

Abstract
Regulation of hormonal, insulin/IGF-1 (Ins/IGF-1) signaling activities, and pathways of the intrinsic generation of reactive oxygen species (ROS) play a role in aging and longevity determination. In this review we discuss the cross-talk between these pathways as mechanisms of signaling that may be important factors in the regulation of aging and longevity. The balance of physiological processes controlling the rate of aging and longevity in several mouse mutants suggests the involvement of cross-talk mechanisms of regulation of the insulin/IGF1 signaling pathway vs. the ROS signaling pathways. In mice, modulation of the Ins/IGF-1 signaling pathways resulting from the Prop1df, Pit1dw and Igf1 receptor mutations exemplify the hormonal pathways associated with aging and longevity determination. These pathways are also targets of the ROS-mediated redox pathways. Similarly, the Klotho and p66Shc mutants link regulation of ROS signaling pathways to aging and longevity determination. Both of these models also display altered insulin signaling activity, a characteristic associated with longevity. The Ins/IGF-1 signaling pathway is of particular interest because of its decreased activity due to genetic manipulation vs. its responsiveness to ROS levels.
Keywords: Insulin/IGF-1 signaling, ROS signaling, Aging, Longevity
1. Introduction
The striking similarity of the molecular and physiological processes of aging and longevity determination are tightly conserved throughout evolution and involve the insulin-like hormone receptor and its downstream effector molecules. Similarly, the activities of certain signaling pathways that regulate the intrinsic generation of reactive oxygen species (ROS) also play a major role in determination of aging and longevity. Integration of the insulin/IGF-1 (Ins/IGF-1) signaling pathway and the second messenger ROS-mediated redox signaling pathways involve regulatory cross-talk processes between these pathways. The molecular mechanisms of signaling via cross-talk may play an important role in the mechanisms of regulation of aging and longevity. Those mouse models of interest include the hypopituitary dwarf mice (Snell and Ames dwarf mice) both of which have altered insulin and IGF-1 levels, the Klotho and p66Shc mutants which serve as examples of the role of redox signaling in aging and longevity determination. A common physiological characteristic shared by these genetic models of delayed aging and extended lifespan includes increased resistance to oxidative stress which suggests the potential importance of cross-talk signaling between these pathways.
The balance of physiological processes controlling the rate of aging in mammals, e.g., development, growth, reproduction, metabolism and resistance to oxidative stress, involves the crosstalk mechanisms of regulation of the Ins/IGF-1 signaling pathway vs. the regulation of ROS signaling pathways (Clancy et al., 2001; Holzenberger et al., 2003; Kenyon, 2005; Tatar et al., 2003). In mice, modulation of the Ins/IGF-1 signaling pathways resulting from the Prop1df, Pit1dw, and Igf1 receptor mutations exemplify the major hormonal pathways that play an important role in aging and longevity determination (Brown-Borg et al., 1996; Coschigano et al., 2000; Flurkey et al., 2001, 2002; Hsieh et al., 2002a,b). These mutations affect the activities of the multiple components of these signaling pathways, i.e., phosphatidylinositol-3-phosphate (PI3K), Akt and Forkhead proteins (Bluher et al., 2003; Holzenberger et al., 2003) which are also targets of the ROS-mediated redox pathways.
