

Abstract
Excess lipid induced metabolic disorders are one of the major existing challenges for the society. Among many different causes of lipid disorders, overproduction and compromised catabolism of triacylglycerol-rich very low density lipoproteins (VLDL) have become increasingly prevalent leading to hyperlipidemia worldwide. This review provides the latest understanding in different aspects of VLDL assembly process, including structure-function relationships within apoB, mutations in APOB causing hypobetalipoproteinemia, significance of modulating microsomal triglyceride-transfer protein activity in VLDL assembly, alterations of VLDL assembly by different fatty acid species, and hepatic proteins involved in vesicular trafficking, and cytosolic lipid droplet metabolism that contribute to VLDL assembly. The role of lipoprotein receptors and exchangeable apolipoproteins that promote or diminish VLDL assembly and secretion is discussed. New understanding on dysregulated insulin signaling as a consequence of excessive triacylglycerol-rich VLDL in the plasma is also presented. It is hoped that a comprehensive view of protein and lipid factors that contribute to molecular and cellular events associated with VLDL assembly and secretion will assist in the identification of pharmaceutical targets to reduce disease complications related to hyperlipidemia.
Introduction
Lipids of dietary origin as well as those stored in the adipose tissues act as energy sources for mammalian cells. Since lipids are hydrophobic in nature, mammals have evolved a mechanism such that the insoluble lipids are made soluble in the form of lipoproteins for transportation and delivery to various organs and tissues by the circulatory system. Formation and secretion of lipoprotein particles is primarily achieved in the liver (as VLDL) and in the intestine (as chylomicrons). The process involved in the assembly and secretion of hepatic VLDL or intestinal chylomicrons is complex and has been studied extensively for the past 2-3 decades. Lipid and protein factors that affect various steps during the assembly and secretion of VLDL and chylomicrons have been identified. The assembly process of hepatic VLDL is initiated in the endoplasmic reticulum (ER) as soon as apoB-100 is translated and translocated into the lumenal side where the elongating apoB-100 polypeptide chain recruits various lipids co-translationally. Each VLDL is composed of one molecule of apoB-100, multiple copies of other apolipoproteins, together with varied amounts of triacylglycerol (TAG) and cholesteryl esters, depending upon the size of resulting particles. Cellular and molecular mechanisms by which different lipid and protein components are brought together for VLDL assembly are not fully understood and remain to be defined. A protein factor other than apoB that is absolutely required for VLDL assembly is the microsomal triglyceride-transfer protein (MTP). The obligatory role of MTP in VLDL assembly/secretion is exemplified by human familial abetalipoproteinemia, characterized by nearly a complete absence of apoB-containing lipoproteins including VLDL (and chylomicrons as well). Available evidence indicates that among different lipid and protein constituents of VLDL, the availability of functional apoB-100 and TAG are by far the most critical for the assembly of secretion-competent VLDL within the ER lumen. An array of protein factors involved in secretory protein translation and translocation across the ER membrane are responsible for initial apoB-100 folding to attain lipid-binding capability within the microsomal lumen. Pathological conditions that disfavor apoB-100 folding or binding of lipids to apoB will result in aborted VLDL assembly and premature intracellular degradation of apoB-100 during or after translation.
Structural and functional elements within apoB-100
The human APOB gene, located on the distal end of the short arm of chromosome 2 (2p23-2p24), encodes a ~20 kb mRNA that is translated into the full-length apoB-100 consisting of 4,536 amino acids in the liver [1-3]. A truncated form of apoB, known as apoB-48, represents the N-terminal 48% of apoB-100 and is produced in the intestine by an mRNA editing mechanism [4]. In humans, apoB-100 and apoB-48 are obligatory proteins for the assembly of respective hepatic VLDL and intestinal chylomicrons [5]. In mouse and rat, the liver synthesizes apoB-48 in addition to apoB-100 [6]. Because of their enormous size, extreme hydrophobicity along with varied extents in lipid-binding, the 3-D structure of apoB-100 or apoB-48 has not been solved at the atomic level. However, attempts have been made, using various algorithms, to compute the structures of various domains of apoB-100. The modeled human apoB-100 molecule is composed of five domains enriched with alternating amphipathic α-helices and amphipathic β-strands, designated βα1-β1-α2-β2-α3 [7]. Various domains and their approximate locations in apoB-100 are depicted in Fig. 1A. Moreover, based on the homology to lamprey lipovitellin, a modeled structure for the N-terminal ~930 amino acids of human apoB-100 has been proposed [8,9]. This model predicts a βα1 domain structure consisting of β-barrel (the first 264 residues) and α-helical bundle (residues 292-593), followed by two amphipathic β-sheets termed C sheet (residues 611-782) and A sheet (residues 783-930), respectively, that may form a lipid-binding pocket [10]. Scanning transmission electron microscopy studies have provided direct evidence that nanogold-labeled apoB fragment (apoB6.4-17) interacted with lipids [11]. A model of human apoB-100 associated with low density lipoprotein (LDL) has been obtained using images captured by electron cryomicroscopy, in which a single apoB-100 molecule with its α-helix and β-sheet rich domains across the LDL surface is proposed [12].
Figure 1.
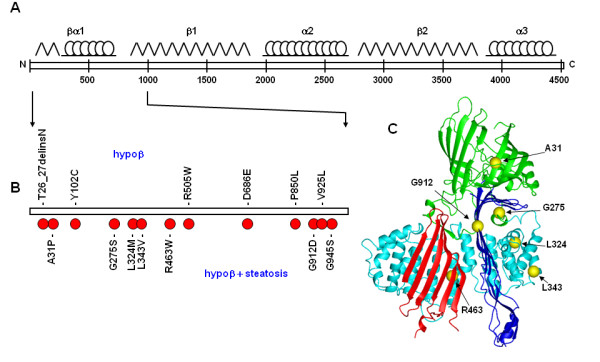
Model of the N-terminal of apoB and positions of FHBL mutations. A, schematic diagram of apoB-100, with predicted locations of βα1, β1, α2, β2, and α3 domains are shown on the top. B, positions of non-truncating FHBL mutations within the N-terminal 1,000 amino acids of apoB-100. Mutations associated with phenotype of hypobetalipoproteinemia (hypoβ) are shown on the top, whereas mutations associated with phenotype of both hypoβ and hepatic steatosis are shown below. C, proposed homologous model of the N-terminal ~930 amino acids of apoB. The β-barrel, α-helical, and β sheet (C-sheet and A-sheet) are highlighted in green, cyan, red, and blue, respectively. Locations of Ala31, Gly275, Leu324, Leu343, Arg463, and Gly912 within the modeled βα1 domain are shown as yellow spheres.
The N-terminal region of apoB contains several binding sites for MTP, an apoB-specific molecular chaperone essential for apoB-100 to fold properly during VLDL assembly/secretion [13,14]. Multiple MTP-binding sites have been identified within the N-terminal βα1 domain of apoB [13,15,16], and the affinity of MTP-binding is inversely related to the apoB polypeptide length [17]. Apart from binding to MTP, the βα1 domain of apoB-100 also binds to scavenger receptors in human macrophages [18] and to lipoprotein lipase [19]. The β1 domain (amino acids 827-1880) of apoB-100 is predominantly made of amphipathic β strands and is involved in irreversible lipid-binding [20,21]. The β2 domain located at the C-terminal half of apoB-100 possesses LDL receptor-binding property [22,23]. The role of the intervening amphipathic α-helix enriched domain α2 and the C-terminal α3 of apoB-100 may represent a flexible region that imparts elasticity to the molecule allowing recruitment of various amounts of core lipids [24].
The amino acid sequences within apoB-100 essential for lipoprotein assembly and secretion have been investigated extensively using two approaches, namely truncation mutagenesis and chimeric protein expression analysis. Incremental truncation of human apoB polypeptide from the carboxyl terminus successively decreased its ability to form buoyant lipoprotein particles [25,26]. Under lipid rich conditions, the ability of apoB to assemble VLDL resides at the length transition between the N-terminal ~30% and ~40% of the polypeptide, a region enriched with amphipathic β strands (i.e. the β1 domain) [26]. Studies with chimera proteins, in which segments of apoB derived from the β1 domain were fused with apoA-I, have shown that inclusion of the amphipathic β strands conferred the ability to assemble VLDL [26]. The amino acid sequences within the β1 domain also influence the rate of apoB translocation across the ER membrane and its susceptibility to proteasomal degradation [27] (for apoB degradation, see below). These studies suggest that the amino acid sequences within the β1 domain are important for lipid-binding in the VLDL assembly process.
Human apoB-100 undergoes several posttranslational modifications, including disulfide bond formation, N-linked glycosylation, and palmitoylation. Mutational analysis has shown that among six disulfide linkages within the βα1 domain, the two involving Cys51/Cys70 and Cys218/Cys234 are essential for apoB-lipoprotein assembly and secretion [28,29]. Results from the cysteine mutagenesis studies corroborate with data obtained from dithiothreitol treatment experiment in which abolishing apoB disulfide bond formation with the reducing agent resulted in decreased apoB secretion [30].
There are 20 potential N-linked glycosylation sites within human apoB-100, of which 16 Asn residues are conjugated with oligosaccharides in apoB-100 associated with plasma LDL [31]. The requirement of Asn158, Asn956, Asn1341, Asn1350, and Asn1496 residues within the amino terminus of apoB-100 has been determined by mutagenesis experiments; results from these studies suggested that the loss of N-glycans, particularly at Asn1496, resulted in decreased stability of apoB and reduced secretion of TAG-rich lipoproteins [32]. Thus, in addition to lipid binding sequences of apoB, the N-linked oligosaccharides conjugated to apoB also play a role conferring the posttranslational stability and the ability to assemble lipoproteins.
Palmitoylation of human apoB is observed in plasma LDL [33] as well as apoB found in cultured hepatic cell lines [34,35]. The 16-carbon fatty acid palmitate is covalently linked to cysteine residues via thioester bonds. Mutagenesis studies of the four cysteine residues Cys1085, Cys1396, Cys1478, and Cys1635 suggest that the lack of palmitoylation within the N-terminal region of apoB does not compromise the ability of apoB-48 to assemble lipoproteins [36]. However, decreased secretion of the short truncation mutant apoB-29 was observed as a result of lack of palmitoylation [35]. It is likely that an interplay exists between the length of amphipathic lipid-binding sequences and palmitoylation of apoB that regulate TAG-rich lipoprotein assembly and secretion.
Many mutations in the APOB gene have been characterized and found to affect the plasma concentrations of apoB, TAG and cholesterol. The most characterized APOB mutations are the ones found in familial hypobetalipoproteinemia (FHBL) [37]. FHBL is inherited in a Mendelian fashion as an autosomal dominant trait. Heterozygote FHBL individuals invariably have plasma cholesterol, TAG, LDL-cholesterol, and apoB at levels approximately 1/3 of normal, whereas homozygote FHBL subjects have barely detectable levels of apoB. Individuals with FHBL have reduced risk of cardiovascular diseases presumably owing to low plasma apoB and cholesterol concentrations [38]. Various truncated forms of apoB, such as apoB-32 [39], apoB-55 [40], apoB-61 [41], apoB-75 [42], and apoB-83 [43] to name a few, are found in human subjects displaying FHBL phenotype as a result of nonsense mutations occurring throughout the apoB polypeptide. To date, novel truncating mutations within APOB are continually being identified in FBHL patients devoid of cardiovascular diseases [44]. Cell culture and transgenic mouse studies with a variety of truncated apoB forms (ranging from apoB-15 to apoB-94) showed that most of the C-terminally truncated apoBs were secreted as efficiently as normal apoB-100 or apoB-48 [25,26,45,46].
Non-truncating mutations in APOB also cause FHBL; most of these nonsynoymous mutations occur within the βα1 domain of apoB [47-49]. Biochemical analysis has shown that unlike the apoB truncation mutants whose secretion efficiency was normal, the nonsynoymous apoB mutants exhibited markedly decreased secretion from transfected cells and abnormal retention of the mutant proteins within the ER or Golgi [49,50]. Twelve non-truncating mutations within the N-terminal 1,000 amino acids of apoB-100, some of them associated with hypobetalipoproteinemia and some confounded with hepatic steatosis, are shown in Fig. 1B. Table 1 summarizes the seven non-truncating apoB mutations that have been biochemically characterized. For example, the mutant A31P apoB proteins identified in an Italian FHBL subject showed severely impaired secretion and augmented intracellular degradation by proteasomes and autophagy [50]. The identification of missense mutations within the βα1 domain of apoB indicates the structural and functional importance of this domain, and also provides additional explanation for the early observations that recombinant apoB segments lacking the N-terminal 1,000 amino acids were either secreted poorly or not secreted at all [25,26]. Position of the amino acids corresponding to FHBL mutations within the predicted βα1 domain of apoB is depicted in Fig. 1C.
Table 1.
