med Trop Sao Paulo. 1995;37:397–406. doi: 10.1590/s0036-46651995000500004. [DOI] [PubMed] [Google Scholar]
- 10.Lehrnbecher T, Groll AH, Chanock SJ. Treatment of fungal infections in neutropenic children. Curr Opin Pediatr. 1999;11:47–55. doi: 10.1097/00008480-199902000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Hübel K, Dale DC, Engert A, Liles WC. Current status of granulocyte (neutrophil) transfusion therapy for infectious diseases. J Infect Dis. 2001;183:321–8. doi: 10.1086/317943. [DOI] [PubMed] [Google Scholar]
- 12.Alavi JB, Root RK, Djerassi I, et al. A randomized clinical trial of granulocyte transfusions for infection in acute leukemia. N Engl J Med. 1977;296:706–11. doi: 10.1056/NEJM197703312961302. [DOI] [PubMed] [Google Scholar]
- 13.Herzig RH, Herzig GP, Graw RG, Jr, Bull MI, Ray KK. Successful granulocyte transfusion therapy for gram-negative septicemia: a prospectively randomized controlled study. N Engl J Med. 1977;296:701–5. doi: 10.1056/NEJM197703312961301. [DOI] [PubMed] [Google Scholar]
- 14.Athens JW, Haab OP, Raab SO, et al. Leukokinetic studies. XI. Blood granulocyte kinetics in polycythemia vera, infection, and myelofibrosis. J Clin Invest. 1965;44:778–88. doi: 10.1172/JCI105190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marsh JC, Boggs DR, Cartwright GE, Wintrobe MM. Neutrophil kinetics in acute infection. J Clin Invest. 1967;46:1943–53. doi: 10.1172/JCI105684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. J Clin Invest. 1976;58:705–15. doi: 10.1172/JCI108517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huestis DW, Glasser L. The neutrophil in transfusion medicine. Transfusion. 1994;34:630–46. doi: 10.1046/j.1537-2995.1994.34794330020.x. [DOI] [PubMed] [Google Scholar]
- 18.Glasser L. Effect of storage on normal neutrophils collected by discontinuous-flow centrifugation leukapheresis. Blood. 1977;50:1145–50. [PubMed] [Google Scholar]
- 19.Safdar A, Hanna HA, Boktour M, et al. Impact of high-dose granulocyte transfusions in patients with cancer with candidemia: retrospective case-control analysis of 491 episodes of Candida species bloodstream infections. Cancer. 2004;101:2859–65. doi: 10.1002/cncr.20710. [DOI] [PubMed] [Google Scholar]
- 20.Spellberg BJ, Collins M, Avanesian V, et al. Optimization of a myeloid cell transfusion strategy for infected neutropenic hosts. J Leukoc Biol. 2006;81:632–41. doi: 10.1189/jlb.0906549. [DOI] [PubMed] [Google Scholar]
- 21.Spellberg BJ, Collins M, French SW, Edwards JE, Jr, Fu Y, Ibrahim AS. A phagocytic cell line markedly improves survival of infected neutropenic mice. J Leukoc Biol. 2005;78:338–44. doi: 10.1189/jlb.0205072. [DOI] [PubMed] [Google Scholar]
- 22.Spellberg BJ, Johnston D, Phan QT, et al. Parenchymal organ, and not splenic, immunity correlates with host survival during disseminated candidiasis. Infect Immun. 2003;71:5756–64. doi: 10.1128/IAI.71.10.5756-5764.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van t Wout JW, Mattie H, van Furth R. Comparison of the efficacies of amphotericin B, fluconazole, and itraconazole against a systemic Candida albicans infection in normal and neutropenic mice. Antimicrob Agents Chemother. 1989;33:147–51. doi: 10.1128/aac.33.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheppard DC, Rieg G, Chiang LY, Filler SG, Edwards JE, Jr, Ibrahim AS. Novel inhalational murine model of invasive pulmonary aspergillosis. Antimicrob Agents Chemother. 2004;48:1908–11. doi: 10.1128/AAC.48.5.1908-1911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rieg G, Spellberg B, Schwartz J, et al. Antifungal prophylaxis is effective against murine invasive pulmonary aspergillosis. Antimicrob Agents Chemother. 2006;50:2895–6. doi: 10.1128/AAC.00299-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrottet N, Decosterd LA, Meylan P, Pascual M, Biollaz J, Buclin T. Valganciclovir in adult solid organ transplant recipients: pharmacokinetic and pharmacodynamic characteristics and clinical interpretation of plasma concentration measurements. Clin Pharmacokinet. 2009;48:399–418. doi: 10.2165/00003088-200948060-00006. [DOI] [PubMed] [Google Scholar]
- 27.Winston DJ, Baden LR, Gabriel DA, et al. Pharmacokinetics of ganciclovir after oral valganciclovir versus intravenous ganciclovir in allogeneic stem cell transplant patients with graft-versus-host disease of the gastrointestinal tract. Biol Blood Marrow Transplant. 2006;12:635–40. doi: 10.1016/j.bbmt.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 28.Ohta T, Masutomi N, Tsutsui N, et al. Metabolomics analysis of plasma and urine in rats treated with fenofibrate and phenobarbital. Program and abstracts of the 47th annual meeting of the Society of Toxicology (Seattle); Reston, VA. 2008. [Google Scholar]
- 29.Rosenstock M, Boudonck K. Metabolomic analysis of urine and kidney tissue in rats treated with valproate. Advances in Metabolic Profiling, European Biomarkers Summit and Proteomics Europe (Lisbon, Portugal) 2008 [Google Scholar]
- 30.Mohney RP, Zhou H, Liu L, Mitchell M, Guha C. Metabolomic profiles of liver and plasma following whole-liver irradiation in mice. Program and abstracts of the 47th annual meeting of the Society of Toxicology (Seattle); Reston, VA. 2008. [Google Scholar]
- 31.van Bekkum DW. Use and abuse of hemopoietic cell grafts in immune deficiency diseases. Transplant Rev. 1972;9:3–53. doi: 10.1111/j.1600-065x.1972.tb01560.x. [DOI] [PubMed] [Google Scholar]
- 32.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10:535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 33.Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann JL, Klatzmann D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proc Natl Acad Sci U S A. 1993;90:7024–8. doi: 10.1073/pnas.90.15.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasenburg A, Tong XW, Rojas-Martinez A, et al. Thymidine kinase (TK) gene therapy of solid tumors: valacyclovir facilitates outpatient treatment. Anticancer Res. 1999;19:2163–5. [PubMed] [Google Scholar]
- 35.Tong X, Engehausen DG, Freund CT, et al. Comparison of long-term survival of cytomegalovirus promotre versus Rous Sarcoma virus promoter-driven thymidine kinase gene therapy in nude mice bearing human ovarian cancer. Hybridoma. 1999;18:93–7. doi: 10.1089/hyb.1999.18.93. [DOI] [PubMed] [Google Scholar]
- 36.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–2. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 37.Sterman DH, Treat J, Litzky LA, et al. Adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir gene therapy in patients with localized malignancy: results of a phase I clinical trial in malignant mesothelioma. Hum Gene Ther. 1998;9:1083–92. doi: 10.1089/hum.1998.9.7-1083. [DOI] [PubMed] [Google Scholar]