On the other hand, the Klotho and p66Shc mutants link the role of oxidative stress and regulation of ROS signaling pathways to aging and longevity determination. For example, overexpression of Klotho in mice extends lifespan and displays decreased insulin signaling activity, a characteristic associated with longevity (Kurosu et al., 2005). In contrast, deletion of Klotho results in a phenotype suggestive of accelerated aging, which includes increased oxidative stress that adversely affects the Ins/IGF-1 signaling pathway. Overexpression of klotho extends lifespan suggesting that this gene functions as a suppressor of aging in mammals (Kurosu et al., 2005). Klotho decreases insulin signaling activity by suppressing tyrosine phosphorylation of the insulin and IGF-1 receptors. This reduces IRS protein association with PI3K, thereby inhibiting insulin and IGF-1 signaling. Klotho-induced inhibition of Ins/IGF-1 signaling is also associated with increased resistance to oxidative stress which potentially contributes to its anti-aging properties. The mechanism of this resistance to oxidative stress involves: (a) binding of Klotho to cell-surface receptor; (b) signaling the inhibition of FOXO phosphorylation which provides the nuclear localization of this transcription factor; (c) FOXO binds to the MnSOD promoter which induces its expression and enhances the removal of ROS; (d) this confers resistance to oxidative stress (Yamamoto et al., 2005). Targeted disruption of the p66Shc gene, which results in a 30% increase in lifespan also confers an increased resistance to exogenous oxidative stress and lowers the level of oxidative burden in vivo (Migliaccio et al., 1999; Nemoto and Finkel, 2002; Trinei et al., 2002). The p66Shc protein belongs to a family of adaptor molecules that regulate protein–protein interaction for a number of cell surface receptors, including the insulin receptor (IR). This is based on its regulation of mammalian Forkhead transcription factor activation that in turn may lead to increased anti-oxidant activities such as catalase and superoxide dismutase (Kops et al., 2002; Nemoto and Finkel, 2002). Some of the physiological characteristics of the long-lived mice lacking p66Shc correlate with decreased mitochondrial function that includes mitochondrial generated ROS (Giorgio et al., 2005). This is based on the observation that p66Shc is a redox enzyme that generates ROS (H2O2) as signaling molecules for apoptosis (Giorgio et al., 2005). The mechanism of this function invokes the use of reducing equivalents of the mitochondrial electron transport chain (ETC) through the oxidation of cytochrome c. Thus, the p66Shc system provides an alternative redox process associated with mitochondrial ETC function at complex III which generates proapoptotic ROS in response to stress signals. Importantly initiation of p66Shc activity is linked to the IRβ-subunit via the JM domain, the details of which are discussed in Section 8. Thus, in the animals whose aging and longevity are altered by genetic manipulation of ROS production, the Ins/IGF-1 pathway also appears to be affected.
Interestingly, the Ins/IGF-1 signaling pathway initiated by ligand–receptor complex formation can be bypassed by ROS (Balaban et al., 2005), a response that mimics the downstream targeting of the ligand–receptor activation, suggesting that ROS are potential physiological factors that may influence longevity determination vs. acceleration of aging. The cross-talk resulting from specific levels and duration of exposure to ROS may play a critical role in the nature of the response to ROS, i.e., a balance between the delay vs. acceleration of the development of aging characteristics. In this context, the levels and source of radical ROS activities of redox-sensitive signaling pathways are an integral part of both normal physiological as well as pathological processes (Droge, 2005a,b). The Ins/IGF-1 signaling pathway is of particular interest because of its decreased activity due to genetic manipulation vs. its responsiveness to ROS levels, i.e., is there a difference between the insulin-IR/IGF-1-IGF-1R-activation vs. the ROS-mediated bypass activation.
2. Cross-talk between Ins/IGF-1 and ROS-redox pathways in Snell and Ames dwarf mice
Mutation analyses in the Snell (Pit-1dw/dw) and Ames (Prop1df/df) long-lived mice have identified signaling pathways that regulate longevity via hormone signal transduction (Bartke et al., 1998; Brown-Borg et al., 1996; Coschigano et al., 2000; Flurkey et al., 2001, 2002; Hsieh et al., 2002a,b). In these mice the GH deficiency alters the levels of circulating insulin and IGF-1 thereby resulting in decreased activity of their pathway. Thus, the reduction-of-function of the Ins/IGF-1 signaling pathway is an important physiological characteristic associated with extended lifespan.