Biochemically characterized APOB missense mutations in FHBL
| Mutation | Predicted location | Hepatosteatosis | Secretion | Retention | Reference |
|---|---|---|---|---|---|
| A31P | β-barrel | yes | <5% normal | Golgi | [50] |
| G275S | α-helix | yes | 60% normal | n.d. | [50] |
| L324M | α-helix | yes | 50% normal | n.d. | [50] |
| L343V | α-helix | yes | 50% normal | ER | [48] |
| R463W | α-helix | yes | 50% normal | ER | [47] |
| G912D | β-sheet | yes | nearly normal | n.d. | [50] |
| G945S | β-sheet | yes | 50% normal | n.d. | [50] |
n.d, not determined
MTP and VLDL assembly
MTP is a heterodimer consisting of a 97-kDa lipid-binding and -transfer subunit [51] and a 55-kDa protein disulfide isomerase (PDI) [52] that is not required for lipid-transfer activity [53]. MTP is predominantly expressed in the hepatocytes and enterocytes where it transfers neutral lipids required for the assembly of apoB containing lipoproteins such as VLDL in the liver and chylomicrons in the intestine. Mutations leading to loss of MTP activity is linked to familial abetalipoproteinemia [54] in which the affected individuals have undetectable levels of apoB in the plasma. Structural studies of various domains within MTP have revealed a lipid-binding cavity resembling those found in intracellular lipid-binding proteins. The entrance of the cavity contains two conserved helices (helix-A: amino acids 725-736 and helix-B: amino acids 781-786). Mutational analysis of the helices has shown that helix A is required for the acquisition of lipids from phospholipid membranes whereas helix B plays a role in transferring lipids to the lipid-binding cavity. Mutations introduced in either of the helices resulted in abolition of lipid binding, which provides an explanation for abetalipoproteinemia found in humans carrying point mutations in the MTTP gene [55].
Availability of lipids in the vicinity of MTP is crucial for proper acquisition and transfer of lipids to the site of VLDL assembly within the ER. Lack of sufficient lipids supply compromises VLDL assembly and maturation, resulting in poorly lipidated apoB polypeptides that are prone to co- or post-translational degradation [56]. Inhibition of MTP with chemical inhibitor results in a similar outcome leading to the failure of VLDL assembly [57]. Studies with rat hepatoma McA-RH7777 cells have suggested that the early stage of lipid assembly initiated during apoB translation and translocation is facilitated by the MTP activity. However, during the later stage of VLDL assembly where bulk TAG is incorporated, the activity of MTP appears not essential [58]. This stage-dependent feature of MTP activity during VLDL assembly has also been observed in human hepatoblastoma HepG2 cells [59] and for the assembly of VLDL by McA-RH7777 cells transfected with recombinant human apoB-48 [60]. Graded inactivation of MTP with increasing doses of MTP inhibitor has revealed that the assembly and secretion of TAG-rich VLDL1 (Sf > 100) and VLDL2 (Sf 20-100) are more dependent on MTP activity than TAG-poor particles such as intermediate density lipoproteins (IDL) and LDL [57]. Moreover, the requirement of MTP activity is more pronounced for apoB-100 than for apoB-48, thus MTP inhibition effectively decreases apoB-100 and TAG secretion but has little effect on secretion of lipid-poor apoB-48 [61]. Lipid pulse-chase analysis carried out in rat primary hepatocytes after MTP activity was inhibited revealed delayed removal of TAG from the membranes of ER and Golgi. Inactivation of MTP did not affect lipolysis or subsequent re-esterification of TAG but the re-esterified TAG was not secreted as VLDL [62]. These cell culture studies suggest that MTP facilitates recruitment of TAG from the ER/Golgi membranes during VLDL assembly without affecting TAG biosynthesis.
In addition to cell culture studies, the role of MTP in VLDL assembly and secretion has also been investigated using genetically modified mice. The liver specific Mttp knockout mice displayed a phenotype equivalent to the human abetalipoproteinemia (i.e. absence of plasma apoB-100 and low levels of apoB-48) and were resistant to hypercholesterolemia induced by high cholesterol diet [63]. Ultrastructural analysis of liver specimen of the Mttp knockout mice has revealed absence of lipid particles within the ER/Golgi lumen, whereas in wildtype control mice the lipidic bodies of VLDL size were readily observable [64]. These results raise the possibility that the MTP activity is required for mobilization and partitioning of TAG substrates into the microsomal lumen for VLDL assembly.
Regulation of MTP expression is achieved by multiple factors that are closely related to fatty acid metabolism and changes in MTP expression is invariably associated with altered hepatic VLDL production. Studies with L35 cells, derived as a single cell clone from the rat hepatoma FAO cells, have shown that the inability of these cells to assemble or secrete VLDL resulted from co-repression of MTP and L-FABP (liver fatty acid binding protein) by chicken ovalbumin upstream promoter transcription factor II that occupied the DR1 promoter region of L-FABP and Mttp. Under these conditions, although VLDL secretion was blocked, the liver did not develop steatosis. In parental FAO cells, the DR1 promoter region of L-FABP and Mttp was occupied by peroxisome proliferator-activated receptor α (PPARα)-retinoid × receptor α (RXRα), and the transcription of L-FABP and MTP were normal, hence the assembly and secretion of VLDL was ensured [65]. In the leptin-deficient obese ob/ob mice, hepatosteatosis and insulin resistance (partly as a result of excessive fatty acid influx into the liver) were ameliorated by deletion of an orphan receptor small heterodimer partner (SHP) [66]. This study has revealed that SHP acts as a repressor of MTP mRNA transcription by liver receptor homolog-1 and therefore decreases MTP activity and VLDL secretion in ob/ob mice [66]. The MTP expression is also regulated by FoxO1, a transcription factor that plays a role in muscle cell growth and differentiation [67]. Studies with HepG2 cells have suggested that FoxO1 could bind to Mttp promoter region and directly stimulate MTP expression. It was also noticed that insulin treatment suppressed FoxO1 mediated MTP expression, and the insulin effect could be abolished by deleting or mutating the FoxO1 binding site within the Mttp promoter [68]. In addition, MTP expression can also be activated by Foxa2 together with PPARγ coactivator β (PGC-1β), resulting in increased VLDL secretion [69].
In summary, cell culture and transgenic mouse studies indicate that alteration in MTP activity has a direct effect on VLDL production. Attenuating MTP expression and/or activity without eliciting hepatosteatosis has been a long sought-after strategy in treating dyslipidemia related to overproduction of VLDL.
Fatty acid substrates affecting VLDL assembly and secretion
TAG utilized for VLDL assembly and secretion originates from multiple biosynthesis pathways. Fatty acyl chains used for TAG synthesis can be derived from de novo synthesis or from hydrolysis of existing TAG, presumably catalyzed by triacylglycerol hydrolase in the liver [70]. These fatty acyl chains are also used for phospholipid biosynthesis. In addition, hydrolysis of existing phospholipids associated with intracellular membranes [71,72] or exogenous phospholipids associated with high density lipoproteins (HDL) [73], also provides fatty acyl chains for TAG synthesis and secretion.
Comparative analyses have shown that the chemical nature of fatty acids, such as the acyl chain length and the extent of unsaturation (i.e. the number and position of double bonds) of acyl chains, have a profound impact on VLDL assembly and secretion. For instance, studies with chicken primary hepatocytes treated with different medium-chain fatty acids have shown that octanoate (8:0), decanoate (10:0), or dodecanoate (12:0) significantly decreased VLDL-apoB secretion compared to palmitate (16:0) [74]. The inhibitory effect of octanoate (usually present in coconut and breast milk) on VLDL was linked to suppression of apoB gene expression and apoB100 protein synthesis without affecting intracellular degradation of apoB [75].
Contrasting saturated fatty acids with mono- or poly-unsaturated fatty acids has shown that certain fatty acid species are preferentially utilized over others for VLDL assembly and secretion. McA-RH7777 cells, when cultured in the presence of exogenous oleate (18:1 n-9), secreted TAG-rich VLDL many fold higher as compared to no oleate supplementation [60,70,76,77]. However, when the cells were treated with poly-unsaturated fatty acids such as eicosapentaenoic acid (20:5 n-3), assembly and secretion of TAG-rich VLDL was significantly decreased [72]. The impaired secretion of apoB in cells treated with docosahexaenoic acid (22:6 n-3) was associated with increased oxidation, aggregation and autophagic degradation of apoB [78]. In rat primary hepatocytes, treatment with chylomicron remnants rich in saturated fatty acids or n-6 polyunsaturated fatty acids were more potent in promoting TAG-rich VLDL secretion than those rich in monounsaturated fatty acids [79]. HepG2 cells treated with a mixture of conjugated linoleic acids synthesized and secreted significantly less apoB-100 as compared to treatment with saturated or polyunsaturated fatty acids [80]. These studies suggest strongly that the characteristics of different fatty acids have distinct impacts on hepatic VLDL secretion.
The differential effects of fatty acid feeding on VLDL production has also been observed in vivo in animal studies. For example, transgenic mice lacking LDL receptor (Ldlr-/-) and expressing only apoB-100 responded differently in VLDL production to diet supplemented with echium oil (rich in 18:4 n-3), fish oil (rich in 20:5 n-3 and 22:6 n-3) or palm oil (enriched in 16:0) [81]. Feeding mice with diet supplemented with or without essential fatty acids also resulted in production of VLDL particles with different size and different catabolic rates [82].
Mechanisms by which certain fatty acid species preferentially promote VLDL secretion remain to be defined. Different fatty acid species may affect transcription of genes involved in TAG synthesis [81], which in turn may affect VLDL assembly and secretion. Acutely, treatment with different fatty acids may generate TAG molecules that are differentially utilized for VLDL assembly and secretion. For examples, studies with McA-RH7777 cells have shown that TAG derived from oleic acid (18:1 n-9) was partitioned into microsomes and was effectively secreted as VLDL, whereas TAG derived from eicosapentaenoic acid (20:5 n-3) was stored in the cytosol and was poorly secreted [72]. The discriminative usage of certain TAG molecules for VLDL assembly and secretion may be attributable to compartmentalization [72]. However as discussed below, because fatty acyl chains are also substrates for membrane phospholipid biogenesis, it is possible that a membrane milieu that supports assembly, trafficking, and maturation of TAG-rich VLDL is imparted by certain fatty acid species.
Besides the chemical nature of fatty acid species, the duration and concentration of fatty acid treatment also influence VLDL assembly and secretion. This could be as a result of either fatty acid-induced lipotoxicity or fatty acid-mediated transcription activation of lipid metabolism genes. Feeding Ldlr-/- mice with conjugated linoleic acid (10trans,12cis-18:2) for a short period of time resulted in hypertriglyceridemia with elevated plasma VLDL and HDL. However, long term feeding with the same fatty acid decreased plasma VLDL associated TAG, probably attributable to upregulated expression of hepatic VLDL receptor, lipoprotein lipase, and fatty acid translocase that could assist in the clearance of lipoproteins from circulation [83]. Prolonged incubation of McA-RH7777 cells with high concentrations of oleic acid also resulted in decreased apoB-100 secretion and increased apo-B100 degradation through proteasomal and non-proteasomal pathways, as a result of ER stress associated with massive accumulation of intracellular TAG [84]. Thus, overloading of lipids to the extent that exceeds the capacity of hepatocytes to effectively assemble and secrete VLDL would lead to impaired TAG secretion and cause hepatosteatosis. In addition to fatty acid-associated lipotoxicity, it has been reported recently that hepatic overexpression of apoB also induce hepatic ER stress and insulin resistance [85]. The molecular mechanisms by which excess fatty acid influx and overproduction of apoB-100 lead to ER stress and altered insulin signal transduction merits further investigation.
Glycerolipid biogenesis and VLDL assembly and secretion
Identification and characterization of glycerolipid synthesis gene products have accelerated our understanding on the impact of hepatic lipid synthesis and/or their availability on VLDL assembly and secretion. The rate limiting step in the de novo biosynthesis of TAG and phospholipids is catalyzed by phosphatidate phosphatase-1 (PAP-1) [86], which converts phosphatidate to diacylglycerol that is subsequently utilized for the synthesis of TAG and phospholipids such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE). In mammals, PAP-1 is encoded by the lipin gene family consisting of lipin-1, -2 and -3 [87,88]. In humans, lipin-1 is highly expressed in skeletal muscles and adipose tissues; lipin-2 and -3 are expressed in brain, liver, jejunum, placenta, and adipose tissues [88]. The Lpin1 gene encodes two alternatively spliced isoforms lipin-1α and -1β [89], and the expression of Lpin1 is upregulated by glucocorticoids and suppressed by insulin [90]. Transient expression of lipin-1α or -1β in McA-RH7777 cells resulted in increased synthesis and secretion glycerolipids under basal or lipid-rich conditions [91]. Secretion of TAG as VLDL1 was increased upon lipin-1α or -1β expression when cells were cultured under lipid-rich conditions [91]. These results were not in accord with studies of hepatocytes isolated from the Lipn1-deficiency fld (fatty liver lipodystrophy) mice, in which the rate of TAG synthesis was not changed in the hepatocytes isolated from adult mice, however, the rate of TAG synthesis increased in the hepatocytes isolated from 14-day-old fld mice. Adenovirus mediated Lipin-1 overexpression in the hepatocytes isolated from adult fld mice resulted in the suppression of VLDL-TAG secretion [92]. Moreover, adenovirus-mediated lipin-1 overexpression in the obese, insulin-resistant UCP-DTA mice resulted in decreased the fasting plasma TAG concentration [92]. The reason for the discrepancy between these studies regarding the role of lipin-1 in VLDL secretion remains to be explained. It has been shown that compartmentalization of lipin-1, in addition to its PAP1 activity, also plays a role in VLDL assembly and secretion [91]. Expression of a mutant form of lipin-1α, in which the nuclear localization signal sequence was removed, resulted in cytosolic presentation of the protein and diminished stimulation of VLDL1 secretion [91]. Nuclear localization of lipin-1 is probably important for transactivation of gene expression, as lipin-1 is known to act as a transcription activator [93]. The dual function of lipin-1 and its variable subcellular compartmentalization with respect to VLDL assembly and secretion requires further investigation.
The final step in the de novo synthesis of TAG is catalyzed by acyl-CoA:diacylglycerol acyltransferase (DGAT). Two DGAT genes encode the respective DGAT1 and DGAT2, which are structurally unrelated and show hepatic expression [94,95]. The DGAT1 knockout mouse has a normal fasting plasma TAG level despite reduced hepatic TAG [96]. Adenovirus mediated expression of DGAT1 in mice increased TAG in the liver but did not increase VLDL production [97]. However, overexpression of human DGAT1 in McA-RH7777 cells resulted in increased synthesis, cellular accumulation, and secretion of TAG as VLDL [98,99]. Concomitantly, intracellular degradation of apoB was decreased in DGAT1 overexpressing cells [98]. Overexpression of DGAT2 in McA-RH7777 cells also resulted in increased secretion of TAG and apoB [98], whereas knockdown of DGAT2 in mice with antisense oligonucleotide decreased TAG and apoB secretion as VLDL [100]. These studies suggest that although TAG synthesis is important for promoting VLDL production, the increased hepatic TAG may be compartmentalized and thus not necessarily be available for VLDL assembly or secretion.
Hepatic VLDL assembly and secretion is also profoundly influenced by alterations in the de novo biosynthesis of phospholipids, such as PE and PC. Two pathways are involved in hepatic PC synthesis; the CDP-choline pathway contributes approximately 70% of total hepatic PC synthesis, whereas the remainder 30% is synthesized through the PE methylation pathway. The PE methylation pathway is catalyzed by PE N-methyltransferase (PEMT), a product of the Pemt2 gene. The Pemt2-/- mouse had normal liver PC content as a result of compensatory upregulation of CTP:phosphocholine cytidylyltransferase (CCT) activity; hence these mice did not display abnormal plasma lipid levels compared to control mice [101]. It appears that as long as CCT activity is intact, hepatic PC synthesis is uncompromised when the PEMT activity is absent. Lack of PEMT in the Ldlr-/- background, however, resulted in a reduction of atherosclerosis incidence when the mice were fed with a high fat diet for 16 weeks [102]. These reduced atherosclerotic lesion development was attributed to lowered phospholipid content in apoB-containing lipoproteins (e.g. VLDL, IDL, and LDL) and reduced secretion of VLDL [102].
The rate limiting step in the CDP-choline pathway for PC synthesis is catalyzed by CCT. At least three CCT isoforms are known in mammals and they are encoded by two genes, namely Pcyt1α for CCTα and Pcyt1b for differentially spliced transcripts CCTβ2 and CCTβ3 [103]. It has been shown that plasma levels of HDL and VLDL were markedly reduced in mice lacking hepatic CCTα expression [104], indicating that the activity of hepatic CCT is an important determinant of VLDL and HDL in vivo. However, reconstituted CCTα in the knock-out hepatocytes by ex vivo infection with adenovirus encoding CCTα failed to stimulate VLDL secretion even though cellular PC levels returned to normal [105]. In contrary, adenoviral delivery of CCTα into knock-out mice normalized plasma HDL and VLDL [105]. The CCTα activity is thus required for lipoprotein metabolism in vivo.
Hepatic PE synthesis is likewise achieved through the CDP-ethanolamine pathway, in which CTP:phosphoethanolamine cytidylyltransferase (encoded by Pcyt2) catalyzes the rate regulatory step for the formation of CDP-ethanolamine [106]. Reduced CDP-ethanolamine synthesis in Pcyt2+/- mice resulted in elevated plasma VLDL (at 32-36 weeks old), as well as progressive development of hepatosteatosis, obesity, and insulin resistance [107]. The increased hepatic TAG synthesis and secretion in Pcyt2+/- mice are probably attributable to increased availability of diacylglycerol (as a result of limited CDP-ethanolamine) [107].
Because phospholipid synthesis not only supplies lipid substrates for VLDL but also contributes to the biogenesis and maintenance of ER/Golgi membranes, it impacts both the cargo and trafficking machinery for VLDL assembly and secretion. Studies with genetically modified mice lacking both PEMT and multiple drug-resistant protein 2 have suggested that the PE/PC ratio is a key regulator of cell membrane integrity in the liver and plays a role in the progression of steatosis into steatohepatitis [108]. It remains to be determined whether or not the composition of phospholipid species in the ER/Golgi membranes affects VLDL assembly/secretion.
VLDL assembly and secretion influenced by vesicular trafficking factors
Protein factors involved in intracellular vesicular trafficking, primarily the small GTPase proteins, play a profound role in VLDL assembly and secretion. The requirement of COPII coated vesicles for apoB exiting from the ER has been demonstrated by an in vitro ER budding assay [109]. Formation of COPII coated vesicles is initiated by the GTPase Sar1 protein. Deficiency of Sar1 has been linked to intestinal apoB-lipoprotein secretion disorder known as chylomicron retention disease in humans [110]. Expression of a GDP-restricted mutant (T39N) of the Sar1 protein in McA-RH7777 cells prevented ER exit of apoB [109]. The anterograde transport from the Golgi apparatus is driven by the small GTPase, ADP ribosylation factor 1 (ARF1). Expression of a dominant-negative T31N mutant of ARF1 in McA-RH7777 cells resulted 80% reduction in the assembly of apoB-100 VLDL1 along with the loss of COPI from the Golgi apparatus. Overexpression of ARF1 in the cells resulted in an oleate dose dependent increase in VLDL1 secretion with a concomitant decrease in VLDL2 secretion [111].
The activities of two phospholipases have been shown to play a role in VLDL maturation, the calcium independent phospholipase A2 (iPLA2β) and phospholipase D (PLD1) [71,72,112]. The iPLA2β is an intracellular protein that does not have a Ca2+-dependent lipid-binding domain but contains ankyrin repeats that may mediate membrane binding. Inhibition of iPLA2β with chemical inhibitors or antisense RNA interfered with the formation of TAG-rich VLDL1 but not dense VLDL particles such as VLDL2 [71,72]. The PLD1 catalyzes the hydrolysis of PC to produce PA and choline and is activated by ARF1. Inactivation of PLD1 activity (and thus the formation of PA) in cultured hepatic cells using chemical inhibitors also blocked VLDL formation [71,72]. Likewise, expression of aberrant ARF1 that lost PLD1-activation function [111] or treatment with brefeldin A (an inhibitor of ARF1) [56,113] effectively prevented TAG-rich VLDL secretion with little effect on the secretion of denser particles. These results suggest that maturation of VLDL probably not only requires sufficient lipid substrate availability, but also depends upon the protein factors that effectively mediate the fusion between lipid droplets and apoB to form TAG-rich VLDL. The fusion events presumably require coordinated synthesis and fusion of vesicle themselves, as well as synthesis and fusion of cargo (i.e. lipid droplets and VLDL precursors) within the vesicles. Factors within the microsomal lumen that are responsible for cargo fusion during the final step of VLDL1 maturation remain to be determined.
Cytosolic lipid droplet-associated proteins affecting VLDL assembly and secretion
Multiple protein factors that are found in association with cytosolic lipid droplets also influence VLDL assembly and secretion. Adipocyte differentiation-related protein (ADRP) is the major protein associated with cytosolic lipid droplets. Overexpression of ADRP in McA-RH7777 cells resulted in increased accumulation of cytosolic lipid droplets and a corresponding decrease in VLDL secretion [114]. Although the molecular mechanism responsible for the inhibitory effect of ADRP expression on VLDL secretion remains to be defined, it is possible that enlargement of cytosolic lipid droplets may diminish the microsomal TAG pool for VLDL secretion.
Recently, another lipid droplet associated protein CideB has been suggested to play a role in VLDL assembly and secretion [115]. CideB is a member of Cide (cell death-inducing DFFA45 (DNA fragmentation factor 45)-like effector) family that also includes CideA, and CideC (or Fsp27). While CideA is expressed at high levels in brown adipose tissue, CideB mRNA and proteins are detected in various tissues with the highest level of expression in the liver. Hepatic CideB exists as a smooth ER- and lipid droplet-associated protein. Mice deficient in CideA or CideB are resistant to high-fat diet-induced obesity and diabetes [116]. The role of CideB in regulating lipid homeostasis has been studied with the Cideb-/- mice. Compared with wildtype littermates, the Cideb-/- mice exhibited an increase in hepatic TAG content and reduced VLDL secretion [115]. Yeast two hybrid and co-immunoprecipitation experiments have shown a physical interaction between CideB and apoB [115]. These data suggest that the cytosolic lipids droplets are functionally in close contact with the ER where initial VLDL assembly takes place.
Non-apoB apolipoproteins affecting VLDL assembly and secretion
In addition to apoB-100, hepatic VLDL particles also contain other apolipoproteins such as apoE and apoC. ApoE (299 amino acids) is a major protein constituent of TAG-rich lipoproteins including VLDL and chylomicrons. Experimental evidences obtained by several laboratories working with apoE overexpressing transgenic mice [117-119] or McA-RH7777 cells [120] have suggested that apoE plays a role in the formation of fully lipidated VLDL. The underlying mechanisms by which apoE expression promotes VLDL assembly and secretion were unclear. Recently, a detailed analysis conducted using McA-RH7777 cells treated with apoE specific siRNA or primary hepatocytes isolated from apoE-/- mice have shown that the assembly (within the Golgi apparatus) or secretion of VLDL was independent of apoE expression [121]. Thus, the VLDL-associated apoE in the Golgi apparatus and media may not drive the formation of fully lipidated VLDL.
Apolipoprotein C-III is a small (79 amino acids) exchangeable apolipoprotein composed of multiple amphipathic α-helices, and is expressed mainly in the liver [122]. It has been reported recently that overexpression of human apoC-III in McA-RH7777 cells resulted in the overproduction and secretion of VLDL-TAG and VLDL-apoB under lipid rich conditions [76]. Overexpression of apoC-III also resulted in increased activity and expression of MTP. The ability of apoC-III to stimulate hepatic VLDL assembly and secretion was abolished by a naturally occurring mutation Ala23Thr [123] that was identified in human subjects with hypotriglyceridemia [124]. Thus apoC-III, a component of VLDL and HDL, appears to play an intracellular role in stimulating VLDL assembly and secretion [76,123]. Mechanisms by which apoC-III exerts the stimulatory effect on VLDL assembly and secretion are unclear.
The human APOA5 gene is a part of the apolipoprotein gene cluster that contains APOA1, APOC3, and APOA4 on chromosome 11 (11q23) [125]. Initial studies has revealed association of single nucleotide polymorphisms within the APOA5 locus with plasma TAG and VLDL in humans, and the effect is not related to the neighboring APOC3 gene markers [125]. Mice expressing the APOA5 transgene displayed a 65% decrease in plasma TAG levels; whereas apoa5 knock-out mice showed a 4-fold increase in plasma TAG concentration [125]. Thus, apoA-V (343 amino acids) has been viewed as a candidate gene regulating plasma TAG concentrations [126]. Overexpression of murine apoA-V in C57Bl/6 mice through adenovirus-mediated gene transfer decreased VLDL production rate in a dose-dependent manner by impairing apoB lipidation [127]. In the same mouse model, overexpression of apoA-V also resulted in decreased plasma TAG by enhancing lipoprotein lipase-mediated clearance of TAG-rich lipoproteins [127]. Transfection studies showed that recombinant apoA-V expressed in McA-RH7777 cells was unexpectedly associated with cytosolic lipid droplets, despite the fact that apoA-V possesses the signal peptide and is a secretory protein [128]. The mechanisms whereby apoA-V expression attenuates VLDL production remain to be defined.
Lipoprotein receptors affecting VLDL assembly and secretion
Intracellular degradation of newly synthesized apoB-100 diminishes overall assembly and secretion of VLDL. The LDL receptor, a ubiquitously expressed protein responsible for the clearance of cholesterol-rich lipoproteins from blood stream through its ligands apoB-100 and apoE, promotes intracellular degradation of apoB-100 resulting in decreased VLDL secretion [129]. The loss of LDL receptor activity in the liver cells leads to increased secretion of VLDL particles, and the particles secreted are small with reduced TAG content [130,131]. Thus, the LDL receptor mediated apoB-100 degradation appears to preferentially target underlipidated particles. Mechanisms responsible for the LDL receptor mediated apoB-100 degradation may involve (i) rapid reuptake of nascent VLDL particles on the cell surface [129,132] and (ii) intracellular targeting of nascent VLDL particles to degradation [129,133]. Both mechanisms appear to require exit of apoB-100 from the ER and interaction of apoE or apoB-100 with the LDL receptor [134].