3. Redox modulation of Ins/IGF-1 signaling
The preliminary response to the IR-mediated activation stimulates ROS production in a variety of cell types (Fig. 1). In adipocytes, it induces NAD(P)H oxidase activity that generates H2O2 (Droge, 2005a,b; Goldstein et al., 2005; Mahadev et al., 2001a,b). This activation of endogenous H2O2 plays a role in the autophosphorylation of the insulin receptor β-subunit (IRβ). There are both positive and negative autoregulatory roles of ROS in the regulation of Ins/IGF-1 signaling. The positive autoregulatory mechanism implicates the NAD(P)H oxidase system, which is the target of insulin-stimulated cellular H2O2 production (Droge, 2005a,b). The negative regulation involves the inhibition of response to insulin.
Fig. 1.
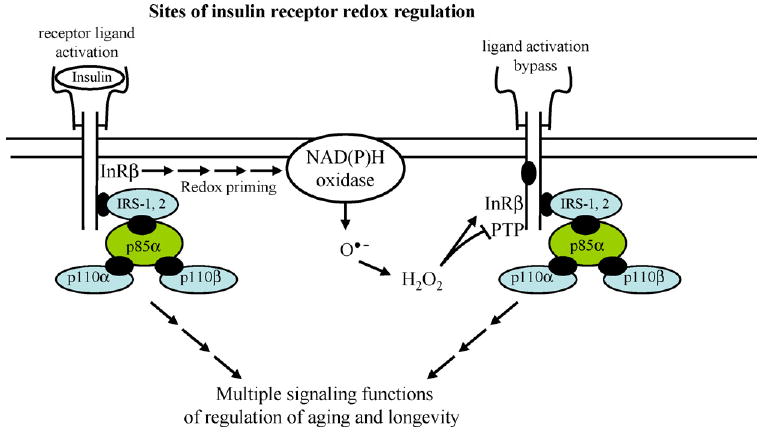
The interactions of the insulin receptor linked-redox signaling pathways. This scheme shows the major interactions of insulin signaling involving the activation of the receptor tyrosine kinase cascade. This leads to the initial redox-mediated priming of the receptor tyrosine phosphorylation and its IRS proteins. The scheme also links the ligand activation bypass of insulin signaling mediated by reactive oxygen species. The steady-state level of tyrosine phosphorylation of these proteins is regulated by PTPases, a family of enzymes that constitutes one of the key targets of regulation by reactive oxygen species.
3.1. The autoregulatory loop–the primer effect
Mild intracellular oxidative conditions enhance the activation of the IRβ-subunit, suggesting that optimal insulin responsiveness involves a “redox priming” of the β-subunit (Goldstein et al., 2005; Schmid et al., 1998, 1999). Thus, the cross-talk between Ins/IGF-1 and ROS signaling is initiated by the insulin-mediated generation of low levels of endogenous H2O2 (Mahadev et al., 2001a,b). This is essential for initial tyrosine phosphorylation of the IR and insulin receptor substrate (IRS) protein(s) due to its reversible oxidative inhibition of protein tyrosine phosphatase which enables IRβ-subunit phosphorylation and facilitates insulin signaling (Mahadev et al., 2001a,b; May et al., 1979; Meng, 2002). This level of cross-talk is the normal physiological process (Fig. 2).
Fig. 2.