Expression and function of LDL receptor are negatively regulated by proprotein convertase subtilisin kexin type 9 (PCSK9) [135,136]. Attempts were made to determine whether the level of PCSK9 expression would affect apoB secretion or intracellular degradation. Transfection studies with McA-RH7777 cells that stably expressed the D374Y mutant form of PCSK9 (identified in human familial hypercholesterolemia) showed that the pathogenic variant expression resulted in increased secretion of apoB-100 lipoproteins (by 2-4-fold) but expression of the wildtype PCSK9 did not increase apoB-100 secretion [137]. These results might be interpreted as reduced degradation of nascent apoB-100 protein mediated by the LDL receptor. Lack of an effect of wildtype PCSK9 expression on apoB-100 secretion was also observed in transfection studies with the human hepatoma HuH7 cells [138]. Likewise, secretion of apoB-100 from primary hepatocytes of wildtype or Pcsk9-/- mice was not significantly different [139]. The possible link between PCSK9, LDL receptor, and hepatic apoB-100 secretion remains to be determined.
Genetic absence of functional ATP binding cassette transporter A1 (ABCA1) in Tangier disease is associated with severely lowered plasma HDL and concomitantly elevated plasma TAG concentrations [140]. This metabolic abnormality has been recapitulated recently in McA-RH7777 cells where the ABCA1 was silenced using siRNA [141]. Unexpectedly, supplementation of culture media of the ABCA1 knockdown cells with nascent large HDL decreased the secretion of buoyant VLDL1 particles [141]. It appears that the large HDL, assembled by hepatic ABCA1, can attenuate VLDL secretion through the phosphoinositide 3 kinase (PI3K)-dependent signaling pathway [141]. This study is reminiscent of a previous observation that the PI3K activity is required for insulin-dependent inhibition of apoB secretion from primary rat hepatocytes [142].
Scavenger receptor B1 (SR-B1) is a receptor for selective uptake of HDL cholesterol and is also known to mediate the catabolism of apoB-containing lipoproteins. Overexpression of SR-B1 in mice via adenovirus-mediated infection resulted in increased plasma concentrations of VLDL-TAG and VLDL-apoB [143]. On the other hand, disruption of the Scarb1 gene expression in mice resulted in reduced VLDL production as well as MTP activity [143], suggesting that hepatic SR-B1 expression is closely linked to VLDL production.
Overall, the involvement of various lipoprotein receptors in regulating VLDL assembly and secretion has gradually been revealed, which introduces another level of complexity to the regulation of hepatic TAG homeostasis. Because these lipoprotein receptors are intimately involved in the metabolism of cholesterol (and phospholipids as well), such as uptake, efflux, and intracellular trafficking, it is possible that hepatic VLDL assembly and secretion are influenced by cellular cholesterol metabolism and related signaling events. The interrelationship between TAG and cholesterol metabolism and its link to VLDL assembly and secretion merits further investigation.
Altered insulin and leptin signaling affecting VLDL assembly and secretion
Hepatic VLDL overproduction and impairment in catabolism/clearance of TAG-rich lipoproteins from circulation represent the two major contributors to hypertriglyceridemia. Many patients with hypertriglyceridemia manifest elevated plasma TAG, accumulation of small dense LDL particles and reduced HDL cholesterol particles, all of which are closely associated with cardiovascular diseases [144]. The other traits associated with hypertriglyceridemia include visceral obesity and insulin resistance, which further exacerbate the aberrant overproduction of VLDL as a result of excess flux of fatty acids (derived partly from lipolysis of plasma TAG-rich lipoproteins) into the liver. Overall, insulin treatment decreases hepatic VLDL production by limiting fatty acid influx into the liver, decreasing the stability of apoB, and promoting the posttranslational degradation of apoB, a process mediated through the PI3-K pathway [145]. Overproduction of hepatic VLDL that results from the loss of insulin responsiveness is often seen in insulin resistance conditions, which is associated with increased posttranslational stability of apoB-100 [145]. The insensitivity of liver cells to insulin mediated suppression of VLDL assembly is observed in fructose-induced insulin resistant hamster model [146], where insulin resistance is accompanied with hepatic inflammation. The fructose-induced, insulin-resistant hamsters exhibited reduced levels of the inhibitor of nuclear factor-κB (IκB) which resulted in a concomitant activation of the inflammatory nuclear factor-κB (NF-κB) cascade. Inhibition of NF-κB cascade with chemical inhibitors also resulted in decreased synthesis of apoB-100 in primary hepatocytes and HepG2 cells, and was probably attributable to the activation of insulin signaling and enhanced proteasomal degradation of apoB-100 [147]. On the other hand, activation of the NF-κB pathway, via adenovirus-mediated IκB kinase overexpression, resulted in increased apoB-100 synthesis as a result of suppressed insulin signaling through the NF-κB pathway [147]. Thus, there is an important link between the inflammatory IκB kinase-NF-κB signaling cascade, insulin signaling, and hepatic apoB100 synthesis and secretion.
The insensitivity of liver cells to insulin-mediated suppression of VLDL assembly is also observed in leptin-deficient obese mouse (ob/ob) [148]. Leptin is an adipose derived hormone that plays a key role in energy intake and expenditure. The leptin-deficient ob/ob mice and the leptin receptor-deficient db/db mice have been used extensively as model systems to study their roles in VLDL assembly and secretion [149]. The in vivo secretion rates of TAG and apoB were reduced in both male and female ob/ob mice as compared to their littermates. However, in db/db obese mice, only male mice showed reduced secretion of TAG. In ob/ob mice and db/db obese females, there was a small increase in apoB-100 secretion and no difference in apoB48 secretion. In these animals, the main cause of dyslipidemia was due to the impaired removal of VLDL from the circulation [149].
The ob/ob and db/db mice have been used to determine diet or protein factors that influence the pathophysiology of lipoprotein metabolism. For instance, the db/db mice have been crossed with mice expressing human cholesteryl ester transfer protein (CETP); the resulting mice displayed lowered levels of VLDL and LDL and became resistant to diet-induced atherosclerosis as compared to controls [150]. These results ascribe an anti-atherogenic role for CETP under diabetic obese conditions. The ob/ob mice crossed into the LDL receptor knockout background (Ldlr-/-) showed severe hyperlipidemia and spontaneous atherosclerosis, which was associated with increased hepatic TAG production, delayed VLDL clearance, and decreased hepatic uptake of LDL [151]. The db/db mice have also been used as a model to study diet-induced nonalcoholic steatohepatitis (NASH). Feeding db/db mice with methionine- and choline-deficient (MCD) diet induced liver injury [152,153], and were probably attributable to impaired hepatic secretion of VLDL [154]. The MCD-fed db/db mice developed hepatic steatosis and displayed insulin resistance that is severer than MCD-fed control mice [153]. These studies indicate that increased fatty acid uptake together with decreased secretion of VLDL represent the major insult that lead to hepatic TAG accumulation under MCD diet conditions.
Liver × receptor α (LXRα), the master regulator of lipid metabolism [155], also regulates hepatic VLDL production. Activation of LXR in hamsters fed with an LXR agonist resulted in markedly increased plasma TAG and VLDL and enhanced expression of sterol response element binding protein-1c (SREBP-1c) and its target lipogenesis genes including steroyl CoA desaturase and fatty acid synthase [156]. Furthermore, LXR activation also led to enhanced stability of newly synthesized apoB and increased secretion of TAG-rich VLDL-apoB. Increased stability of apoB-100 was probably achieved through attenuated insulin receptor and insulin receptor substrate-1 tyrosine phosphorylation and concomitant increases in protein tyrosine phosphatase 1B [156].
Studies with rats have shown that acute leptin treatment, like that of insulin, lowered plasma VLDL-associated TAG [157,158]. However, the molecular mechanism responsible for the leptin-induced hypotriglyceridemia may be distinct from that of insulin. The leptin treatment resulted in decreased lipogenesis and enhanced β-oxidation, thus limiting lipid substrate for VLDL assembly/secretion without affecting hepatic apoB levels [159,160]. On the other hand, insulin treatment had no effect on β-oxidation but decreased hepatic apoB levels [160], probably owning to enhanced posttranslational degradation of apoB. In summary, hepatic VLDL production is promoted under conditions where post-translational stability of apoB is augmented (e.g. enhanced lipid substrate supply), and its production is suppressed when post-translational degradation of apoB is increased (e.g. decreased lipid substrate availability).
Intracellular degradation of newly synthesized apoB
Intracellular degradation of apoB refers to a process whereby the newly synthesized apoB proteins are degraded prior to secretion. It has been shown that apoB degradation occurs during and after the protein translation, and the co- and post-translational degradation of apoB takes place in both ER and post-ER compartments. Attenuated intracellular degradation of newly synthesized apoB is accompanied with increased VLDL production, as often observed under chronic hyperinsulinemia and insulin resistance conditions [161,162]. Under conditions unfavorable for apoB folding or lipid assembly, the newly synthesized apoB polypeptide undergoes ubiquitin-mediated proteasomal degradation [56,163-165]. Lack of sufficient lipid supply or availability often triggers the degradation process. Under certain conditions, apoB degradation occurs even after an assembly intermediate (i.e. a VLDL precursor) has already been assembled (for extensive review on apoB degradation see [166-169]).
Non-proteasomal degradation pathway for apoB has been described which involves autophagosomes [170,171]. Autophagosomes are membrane structures that encase and target intracellular substrates to lysosomes for disposal, a process termed autophagy [172]. The involvement of autophagy process for apoB degradation appears to be pronounced when ubiquitin/proteasome pathway is inhibited, and the apoB proteins accumulated in the crescent-shaped structures that are in close proximity to cytosolic lipid droplets [173]. It has been postulated that the crescent structures are the sites where proteasomal and autophagosomal pathways converge [173]. The autophagy mediated apoB degradation has also been suggested in cells treated with n-3 fatty acids such as docosahexaenoic acid (22:6 n-3), and under this condition apoB also undergoes aggregation and oxidation [78]. As mentioned earlier, the n-3 fatty acid induced intracellular apoB degradation is probably related to the poor utilization of TAG molecules for VLDL assembly and secretion [72]. Missense mutations within the βα1 domain of apoB also resulted in increased degradation through autophagy [50], which presumably resulted from impaired secretion of the mutant apoB as lipoproteins.
In summary, in addition to the ubiquitin/proteasome pathway presumably responsible for ER-associated degradation of misfolded apoB polypeptides, the autophagosome-mediated apoB and apoB-lipoprotein degradation may represent an alternative pathway (post-ER degradation) for the disposal of aborted assembly intermediates. Since augmented intracellular degradation of apoB and apoB-lipoproteins is a potential means to suppress overproduction of the atherogenic VLDL/LDL, further studies are merited to identify and characterize factors involved in autophagy-mediated apoB degradation.
Concluding remarks and perspectives
The past three decades have witnessed a tremendous advancement in our knowledge and understanding of protein and lipids factors that influence VLDL assembly and secretion. Amino acid sequences within apoB-100, particularly the N-terminal βα1 domain and the β-sheet enriched β1 domain, have been recognized as functional elements governing VLDL assembly/secretion. The role of MTP in facilitating lipid mobilization into the microsomal lumen and that of protein factors (e.g. apoC-III) in promoting bulk TAG incorporation during VLDL maturation is being revealed. In addition, enzymes that are directly involved in hepatic lipogenesis and glycerolipids biogenesis, such as lipin-1 and DGAT among others, have been cloned and characterized. Delineation and characterization of regulatory pathways of these lipid synthesis enzymes have provided molecular explanations for hormonal regulation (e.g. insulin and glucocorticoids) of VLDL assembly and secretion. Moreover, a comprehensive view of the interplay among various transcription factors (e.g. SREBP-1, LXR, PPAR, and PGC-1) in regulating hepatic lipogenesis, β-oxidation, and glycerolipids biogenesis has begun to emerge, and their implications to VLDL assembly/secretion under stress conditions require further investigation. Finally, new mechanisms such as proteasomes and autophagosome that may play a role in intracellular degradation of apoB thus attenuate hepatic VLDL production have been suggested and also merit additional studies. Various protein and lipid factors that participate and influence hepatic VLDL assembly/secretion are depicted in Fig. 2.
Figure 2.
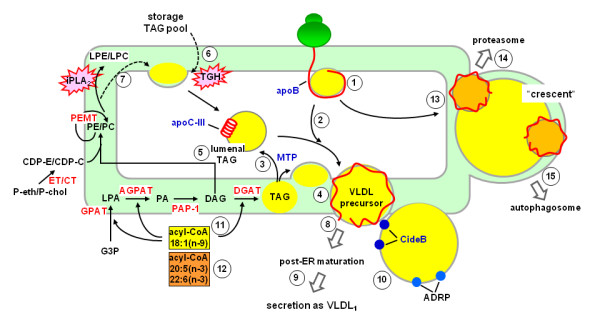
Protein and lipid factors affecting VLDL assembly and secretion. The apoB polypeptide initiates lipid recruitment during translation and translocation (1); this process may or may not require the activity of MTP. The nascent apoB-lipid particle acquires, in a step-wise fashion, additional TAG (2). The activity of MTP is required for partitioning of TAG into the lumen (3) or the membranes of ER microsomes for VLDL assembly (4). The lumenal TAG substrate, referred to as "lumenal lipid droplet", exists in association with non-apoB apolipoproteins such as apoC-III (5). In addition to that synthesized from the de novo pathway (catalyzed by GPAT, AGPAT, PAP-1, and DGAT), the TAG substrate utilized for VLDL assembly is also derived from esterification of fatty acyl chains originated from TGH-mediated hydrolysis of existing storage and lumenal TAG pools (6) or from phospholipid turnover catalyzed by iPLA2 (7). The resulting VLDL precursor exits the ER through coatomer-mediated budding/vesiculation events (8), and maturation of VLDL1 is achieved through ER/Golgi trafficking (9). Proteins associated with cytosolic lipid droplets, such as CideB and ADRP (10), influence VLDL assembly through an unknown mechanism. Molecular species of fatty acids have a profound impact on VLDL production; oleic acid (18:1 n-9) promotes (11) whereas EPA (20:5 n-3) and DHA (22:6 n-3) attenuate (12) VLDL assembly/secretion. Misfolded apoB protein and aborted lipoprotein assembly intermediates are targeted for intracellular degradation (13). Two degradative pathways, namely the ubiquitin/proteasome pathway (14) and autophagy (15), may involve formation of "crescent" structures that contain apoB and are in close association with cytosolic lipid droplets. ADRP, adipocyte differentiation-related protein; AGPAT, acylglycerol-3-phosphate acyltransferase; CDP-C, CDP-choline; CDP-E, CDP-ethanolamine; CT, CTP:phosphocholine cytidylyltransferase; DAG, diacylglycerol; DGAT, acyl-CoA:diacylglycerol acyltransferase; ET, CTP:phosphoethanolamine cytidylyltransferase; G-3-P, glycerol-3-phosphate; GPAT, glycerol-3-phosphate acyltransferase; LPA, lysophosphatidate; LPC, lysophophatidylcholine; LPE, lysophosphatidylethanolamine; MTP, microsomal triglyceride-transfer protein; PAP-1, phosphatidate phosphatase-1; PC, phosphatidylcholine; P-cho, phosphocholine; PE, phosphatidylethanolamine; P-eth, phosphoethanolamine; TAG, triacylglycerol; VLDL, very low density lipoproteins.
Other than understanding the biosynthesis of lipid substrates required for VLDL assembly and secretion, a great deal of knowledge has also accumulated with respect to the temporal and spatial events associated with lipid incorporation into VLDL. It is clear that VLDL precursors are assembled within the ER and the maturation of TAG-rich VLDL1 take place in post-ER compartments. However, several questions remain unanswered. For example, it is unclear about the biochemical origin(s) of TAG or the relative contribution between the de novo synthesis and the hydrolysis/re-esterification pathways during VLDL maturation under normal or stress conditions. It is also unclear what protein factors or membrane structures are required for the mobilization of lumenal TAG substrates or their delivery to VLDL precursors. Although increasing evidence suggests that protein factors associated with cytosolic lipid droplets (e.g. CideB and ADRP) may participate or influence lumenal VLDL assembly, the cellular mechanisms through which the cytosolic and lumenal components communicate remains an enigma. Finally, knowledge gained from cell culture and animal studies remains to be verified in humans with advanced in vivo technologies to validate the pathophysiological relevance.
List of abbreviations used
ABCA1: ATP binding cassette transporter A1; ADRP: adipocyte differentiation-related protein; AGPAT: 1-acylglycerol-3-phosphate O-acyltransferase; ARF1: ADP ribosylation factor 1; CCT: CTP:phosphocholine cytidylyltransferase; CETP: cholesteryl ester transfer protein; DGAT: acyl-CoA:diacylglycerol acyltransferase; DHA: docosahexaenoic acid; EPA: eicosapentaenoic acid; ER: endoplasmic reticulum; FHBL: familial hypobetalipoproteinemia; GPAT: glycerol-3-phosphate acyltransferase; HDL: high density lipoproteins; IDL: intermediate density lipoproteins; IκB: inhibitor of nuclear factor-κB; LDL: low density lipoproteins; L-FABP: liver fatty acid binding protein; LPA: lysophosphatidate; LXRα: liver × receptor α; MCD: methionine- and choline-deficient; MTP: microsomal triglyceride-transfer protein; NF-κB: nuclear factor-κB; PA: phosphatidate; PAP-1: phosphatidate phosphatase-1; PC: phosphatidylcholine; PCSK9: proprotein convertase subtilisin kexin type 9; PDI: protein disulfide isomerase; PE: phosphatidylethanolamine; PEMT: PE N-methyltransferase; PGC: peroxisome proliferator-activated receptor gamma coactivator; PI3K: phosphoinositide 3 kinase; PPAR: peroxisome proliferator-activated receptor; SHP: small heterodimer partner; SR-B1: scavenger receptor B1; SREBP: sterol regulatory element binding protein; TAG: triacylglycerol; TGH: triacylglycerol hydrolase; VLDL: very low density lipoproteins.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both MS and ZY drafted, edited, and final approved the manuscript.
Authors' information
Meenakshi Sundaram, PhD, Research Associate, Department of Biochemistry, Microbiology and Immunology, University of Ottawa
Zemin Yao, PhD, Professor and Chair, Department of Biochemistry, Microbiology and Immunology, Ottawa Institute of Systems Biology, University of Ottawa
Contributor Information
Meenakshi Sundaram, Email: msundara@uottawa.ca.
Zemin Yao, Email: zyao@uottawa.ca.
Acknowledgements
This work is supported by Canadian Institutes of Health Research Grant NMD-15486. Z.Y. was a recipient of the Career Investigator award from the Heart and Stroke Foundation of Ontario.
References
- Law SW, Lackner KJ, Hospattankar AV, Anchors JM, Sakaguchi AY, Naylor SL, Brewer HB Jr. Human apolipoprotein B-100: cloning, analysis of liver mRNA, and assignment of the gene to chromosome 2. Proc Natl Acad Sci USA. 1985;82:8340–8344. doi: 10.1073/pnas.82.24.8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ, West R, Mehrabian M, Reuben MA, LeBoeuf RC, Kaptein JS, Johnson DF, Schumaker VN, Yuhasz MP, Schotz MC. Cloning and expression of apolipoprotein B, the major protein of low and very low density lipoproteins. Proc Natl Acad Sci USA. 1985;82:4597–4601. doi: 10.1073/pnas.82.14.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb SS, Disteche C, Motulsky AG, Lebo RV, Kan YW. Chromosomal localization of the human apolipoprotein B gene and detection of homologous RNA in monkey intestine. Proc Natl Acad Sci USA. 1986;83:419–422. doi: 10.1073/pnas.83.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, Weng SA, Silberman SR, Cai SJ, Deslypere JP, Rosseneu M. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238:363–366. doi: 10.1126/science.3659919. [DOI] [PubMed] [Google Scholar]
- Kane JP, Hardman DA, Paulus HE. Heterogeneity of apolipoprotein B: isolation of a new species from human chylomicrons. Proc Natl Acad Sci USA. 1980;77:2465–2469. doi: 10.1073/pnas.77.5.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennyson GE, Sabatos CA, Higuchi K, Meglin N, Brewer HB Jr. Expression of apolipoprotein B mRNAs encoding higher- and lower-molecular weight isoproteins in rat liver and intestine. Proc Natl Acad Sci USA. 1989;86:500–504. doi: 10.1073/pnas.86.2.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, De LH, Dashti N. Structure of apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001;42:1346–1367. [PubMed] [Google Scholar]
- Mann CJ, Anderson TA, Read J, Chester SA, Harrison GB, Kochl S, Ritchie PJ, Bradbury P, Hussain FS, Amey J. The structure of vitellogenin provides a molecular model for the assembly and secretion of atherogenic lipoproteins. J Mol Biol. 1999;285:391–408. doi: 10.1006/jmbi.1998.2298. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Gantz D, Bullitt E, McKnight CJ. Defining lipid-interacting domains in the N-terminal region of apolipoprotein B. Biochemistry. 2006;45:11799–11808. doi: 10.1021/bi060600w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashti N, Gandhi M, Liu X, Lin X, Segrest JP. The N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assembly. Biochemistry. 2002;41:6978–6987. doi: 10.1021/bi011757l. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Simon MN, Wall JS, McKnight CJ. Structural analysis of reconstituted lipoproteins containing the N-terminal domain of apolipoprotein B. Biophys J. 2007;92:4097–4108. doi: 10.1529/biophysj.106.101105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Rudenko G, Ludtke SJ, Deisenhofer J, Chiu W, Pownall HJ. Model of human low-density lipoprotein and bound receptor based on cryoEM. Proc Natl Acad Sci USA. 2010;107:1059–1064. doi: 10.1073/pnas.0908004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain MM, Bakillah A, Nayak N, Shelness GS. Amino acids 430-570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J Biol Chem. 1998;273:25612–25615. doi: 10.1074/jbc.273.40.25612. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, Dashti N. N-terminal domain of apolipoprotein B has structural homology to lipovitellin and microsomal triglyceride transfer protein: a "lipid pocket" model for self-assembly of apob-containing lipoprotein particles. J Lipid Res. 1999;40:1401–1416. [PubMed] [Google Scholar]
- Liang J, Ginsberg HN. Microsomal triglyceride transfer protein binding and lipid transfer activities are independent of each other, but both are required for secretion of apolipoprotein B lipoproteins from liver cells. J Biol Chem. 2001;276:28606–28612. doi: 10.1074/jbc.M100294200. [DOI] [PubMed] [Google Scholar]
- Bradbury P, Mann CJ, Kochl S, Anderson TA, Chester SA, Hancock JM, Ritchie PJ, Amey J, Harrison GB, Levitt DG. A common binding site on the microsomal triglyceride transfer protein for apolipoprotein B and protein disulfide isomerase. J Biol Chem. 1999;274:3159–3164. doi: 10.1074/jbc.274.5.3159. [DOI] [PubMed] [Google Scholar]
- Hussain MM, Bakillah A, Jamil H. Apolipoprotein B binding to microsomal triglyceride transfer protein decreases with increases in length and lipidation: implications in lipoprotein biosynthesis. Biochemistry. 1997;36:13060–13067. doi: 10.1021/bi971395a. [DOI] [PubMed] [Google Scholar]
- Kreuzer J, White AL, Knott TJ, Jien ML, Mehrabian M, Scott J, Young SG, Haberland ME. Amino terminus of apolipoprotein B suffices to produce recognition of malondialdehyde-modified low density lipoprotein by the scavenger receptor of human monocyte-macrophages. J Lipid Res. 1997;38:324–342. [PubMed] [Google Scholar]
- Sivaram P, Choi SY, Curtiss LK, Goldberg IJ. An amino-terminal fragment of apolipoprotein B binds to lipoprotein lipase and may facilitate its binding to endothelial cells. J Biol Chem. 1994;269:9409–9412. [PubMed] [Google Scholar]
- Shelness GS, Hou L, Ledford AS, Parks JS, Weinberg RB. Identification of the lipoprotein initiating domain of apolipoprotein B. J Biol Chem. 2003;278:44702–44707. doi: 10.1074/jbc.M307562200. [DOI] [PubMed] [Google Scholar]
- Wang L, Martin DD, Genter E, Wang J, McLeod RS, Small DM. Surface study of apoB1694-1880, a sequence that can anchor apoB to lipoproteins and make it nonexchangeable. J Lipid Res. 2009;50:1340–1352. doi: 10.1194/jlr.M900040-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospattankar AV, Law SW, Lackner K, Brewer HB Jr. Identification of low density lipoprotein receptor binding domains of human apolipoprotein B-100: a proposed consensus LDL receptor binding sequence of apoB-100. Biochem Biophys Res Commun. 1986;139:1078–1085. doi: 10.1016/S0006-291X(86)80287-X. [DOI] [PubMed] [Google Scholar]
- Yang CY, Chen SH, Gianturco SH, Bradley WA, Sparrow JT, Tanimura M, Li WH, Sparrow DA, DeLoof H, Rosseneu M. Sequence, structure, receptor-binding domains and internal repeats of human apolipoprotein B-100. Nature. 1986;323:738–742. doi: 10.1038/323738a0. [DOI] [PubMed] [Google Scholar]