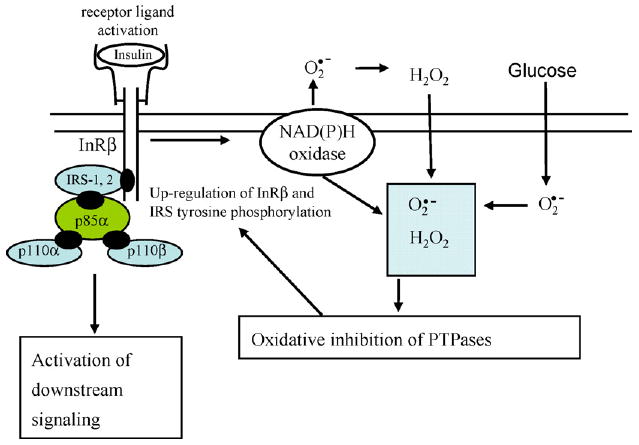
The pathway of insulin-stimulated production of reactive oxygen species. The generation of cellular reactive oxygen species (super oxide or H2O2), initiated by insulin is coupled to the NADPH oxidase system. This pathway involves the activation of the catalytic subunit homologue, Nox 4 (Goldstein et al., 2005). Superoxide generated by the NADPH oxidase system is converted to H2O2 and plays a role in modifying the catalytic activity of thiol-dependent regulatory proteins. The generation of reactive oxygen species by insulin may be enhanced by high glucose levels that increase mitochondrial superoxide production and may also activate the NADPH oxidase system. (This figure is a modification from Goldstein et al., 2005.)
3.2. Potential redox targets in Ins/IGF-1 signaling–the effects of exogenous ROS
Using rat adipocytes in culture it has been shown that exogenous H2O2, modulates insulin signaling by stimulation of IR phosphorylation and tyrosine kinase activity. This response to H2O2 is not a direct effect on the ligand binding domain of the receptor, but mimics the physiological responses to insulin, such as glucose transport (Hayes and Lockwood, 1987), glycogen synthesis (Lawrence and Larner, 1978), lipogenesis (May and de Haen, 1979) lypolysis (Little and de Haen, 1980) and phosphoenolpyruvate carboxykinase gene expression (Sutherland et al., 1997). On the other hand, long-term exposure to exogenous H2O2 exerts an inhibitory effect on insulin action by decreasing insulin-induced glucose transport and GLUT-4 translocation (Rudich et al., 1998). This is achieved by the phosphorylation of specific serine residues of IRS-1 and IRS-2, a mechanism that attenuates the activation of PI3K and Akt, and may be a part of the mechanism of insulin resistance (Werner et al., 2004; White, 2003). These studies suggest that low levels of endogenously generated H2O2 function as an initiator or “primer” of IRβ-subunit tyrosine phosphorylation (Mahadev et al., 2001a,b; May and de Haen, 1979; Schmid et al., 1998), and subsequent downstream signaling, whereas higher concentrations and longer exposure to exogenous H2O2 attenuate the insulin response (Gardner et al., 2003; Potashnik et al., 2003; Rudich et al., 1998). These observations suggest that the differences in the chronic age-associated exposure to increased endogenous oxidative stress (H2O2) may affect the activity of the Ins/IGF-1 pathway, as does the genetically mediated down-regulation associated with longevity. However, the mechanism of attenuation of the pathway by oxidative stress involves specific phosphorylation of the IRβ-subunit and IRS proteins and is often associated with pathological conditions while the genetic manipulation, such as those of the dwarf mice, involves attenuation of the Ins/IGF-1 pathway activity which is associated with longevity.
3.3. Differences in regulation of Ins/IGF–possible acceleration of aging characteristics
The IRβ-chain contains multiple sites of tyrosine phosphorylation, i.e., at Tyr1158, Tyr1162 and Tyr1163, that are targets of oxidants such as H2O2, and the tyrosine phosphatase inhibitors, vanadate and pervanadate, which exert insulin-like effects in the absence of insulin (Droge, 2005a,b). This enhancement is mediated by two different mechanisms shown in Fig. 2. One of these mechanisms involves the direct enhancement of IRβ tyrosine kinase activity by insulin, while the second involves the activation that bypasses insulin-IR binding that triggers tyrosine phosphorylation (Fig. 2). Both of these actions are mediated by the oxidative inhibition of tyrosine phosphatases, which function as negative regulators. The redox regulation of the tyrosine phosphatases involves the reversible oxidation of the cysteine residue in the catalytic site to sulfenic acid and sulfenylamide derivatives. Furthermore, H2O2 interacts directly with the redox-sensitive cysteine of the IR kinase domain and modulates its kinetic properties; this involves the bypassing interaction that occurs independently of insulin–receptor ligand formation (Droge, 2005a,b).