- Wang L, Walsh MT, Small DM. Apolipoprotein B is conformationally flexible but anchored at a triolein/water interface: a possible model for lipoprotein surfaces. Proc Natl Acad Sci USA. 2006;103:6871–6876. doi: 10.1073/pnas.0602213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gretch DG, Sturley SL, Wang L, Lipton BA, Dunning A, Grunwald KA, Wetterau JR, Yao Z, Talmud P, Attie AD. The amino terminus of apolipoprotein B is necessary but not sufficient for microsomal triglyceride transfer protein responsiveness. J Biol Chem. 1996;271:8682–8691. doi: 10.1074/jbc.271.15.8682. [DOI] [PubMed] [Google Scholar]
- McLeod RS, Wang Y, Wang S, Rusinol A, Links P, Yao Z. Apolipoprotein B sequence requirements for hepatic very low density lipoprotein assembly. Evidence that hydrophobic sequences within apolipoprotein B48 mediate lipid recruitment. J Biol Chem. 1996;271:18445–18455. doi: 10.1074/jbc.271.24.14124. [DOI] [PubMed] [Google Scholar]
- Liang J, Wu X, Jiang H, Zhou M, Yang H, Angkeow P, Huang LS, Sturley SL, Ginsberg H. Translocation efficiency, susceptibility to proteasomal degradation, and lipid responsiveness of apolipoprotein B are determined by the presence of beta sheet domains. J Biol Chem. 1998;273:35216–35221. doi: 10.1074/jbc.273.52.35216. [DOI] [PubMed] [Google Scholar]
- Tran K, Boren J, Macri J, Wang Y, McLeod R, Avramoglu RK, Adeli K, Yao Z. Functional analysis of disulfide linkages clustered within the amino terminus of human apolipoprotein B. J Biol Chem. 1998;273:7244–7251. doi: 10.1074/jbc.273.13.7244. [DOI] [PubMed] [Google Scholar]
- Huang XF, Shelness GS. Identification of cysteine pairs within the amino-terminal 5% of apolipoprotein B essential for hepatic lipoprotein assembly and secretion. J Biol Chem. 1997;272:31872–31876. doi: 10.1074/jbc.272.50.31872. [DOI] [PubMed] [Google Scholar]
- Ingram MF, Shelness GS. Apolipoprotein B-100 destined for lipoprotein assembly and intracellular degradation undergoes efficient translocation across the endoplasmic reticulum membrane. J Lipid Res. 1996;37:2202–2214. [PubMed] [Google Scholar]
- Taniguchi T, Ishikawa Y, Tsunemitsu M, Fukuzaki H. The structures of the asparagine-linked sugar chains of human apolipoprotein B-100. Arch Biochem Biophys. 1989;273:197–205. doi: 10.1016/0003-9861(89)90179-3. [DOI] [PubMed] [Google Scholar]
- Vukmirica J, Nishimaki-Mogami T, Tran K, Shan J, McLeod RS, Yuan J, Yao Z. The N-linked oligosaccharides at the amino terminus of human apoB are important for the assembly and secretion of VLDL. J Lipid Res. 2002;43:1496–1507. doi: 10.1194/jlr.M200077-JLR200. [DOI] [PubMed] [Google Scholar]
- Dunphy JT, Linder ME. Signalling functions of protein palmitoylation. Biochim Biophys Acta. 1998;1436:245–261. doi: 10.1016/s0005-2760(98)00130-1. [DOI] [PubMed] [Google Scholar]
- Hoeg JM, Meng MS, Ronan R, Demosky SJ Jr, Fairwell T, Brewer HB Jr. Apolipoprotein B synthesized by Hep G2 cells undergoes fatty acid acylation. J Lipid Res. 1988;29:1215–1220. [PubMed] [Google Scholar]
- Zhao Y, McCabe JB, Vance J, Berthiaume LG. Palmitoylation of apolipoprotein B is required for proper intracellular sorting and transport of cholesteroyl esters and triglycerides. Mol Biol Cell. 2000;11:721–734. doi: 10.1091/mbc.11.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukmirica J, Tran K, Liang X, Shan J, Yuan J, Miskie BA, Hegele RA, Resh MD, Yao Z. Assembly and secretion of very low density lipoproteins containing apolipoprotein B48 in transfected McA-RH7777 cells. Lack of evidence that palmitoylation of apolipoprotein B48 is required for lipoprotein secretion. J Biol Chem. 2003;278:14153–14161. doi: 10.1074/jbc.M211995200. [DOI] [PubMed] [Google Scholar]
- Schonfeld G. Familial hypobetalipoproteinemia: a review. J Lipid Res. 2003;44:878–883. doi: 10.1194/jlr.R300002-JLR200. [DOI] [PubMed] [Google Scholar]
- Heiss G, Tamir I, Davis CE, Tyroler HA, Rifkand BM, Schonfeld G, Jacobs D, Frantz ID Jr. Lipoprotein-cholesterol distributions in selected North American populations: the lipid research clinics program prevalence study. Circulation. 1980;61:302–315. doi: 10.1161/01.cir.61.2.302. [DOI] [PubMed] [Google Scholar]
- McCormick SP, Fellowes AP, Walmsley TA, George PM. Apolipoprotein B-32: a new truncated mutant of human apolipoprotein B capable of forming particles in the low density lipoprotein range. Biochim Biophys Acta. 1992;1138:290–296. doi: 10.1016/0925-4439(92)90006-9. [DOI] [PubMed] [Google Scholar]
- Wagner RD, Krul ES, Tang J, Parhofer KG, Garlock K, Talmud P, Schonfeld G. ApoB-54.8, a truncated apolipoprotein found primarily in VLDL, is associated with a nonsense mutation in the apoB gene and hypobetalipoproteinemia. J Lipid Res. 1991;32:1001–1011. [PubMed] [Google Scholar]
- Pullinger CR, Hillas E, Hardman DA, Chen GC, Naya-Vigne JM, Iwasa JA, Hamilton RL, Lalouel JM, Williams RR, Kane JP. Two apolipoprotein B gene defects in a kindred with hypobetalipoproteinemia, one of which results in a truncated variant, apoB-61, in VLDL and LDL. J Lipid Res. 1992;33:699–710. [PubMed] [Google Scholar]
- Krul ES, Parhofer KG, Barrett PH, Wagner RD, Schonfeld G. ApoB-75, a truncation of apolipoprotein B associated with familial hypobetalipoproteinemia: genetic and kinetic studies. J Lipid Res. 1992;33:1037–1050. [PubMed] [Google Scholar]
- Farese RV Jr, Garg A, Pierotti VR, Vega GL, Young SG. A truncated species of apolipoprotein B, B-83, associated with hypobetalipoproteinemia. J Lipid Res. 1992;33:569–577. [PubMed] [Google Scholar]
- Fouchier SW, Sankatsing RR, Peter J, Castillo S, Pocovi M, Alonso R, Kastelein JJ, Defesche JC. High frequency of APOB gene mutations causing familial hypobetalipoproteinaemia in patients of Dutch and Spanish descent. J Med Genet. 2005;42:e23. doi: 10.1136/jmg.2004.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fitzgerald RL, Schonfeld G. Hypobetalipoproteinemic mice with a targeted apolipoprotein (Apo) B-27.6-specifying mutation: in vivo evidence for an important role of amino acids 1254-1744 of ApoB in lipid transport and metabolism of the apoB-containing lipoprotein. J Biol Chem. 2002;277:14135–14145. doi: 10.1074/jbc.M200617200. [DOI] [PubMed] [Google Scholar]
- McLeod RS, Zhao Y, Selby SL, Westerlund J, Yao Z. Carboxyl-terminal truncation impairs lipid recruitment by apolipoprotein B100 but does not affect secretion of the truncated apolipoprotein B-containing lipoproteins. J Biol Chem. 1994;269:2852–2862. [PubMed] [Google Scholar]
- Burnett JR, Shan J, Miskie BA, Whitfield AJ, Yuan J, Tran K, McKnight CJ, Hegele RA, Yao Z. A novel nontruncating APOB gene mutation, R463W, causes familial hypobetalipoproteinemia. J Biol Chem. 2003;278:13442–13452. doi: 10.1074/jbc.M300235200. [DOI] [PubMed] [Google Scholar]
- Burnett JR, Zhong S, Jiang ZG, Hooper AJ, Fisher EA, McLeod RS, Zhao Y, Barrett PH, Hegele RA, van Bockxmeer FM. Missense mutations in APOB within the betaalpha1 domain of human APOB-100 result in impaired secretion of ApoB and ApoB-containing lipoproteins in familial hypobetalipoproteinemia. J Biol Chem. 2007;282:24270–24283. doi: 10.1074/jbc.M702442200. [DOI] [PubMed] [Google Scholar]
- Tarugi P, Averna M, Di LE, Cefalu AB, Noto D, Magnolo L, Cattin L, Bertolini S, Calandra S. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007;195:e19–e27. doi: 10.1016/j.atherosclerosis.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Zhong S, Magnolo AL, Sundaram M, Zhou H, Yao EF, Di LE, Loria P, Wang S, Bamji-Mirza M, Wang L. Nonsynonymous mutations within APOB in human familial hypobetalipoproteinemia: evidence for feedback inhibition of lipogenesis and postendoplasmic reticulum degradation of apolipoprotein B. J Biol Chem. 2010;285:6453–6464. doi: 10.1074/jbc.M109.060467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamil H, Dickson JK Jr, Chu CH, Lago MW, Rinehart JK, Biller SA, Gregg RE, Wetterau JR. Microsomal triglyceride transfer protein. Specificity of lipid binding and transport. J Biol Chem. 1995;270:6549–6554. doi: 10.1074/jbc.270.12.6549. [DOI] [PubMed] [Google Scholar]
- Wetterau JR, Combs KA, Spinner SN, Joiner BJ. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J Biol Chem. 1990;265:9800–9807. [PubMed] [Google Scholar]
- Wang L, Fast DG, Attie AD. The enzymatic and non-enzymatic roles of protein-disulfide isomerase in apolipoprotein B secretion. J Biol Chem. 1997;272:27644–27651. doi: 10.1074/jbc.272.44.27644. [DOI] [PubMed] [Google Scholar]
- Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, Schmitz J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- Read J, Anderson TA, Ritchie PJ, Vanloo B, Amey J, Levitt D, Rosseneu M, Scott J, Shoulders CC. A mechanism of membrane neutral lipid acquisition by the microsomal triglyceride transfer protein. J Biol Chem. 2000;275:30372–30377. doi: 10.1074/jbc.C000364200. [DOI] [PubMed] [Google Scholar]
- Zhou M, Fisher EA, Ginsberg HN. Regulated Co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J Biol Chem. 1998;273:24649–24653. doi: 10.1074/jbc.273.38.24649. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tran K, Yao Z. The activity of microsomal triglyceride transfer protein is essential for accumulation of triglyceride within microsomes in McA-RH7777 cells. A unified model for the assembly of very low density lipoproteins. J Biol Chem. 1999;274:27793–27800. doi: 10.1074/jbc.274.39.27793. [DOI] [PubMed] [Google Scholar]
- Rustaeus S, Stillemark P, Lindberg K, Gordon D, Olofsson SO. The microsomal triglyceride transfer protein catalyzes the post-translational assembly of apolipoprotein B-100 very low density lipoprotein in McA-RH7777 cells. J Biol Chem. 1998;273:5196–5203. doi: 10.1074/jbc.273.9.5196. [DOI] [PubMed] [Google Scholar]
- Gordon DA, Jamil H. Progress towards understanding the role of microsomal triglyceride transfer protein in apolipoprotein-B lipoprotein assembly. Biochim Biophys Acta. 2000;1486:72–83. doi: 10.1016/s1388-1981(00)00049-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, McLeod RS, Yao Z. Normal activity of microsomal triglyceride transfer protein is required for the oleate-induced secretion of very low density lipoproteins containing apolipoprotein B from McA-RH7777 cells. J Biol Chem. 1997;272:12272–12278. doi: 10.1074/jbc.272.19.12272. [DOI] [PubMed] [Google Scholar]
- Kulinski A, Rustaeus S, Vance JE. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J Biol Chem. 2002;277:31516–31525. doi: 10.1074/jbc.M202015200. [DOI] [PubMed] [Google Scholar]
- Hebbachi A, Gibbons GF. Inactivation of microsomal triglyceride transfer protein impairs the normal redistribution but not the turnover of newly synthesized glycerolipid in the cytosol, endoplasmic reticulum and Golgi of primary rat hepatocytes. Biochim Biophys Acta. 1999;1441:36–50. doi: 10.1016/s1388-1981(99)00138-9. [DOI] [PubMed] [Google Scholar]
- Chang BH, Liao W, Li L, Nakamuta M, Mack D, Chan L. Liver-specific inactivation of the abetalipoproteinemia gene completely abrogates very low density lipoprotein/low density lipoprotein production in a viable conditional knockout mouse. J Biol Chem. 1999;274:6051–6055. doi: 10.1074/jbc.274.10.6051. [DOI] [PubMed] [Google Scholar]
- Raabe M, Veniant MM, Sullivan MA, Zlot CH, Bjorkegren J, Nielsen LB, Wong JS, Hamilton RL, Young SG. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103:1287–1298. doi: 10.1172/JCI6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann NJ, Kang S, Li AC, Chen AZ, Newberry EP, Davidson NO, Hui ST, Davis RA. Coordinate transcriptional repression of liver fatty acid-binding protein and microsomal triglyceride transfer protein blocks hepatic very low density lipoprotein secretion without hepatosteatosis. J Biol Chem. 2006;281:33066–33077. doi: 10.1074/jbc.M607148200. [DOI] [PubMed] [Google Scholar]
- Huang J, Iqbal J, Saha PK, Liu J, Chan L, Hussain MM, Moore DD, Wang L. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46:147–157. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- Hribal ML, Nakae J, Kitamura T, Shutter JR, Accili D. Regulation of insulin-like growth factor-dependent myoblast differentiation by Foxo forkhead transcription factors. J Cell Biol. 2003;162:535–541. doi: 10.1083/jcb.200212107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, Meseck M, Dong HH. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest. 2008;118:2347–2364. doi: 10.1172/JCI32914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Stoffel M. Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab. 2006;3:99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Dolinsky VW, Douglas DN, Lehner R, Vance DE. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. Biochem J. 2004;378:967–974. doi: 10.1042/BJ20031320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran K, Wang Y, DeLong CJ, Cui Z, Yao Z. The assembly of very low density lipoproteins in rat hepatoma McA-RH7777 cells is inhibited by phospholipase A2 antagonists. J Biol Chem. 2000;275:25023–25030. doi: 10.1074/jbc.M908971199. [DOI] [PubMed] [Google Scholar]
- Tran K, Sun F, Cui Z, Thorne-Tjomsland G, St GC, Lapierre LR, McLeod RS, Jamieson JC, Yao Z. Attenuated secretion of very low density lipoproteins from McA-RH7777 cells treated with eicosapentaenoic acid is associated with impaired utilization of triacylglycerol synthesized via phospholipid remodeling. Biochim Biophys Acta. 2006;1761:463–473. doi: 10.1016/j.bbalip.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Robichaud JC, van d V, Yao Z, Trigatti B, Vance DE. Hepatic uptake and metabolism of phosphatidylcholine associated with high density lipoproteins. Biochim Biophys Acta. 2009;1790:538–551. doi: 10.1016/j.bbagen.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Sato K, Cho Y, Tachibana S, Chiba T, Schneider WJ, Akiba Y. Impairment of VLDL secretion by medium-chain fatty acids in chicken primary hepatocytes is affected by the chain length. J Nutr. 2005;135:1636–1641. doi: 10.1093/jn/135.7.1636. [DOI] [PubMed] [Google Scholar]
- Tachibana S, Sato K, Cho Y, Chiba T, Schneider WJ, Akiba Y. Octanoate reduces very low-density lipoprotein secretion by decreasing the synthesis of apolipoprotein B in primary cultures of chicken hepatocytes. Biochim Biophys Acta. 2005;1737:36–43. doi: 10.1016/j.bbalip.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, Hussain MM, Parks RJ, Wang Y, Yao Z. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res. 2010;51:150–161. doi: 10.1194/M900346-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran K, Thorne-Tjomsland G, DeLong CJ, Cui Z, Shan J, Burton L, Jamieson JC, Yao Z. Intracellular assembly of very low density lipoproteins containing apolipoprotein B100 in rat hepatoma McA-RH7777 cells. J Biol Chem. 2002;277:31187–31200. doi: 10.1074/jbc.M200249200. [DOI] [PubMed] [Google Scholar]
- Pan M, Maitin V, Parathath S, Andreo U, Lin SX, St GC, Yao Z, Maxfield FR, Williams KJ, Fisher EA. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci USA. 2008;105:5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Soldado I, Avella M, Botham KM. Differential influence of different dietary fatty acids on very low-density lipoprotein secretion when delivered to hepatocytes in chylomicron remnants. Metabolism. 2009;58:186–195. doi: 10.1016/j.metabol.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Takechi R, Ho SS. Conjugated linoleic acid suppresses the secretion of atherogenic lipoproteins from human HepG2 liver cells. Clin Chem Lab Med. 2005;43:269–274. doi: 10.1515/CCLM.2005.045. [DOI] [PubMed] [Google Scholar]
- Zhang P, Boudyguina E, Wilson MD, Gebre AK, Parks JS. Echium oil reduces plasma lipids and hepatic lipogenic gene expression in apoB100-only LDL receptor knockout mice. J Nutr Biochem. 2008;19:655–663. doi: 10.1016/j.jnutbio.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Havinga R, Bos T, Bloks VW, Kuipers F, Verkade HJ. Essential fatty acid deficiency in mice is associated with hepatic steatosis and secretion of large VLDL particles. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1150–G1158. doi: 10.1152/ajpgi.00456.2004. [DOI] [PubMed] [Google Scholar]
- Degrace P, Moindrot B, Mohamed I, Gresti J, Du ZY, Chardigny JM, Sebedio JL, Clouet P. Upregulation of liver VLDL receptor and FAT/CD36 expression in LDLR-/- apoB100/100 mice fed trans-10,cis-12 conjugated linoleic acid. J Lipid Res. 2006;47:2647–2655. doi: 10.1194/jlr.M600140-JLR200. [DOI] [PubMed] [Google Scholar]
- Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. doi: 10.1172/JCI32752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Q, Tsai J, Xu E, Qiu W, Bereczki E, Santha M, Adeli K. Apolipoprotein B100 acts as a molecular link between lipid-induced endoplasmic reticulum stress and hepatic insulin resistance. Hepatology. 2009;50:77–84. doi: 10.1002/hep.22960. [DOI] [PubMed] [Google Scholar]
- Carman GM, Han GS. Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem Sci. 2006;31:694–699. doi: 10.1016/j.tibs.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- Peterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem. 2005;280:32883–32889. doi: 10.1074/jbc.M503885200. [DOI] [PubMed] [Google Scholar]
- Manmontri B, Sariahmetoglu M, Donkor J, Bou Khalil M, Sundaram M, Yao Z, Reue K, Lehner R, Brindley DN. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J Lipid Res. 2008;49:1056–1067. doi: 10.1194/jlr.M800013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou Khlail M, Sundaram M, Zhang HY, Links PH, Raven JF, Manmontri B, Sariahmetoglu M, Tran K, Reue K, Brindley DN. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic VLDL. J Lipid Res. 2009;50:47–58. doi: 10.1194/jlr.M800204-JLR200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gropler MC, Norris J, Lawrence JC Jr, Harris TE, Finck BN. Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arterioscler Thromb Vasc Biol. 2008;28:1738–1744. doi: 10.1161/ATVBAHA.108.171538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC Jr, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, Voelker T, Farese RV Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem. 2001;276:38870–38876. doi: 10.1074/jbc.M106219200. [DOI] [PubMed] [Google Scholar]
- Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci USA. 1998;95:13018–13023. doi: 10.1073/pnas.95.22.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV Jr. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- Millar JS, Stone SJ, Tietge UJ, Tow B, Billheimer JT, Wong JS, Hamilton RL, Farese RV Jr, Rader DJ. Short-term overexpression of DGAT1 or DGAT2 increases hepatic triglyceride but not VLDL triglyceride or apoB production. J Lipid Res. 2006;47:2297–2305. doi: 10.1194/jlr.M600213-JLR200. [DOI] [PubMed] [Google Scholar]
- Liang JJ, Oelkers P, Guo C, Chu PC, Dixon JL, Ginsberg HN, Sturley SL. Overexpression of human diacylglycerol acyltransferase 1, acyl-coa:cholesterol acyltransferase 1, or acyl-CoA:cholesterol acyltransferase 2 stimulates secretion of apolipoprotein B-containing lipoproteins in McA-RH7777 cells. J Biol Chem. 2004;279:44938–44944. doi: 10.1074/jbc.M408507200. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Sasaki E, Kakinuma C, Yano T, Miura S, Ezaki O. Increased very low density lipoprotein secretion and gonadal fat mass in mice overexpressing liver DGAT1. J Biol Chem. 2005;280:21506–21514. doi: 10.1074/jbc.M412989200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Millar JS, Cromley DA, Graham M, Crooke R, Billheimer JT, Rader DJ. Knockdown of acyl-CoA:diacylglycerol acyltransferase 2 with antisense oligonucleotide reduces VLDL TG and ApoB secretion in mice. Biochim Biophys Acta. 2008;1781:97–104. doi: 10.1016/j.bbalip.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Walkey CJ, Donohue LR, Bronson R, Agellon LB, Vance DE. Disruption of the murine gene encoding phosphatidylethanolamine N-methyltransferase. Proc Natl Acad Sci USA. 1997;94:12880–12885. doi: 10.1073/pnas.94.24.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Su B, Jacobs RL, Kennedy B, Francis GA, Waddington E, Brosnan JT, Vance JE, Vance DE. Lack of phosphatidylethanolamine N-methyltransferase alters plasma VLDL phospholipids and attenuates atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2009;29:1349–1355. doi: 10.1161/ATVBAHA.109.188672. [DOI] [PubMed] [Google Scholar]
- Vance DE, Vance JE. Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J Lipid Res. 2009;50(Suppl):S132–S137. doi: 10.1194/jlr.R800048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs RL, Devlin C, Tabas I, Vance DE. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem. 2004;279:47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- Jacobs RL, Lingrell S, Zhao Y, Francis GA, Vance DE. Hepatic CTP:phosphocholine cytidylyltransferase-alpha is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J Biol Chem. 2008;283:2147–2155. doi: 10.1074/jbc.M706628200. [DOI] [PubMed] [Google Scholar]
- Poloumienko A, Cote A, Quee AT, Zhu L, Bakovic M. Genomic organization and differential splicing of the mouse and human Pcyt2 genes. Gene. 2004;325:145–155. doi: 10.1016/j.gene.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Hakimuddin F, Bonen A, Bakovic M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J Biol Chem. 2009;284:25704–25713. doi: 10.1074/jbc.M109.023846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, Vance DE. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006;3:321–331. doi: 10.1016/j.cmet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Gusarova V, Brodsky JL, Fisher EA. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J Biol Chem. 2003;278:48051–48058. doi: 10.1074/jbc.M306898200. [DOI] [PubMed] [Google Scholar]
- Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34:29–31. doi: 10.1038/ng1145. [DOI] [PubMed] [Google Scholar]
- Asp L, Magnusson B, Rutberg M, Li L, Boren J, Olofsson SO. Role of ADP ribosylation factor 1 in the assembly and secretion of ApoB-100-containing lipoproteins. Arterioscler Thromb Vasc Biol. 2005;25:566–570. doi: 10.1161/01.ATV.0000154135.21689.47. [DOI] [PubMed] [Google Scholar]
- Asp L, Claesson C, Boren J, Olofsson SO. ADP-ribosylation factor 1 and its activation of phospholipase D are important for the assembly of very low density lipoproteins. J Biol Chem. 2000;275:26285–26292. doi: 10.1074/jbc.M003520200. [DOI] [PubMed] [Google Scholar]
- Rustaeus S, Lindberg K, Boren J, Olofsson SO. Brefeldin A reversibly inhibits the assembly of apoB containing lipoproteins in McA-RH7777 cells. J Biol Chem. 1995;270:28879–28886. doi: 10.1074/jbc.270.48.28879. [DOI] [PubMed] [Google Scholar]
- Magnusson B, Asp L, Bostrom P, Ruiz M, Stillemark-Billton P, Linden D, Boren J, Olofsson SO. Adipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteins. Arterioscler Thromb Vasc Biol. 2006;26:1566–1571. doi: 10.1161/01.ATV.0000223345.11820.da. [DOI] [PubMed] [Google Scholar]
- Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, Li P. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 2009;9:177–190. doi: 10.1016/j.cmet.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Yon TS, Chen Z, Guo K, Ng CP, Ponniah S, Lin SC, Hong W, Li P. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat Genet. 2003;35:49–56. doi: 10.1038/ng1225. [DOI] [PubMed] [Google Scholar]
- Kuipers F, Jong MC, Lin Y, Eck M, Havinga R, Bloks V, Verkade HJ, Hofker MH, Moshage H, Berkel TJ. Impaired secretion of very low density lipoprotein-triglycerides by apolipoprotein E- deficient mouse hepatocytes. J Clin Invest. 1997;100:2915–2922. doi: 10.1172/JCI119841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensenkamp AR, Jong MC, van GH, Van Luyn MJ, Bloks V, Havinga R, Voshol PJ, Hofker MH, van Dijk KW, Havekes LM. Apolipoprotein E participates in the regulation of very low density lipoprotein-triglyceride secretion by the liver. J Biol Chem. 1999;274:35711–35718. doi: 10.1074/jbc.274.50.35711. [DOI] [PubMed] [Google Scholar]
- Maugeais C, Tietge UJ, Tsukamoto K, Glick JM, Rader DJ. Hepatic apolipoprotein E expression promotes very low density lipoprotein-apolipoprotein B production in vivo in mice. J Lipid Res. 2000;41:1673–1679. [PubMed] [Google Scholar]
- Huang Y, Liu XQ, Rall SC Jr, Taylor JM, von EA, Assmann G, Mahley RW. Overexpression and accumulation of apolipoprotein E as a cause of hypertriglyceridemia. J Biol Chem. 1998;273:26388–26393. doi: 10.1074/jbc.273.41.26388. [DOI] [PubMed] [Google Scholar]