Redox-sensitive cysteine and tyrosine residues of signaling proteins play a major role in the mechanism of redox-sensitive signaling, which enable a response to changes in ROS via the status of the thiol/disulfide redox-cycle machinery (Droge, 2005a,b). The Ins/IGF-1 signaling pathway is of particular interest because of the changes in its responsiveness to the ROS environment. Thus, the lower levels of ROS activate while higher ROS levels inhibit its signaling processes. This raises the question of whether the age-associated increase in endogenous ROS production, which enhances aging characteristics is a consequence of altered Ins/IGF-1 signaling. For example, the level of H2O2 impairment of insulin-induced receptor autophosphorylation and down-regulation of target activities (PI3K, Akt, Forkhead transcription factors and GLUT4 translocation) may all be consequences of age-associated elevated intrinsic ROS levels. The ROS-mediated switching of tyrosine vs. serine phosphorylation of IRS-1/IRS-2 in aged tissues may play a significant role in delayed vs. accelerated aging (Gardner et al., 2003). Clearly, the multiple sites of regulation at the IRβ-chain and/or the IRS proteins suggest a sensitive balance between activation of the pathway by tyrosine phosphorylation vs. delay or inhibition of the pathway by serine phosphorylation.
3.4. Insulin-ROS cross-talk at the insulin receptor
The insulin receptor contains information necessary to engage multiple signaling pathways that can be differentially activated (Sattar et al., 2007). For example, insulin activation of the IR-kinase leads to phosphorylation of the p52Shc isoform, which is a splice variant of p66Shc and one of two cytoplasmic adaptor proteins (p52Shc/p46Shc) involved in the intracellular ROS production via signaling from activated tyrosine kinases to Ras (Pelicci et al., 1992). Although p66Shc has the same modular structure as p52Shc/p46Shc, p52Shc is involved in Ras regulation whereas p66Shc regulates ROS metabolism and apoptosis (Migliaccio et al., 1997, 1999; Trinei et al., 2002). The phosphorylated p52Shc binds to the juxta membrane (JM) domain of IRβ-subunit that contains Tyr972 (Gustafson et al., 1995; Sasaoka et al., 1994a,b). Deletion of NPEY972 in the JM domain of the β-subunit prevents phosphorylation of p52Shc, i.e., this mutant shows a loss of insulin-mediated stimulation of p52Shc phosphorylation (Sattar et al., 2007). This suggests that the tyrosine residue of this motif is important for p52Shc phosphorylation (Berhanu et al., 1997). On the other hand, the JM domain-specific amino acids of hIR do not play a critical role in IRS-1 phosphorylation as indicated by the activation of the downstream Akt signaling molecule in the absence of the JM domain. Interestingly, the JM domain separates insulin-stimulated ROS metabolism, and insulin stimulated IRS-1/IRS-2 activation. Both are discrete pathways that play a role in the regulation of aging and longevity.
3.5. Effects of exogenous ROS
At least three downstream components of the insulin signaling pathway are also potential targets of redox regulation: (1) serine protein phosphatase (PP2A), which mediates the negative regulation of Akt by dephosphorylation of Ser473, a redox-sensitive residue that is sensitive to H2O2; (2) MAP kinase phosphatase-1 (MKP-1), which attenuates insulin-stimulated MAP kinase activity, is dependent on reduced thiol for activity (Kusari et al., 1997). In addition, MKP-1 mRNA is increased by ROS (Keyse, 1998); (3) phosphatase and Tenasin homologue (PTEN), which dephosphorylates the 3′-phosphate of inositol phospholipids generated by PI3K, modulates insulin signaling (Ren et al., 1998). PTEN is inactivated by oxidant molecules, which cause a disulfide linkage between two essential cysteine residues in its active sites (Lee et al., 2002). Reduction of H2O2-oxidized PTEN is mediated by thioredoxin, which suggests a mechanism by which the accumulation of 3′-phosphorylated phosphinositides is controlled (Lee et al., 2002).