- Gusarova V, Seo J, Sullivan ML, Watkins SC, Brodsky JL, Fisher EA. Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J Biol Chem. 2007;282:19453–19462. doi: 10.1074/jbc.M700475200. [DOI] [PubMed] [Google Scholar]
- Gangabadage CS, Zdunek J, Tessari M, Nilsson S, Olivecrona G, Wijmenga SS. Structure and dynamics of human apolipoprotein CIII. J Biol Chem. 2008;283:17416–17427. doi: 10.1074/jbc.M800756200. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Zhong S, Bou Khalil M, Zhou H, Jiang ZG, Zhao Y, Iqbal J, Hussain MM, Figeys D, Wang Y. Functional analysis of the missense APOC3 mutation Ala23Thr associated with human hypotriglyceridemia. J Lipid Res. 2010. in press . [DOI] [PMC free article] [PubMed]
- Liu H, Labeur C, Xu CF, Ferrell R, Lins L, Brasseur R, Rosseneu M, Weiss KM, Humphries SE, Talmud PJ. Characterization of the lipid-binding properties and lipoprotein lipase inhibition of a novel apolipoprotein C-III variant Ala23Thr. J Lipid Res. 2000;41:1760–1771. [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- van Dijk KW, Rensen PC, Voshol PJ, Havekes LM. The role and mode of action of apolipoproteins CIII and AV: synergistic actors in triglyceride metabolism? Curr Opin Lipidol. 2004;15:239–246. doi: 10.1097/00041433-200406000-00002. [DOI] [PubMed] [Google Scholar]
- Schaap FG, Rensen PC, Voshol PJ, Vrins C, van d V, Chamuleau RA, Havekes LM, Groen AK, van Dijk KW. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglyceride (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. J Biol Chem. 2004;279:27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- Shu X, Ryan RO, Forte TM. Intracellular lipid droplet targeting by apolipoprotein A-V requires the carboxyl-terminal segment. J Lipid Res. 2008;49:1670–1676. doi: 10.1194/jlr.M800111-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twisk J, Gillian-Daniel DL, Tebon A, Wang L, Barrett PH, Attie AD. The role of the LDL receptor in apolipoprotein B secretion. J Clin Invest. 2000;105:521–532. doi: 10.1172/JCI8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James RW, Martin B, Pometta D, Fruchart JC, Duriez P, Puchois P, Farriaux JP, Tacquet A, Demant T, Clegg RJ. Apolipoprotein B metabolism in homozygous familial hypercholesterolemia. J Lipid Res. 1989;30:159–169. [PubMed] [Google Scholar]
- Teusink B, Mensenkamp AR, van der BH, Kuipers F, van Dijk KW, Havekes LM. Stimulation of the in vivo production of very low density lipoproteins by apolipoprotein E is independent of the presence of the low density lipoprotein receptor. J Biol Chem. 2001;276:40693–40697. doi: 10.1074/jbc.M106396200. [DOI] [PubMed] [Google Scholar]
- Williams KJ, Brocia RW, Fisher EA. The unstirred water layer as a site of control of apolipoprotein B secretion. J Biol Chem. 1990;265:16741–16744. [PubMed] [Google Scholar]
- Jiang XC, Qin S, Qiao C, Kawano K, Lin M, Skold A, Xiao X, Tall AR. Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat Med. 2001;7:847–852. doi: 10.1038/89977. [DOI] [PubMed] [Google Scholar]
- Blasiole DA, Oler AT, Attie AD. Regulation of ApoB secretion by the low density lipoprotein receptor requires exit from the endoplasmic reticulum and interaction with ApoE or ApoB. J Biol Chem. 2008;283:11374–11381. doi: 10.1074/jbc.M710457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci USA. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XM, Eden ER, Tosi I, Neuwirth CK, Wile D, Naoumova RP, Soutar AK. Evidence for effect of mutant PCSK9 on apolipoprotein B secretion as the cause of unusually severe dominant hypercholesterolaemia. Hum Mol Genet. 2005;14:1161–1169. doi: 10.1093/hmg/ddi128. [DOI] [PubMed] [Google Scholar]
- Lalanne F, Lambert G, Amar MJ, Chetiveaux M, Zair Y, Jarnoux AL, Ouguerram K, Friburg J, Seidah NG, Brewer HB Jr. Wild-type PCSK9 inhibits LDL clearance but does not affect apoB-containing lipoprotein production in mouse and cultured cells. J Lipid Res. 2005;46:1312–1319. doi: 10.1194/jlr.M400396-JLR200. [DOI] [PubMed] [Google Scholar]
- Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA. 2005;102:5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clee SM, Kastelein JJ, van DM, Marcil M, Roomp K, Zwarts KY, Collins JA, Roelants R, Tamasawa N, Stulc T. Age and residual cholesterol efflux affect HDL cholesterol levels and coronary artery disease in ABCA1 heterozygotes. J Clin Invest. 2000;106:1263–1270. doi: 10.1172/JCI10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Gebre AK, Seo J, Shelness GS, Parks JS. A novel role for ABCA1-generated large pre-beta migrating nascent HDL in the regulation of hepatic VLDL triglyceride secretion. J Lipid Res. 2010;51:729–742. doi: 10.1194/jlr.M900083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phung TL, Roncone A, Jensen KL, Sparks CE, Sparks JD. Phosphoinositide 3-kinase activity is necessary for insulin-dependent inhibition of apolipoprotein B secretion by rat hepatocytes and localizes to the endoplasmic reticulum. J Biol Chem. 1997;272:30693–30702. doi: 10.1074/jbc.272.49.30693. [DOI] [PubMed] [Google Scholar]
- Wiersma H, Nijstad N, Gautier T, Iqbal J, Kuipers F, Hussain MM, Tietge UJ. Scavenger receptor BI facilitates hepatic very low density lipoprotein production in mice. J Lipid Res. 2010;51:544–553. doi: 10.1194/jlr.M000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002;106:2137–2142. doi: 10.1161/01.CIR.0000035280.64322.31. [DOI] [PubMed] [Google Scholar]
- Meshkani R, Adeli K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin Biochem. 2009;42:1331–1346. doi: 10.1016/j.clinbiochem.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Taghibiglou C, Carpentier A, Van Iderstine SC, Chen B, Rudy D, Aiton A, Lewis GF, Adeli K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem. 2000;275:8416–8425. doi: 10.1074/jbc.275.12.8416. [DOI] [PubMed] [Google Scholar]
- Tsai J, Zhang R, Qiu W, Su Q, Naples M, Adeli K. Inflammatory NF-kappaB activation promotes hepatic apolipoprotein B100 secretion: evidence for a link between hepatic inflammation and lipoprotein production. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1287–G1298. doi: 10.1152/ajpgi.90540.2008. [DOI] [PubMed] [Google Scholar]
- Wiegman CH, Bandsma RH, Ouwens M, Sluijs FH van der, Havinga R, Boer T, Reijngoud DJ, Romijn JA, Kuipers F. Hepatic VLDL production in ob/ob mice is not stimulated by massive de novo lipogenesis but is less sensitive to the suppressive effects of insulin. Diabetes. 2003;52:1081–1089. doi: 10.2337/diabetes.52.5.1081. [DOI] [PubMed] [Google Scholar]
- Li X, Grundy SM, Patel SB. Obesity in db and ob animals leads to impaired hepatic very low density lipoprotein secretion and differential secretion of apolipoprotein B-48 and B-100. J Lipid Res. 1997;38:1277–1288. [PubMed] [Google Scholar]
- MacLean PS, Bower JF, Vadlamudi S, Osborne JN, Bradfield JF, Burden HW, Bensch WH, Kauffman RF, Barakat HA. Cholesteryl ester transfer protein expression prevents diet-induced atherosclerotic lesions in male db/db mice. Arterioscler Thromb Vasc Biol. 2003;23:1412–1415. doi: 10.1161/01.ATV.0000080687.94313.67. [DOI] [PubMed] [Google Scholar]
- Coenen KR, Gruen ML, Hasty AH. Obesity causes very low density lipoprotein clearance defects in low-density lipoprotein receptor-deficient mice. J Nutr Biochem. 2007;18:727–735. doi: 10.1016/j.jnutbio.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Pickens MK, Yan JS, Ng RK, Ogata H, Grenert JP, Beysen C, Turner SM, Maher JJ. Dietary sucrose is essential to the development of liver injury in the MCD model of steatohepatitis. J Lipid Res. 2009;50:2072–2082. doi: 10.1194/jlr.M900022-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49:1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. 1988;263:2998–3004. [PubMed] [Google Scholar]
- Pawar A, Botolin D, Mangelsdorf DJ, Jump DB. The role of liver × receptor-alpha in the fatty acid regulation of hepatic gene expression. J Biol Chem. 2003;278:40736–40743. doi: 10.1074/jbc.M307973200. [DOI] [PubMed] [Google Scholar]
- Basciano H, Miller A, Baker C, Naples M, Adeli K. LXRalpha activation perturbs hepatic insulin signaling and stimulates production of apolipoprotein B-containing lipoproteins. Am J Physiol Gastrointest Liver Physiol. 2009;297:G323–G332. doi: 10.1152/ajpgi.90546.2008. [DOI] [PubMed] [Google Scholar]
- Huang W, Dedousis N, Bandi A, Lopaschuk GD, O'Doherty RM. Liver triglyceride secretion and lipid oxidative metabolism are rapidly altered by leptin in vivo. Endocrinology. 2006;147:1480–1487. doi: 10.1210/en.2005-0731. [DOI] [PubMed] [Google Scholar]
- Huang W, Dedousis N, O'Doherty RM. Hepatic steatosis and plasma dyslipidemia induced by a high-sucrose diet are corrected by an acute leptin infusion. J Appl Physiol. 2007;102:2260–2265. doi: 10.1152/japplphysiol.01449.2006. [DOI] [PubMed] [Google Scholar]
- Huang W, Dedousis N, Bhatt BA, O'Doherty RM. Impaired activation of phosphatidylinositol 3-kinase by leptin is a novel mechanism of hepatic leptin resistance in diet-induced obesity. J Biol Chem. 2004;279:21695–21700. doi: 10.1074/jbc.M401546200. [DOI] [PubMed] [Google Scholar]
- Huang W, Metlakunta A, Dedousis N, Ortmeyer HK, Stefanovic-Racic M, O'Doherty RM. Leptin augments the acute suppressive effects of insulin on hepatic very low-density lipoprotein production in rats. Endocrinology. 2009;150:2169–2174. doi: 10.1210/en.2008-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirieac DV, Collins HL, Cianci J, Sparks JD, Sparks CE. Altered triglyceride-rich lipoprotein production in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. 2004;287:E42–E49. doi: 10.1152/ajpendo.00297.2003. [DOI] [PubMed] [Google Scholar]
- Taghibiglou C, Carpentier A, Van Iderstine SC, Chen B, Rudy D, Aiton A, Lewis GF, Adeli K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem. 2000;275:8416–8425. doi: 10.1074/jbc.275.12.8416. [DOI] [PubMed] [Google Scholar]
- Benoist F, Grand-Perret T. Co-translational degradation of apolipoprotein B100 by the proteasome is prevented by microsomal triglyceride transfer protein. Synchronized translation studies on HepG2 cells treated with an inhibitor of microsomal triglyceride transfer protein. J Biol Chem. 1997;272:20435–20442. doi: 10.1074/jbc.272.33.20435. [DOI] [PubMed] [Google Scholar]
- Fisher EA, Zhou M, Mitchell DM, Wu X, Omura S, Wang H, Goldberg AL, Ginsberg HN. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J Biol Chem. 1997;272:20427–20434. doi: 10.1074/jbc.272.33.20427. [DOI] [PubMed] [Google Scholar]
- Liao W, Yeung SC, Chan L. Proteasome-mediated degradation of apolipoprotein B targets both nascent peptides cotranslationally before translocation and full-length apolipoprotein B after translocation into the endoplasmic reticulum. J Biol Chem. 1998;273:27225–27230. doi: 10.1074/jbc.273.42.27225. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN. Role of lipid synthesis, chaperone proteins and proteasomes in the assembly and secretion of apoprotein B-containing lipoproteins from cultured liver cells. Clin Exp Pharmacol Physiol. 1997;24:A29–A32. doi: 10.1111/j.1440-1681.1997.tb03051.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50(Suppl):S162–S166. doi: 10.1194/jlr.R800090-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelness GS, Ingram MF, Huang XF, DeLozier JA. Apolipoprotein B in the rough endoplasmic reticulum: translation, translocation and the initiation of lipoprotein assembly. J Nutr. 1999;129:456S–462S. doi: 10.1093/jn/129.2.456S. [DOI] [PubMed] [Google Scholar]
- Yao Z, Tran K, McLeod RS. Intracellular degradation of newly synthesized apolipoprotein B. J Lipid Res. 1997;38:1937–1953. [PubMed] [Google Scholar]
- Mortimore GE, Miotto G, Venerando R, Kadowaki M. Autophagy. Subcell Biochem. 1996;27:93–135. doi: 10.1007/978-1-4615-5833-0_4. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Berg TO, Blankson H, Fengsrud M, Holen I, Stromhaug PE. Structural aspects of autophagy. Adv Exp Med Biol. 1996;389:103–111. doi: 10.1007/978-1-4613-0335-0_12. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell. 2006;17:2674–2683. doi: 10.1091/mbc.E05-07-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]