4. Redox-sensitive sites of the Ins/IGF-1 signaling pathways at IRS-1/IRS-2
Activation of the Ins/IGF-1 signaling pathway involves rapid autophosphorylation of two of three regulatory tyrosine residues of the IRβ kinase domain (Erbina et al., 1985; Saltiel and Kahn, 2001; White and Kahn, 1994). This is followed by phosphorylation of the third tyrosyl residue that determines the overall level of IR kinase activity (Hubbard, 1997). Autophosphorylation of the IR tyrosine residues stimulates the docking of proteins of the IRS family at these phosphorylated sites. Thus, phosphorylation of multiple tyrosyl residues of IRS-1/IRS-2 sets up the activation of multiple downstream signaling targets of the pathway (White, 2002).
4.1. The negative regulation of IRS-1/IRS-2 by ROS
Serine phosphorylation is part of the mechanism that involves the ROS-mediated negative regulation of IRS signaling. Although there are over 70 potential serine phosphorylation sites on IRS-1/IRS-2, hyperphosphorylation stimulated by ROS is associated with insulin resistance. These phosphorylations occur at Ser302, Ser307, Ser612 and Ser632 (Furukawa et al., 2005; Hirosumi et al., 2002; Kim et al., 2004; Um et al., 2004; Yu et al., 2002). The mechanism of serine phosphorylation-mediated inhibition of IRS-1/IRS-2 is based on its ability to stimulate dissociation between IRβ-IRS-1 and/or IRS-1-PI3K, thus preventing activation of the downstream PI3K, possibly increasing degradation of the IRS proteins (Egawa et al., 2000; Li et al., 1999; Moeschel et al., 2004; Mothe and Van Obberghen, 1996). This represents another mechanism of regulation of the IRS-1/IGF-1 signaling activity that may involve the decrease in pool levels of specific signaling protein(s).
A similar mechanism occurs in the liver where accumulation of intracellular lipid metabolites activate a serine kinase cascade leading to decreased IR kinase activity that results in (1) lower insulin-stimulated IRS-2 tyrosine phosphorylation; (2) lower IRS-2-associated PI3K activity; (3) lower Akt activity (Hsieh et al., 2002a; Samuel et al., 2004). This results in decreased phosphorylation of FOXO, which allows it to translocate to the nucleus where it activates the transcription of redox enzymes such as MnSOD and rate-controlling enzymes of gluconeogenesis. In general, studies of longevity in Ames and Snell mice and of rodent insulin resistance indicate decreased insulin pathway activity. A difference, however, lies in the level of serine phosphorylation of specific signaling proteins and the consequences of these phosphorylations on pathway activity.
The IRS-1 and IRS-2 proteins, through their docking to activated InsRβ (DuPont et al., 1998; Shimomura et al., 2000; Whitehead et al., 2000), serve as a major crossroads that mediate the downstream signaling cascade via the p85α regulatory and p110α/p110β catalytic subunits of PI3K, and Akt (Nakae et al., 1999; Sun et al., 1995) that links the Ins/IGF-1 signaling to the regulation of Forkhead transcription factors (AFX, FKHR and FKHRL1). These regulate such biological processes as carbohydrate metabolism, cell cycle and apoptosis (Biggs et al., 1999; Cahill et al., 2001; Kops et al., 1999; Rena et al., 1999;), the Ins/IGF-1 downstream targets whose activities regulate lifespan in the long-lived dwarf mice (Bartke et al., 1998; Brown-Borg et al., 1996; Coschigano et al., 2000; Flurkey et al., 2001, 2002).
5. Multiple pathways of PI3K subunit signaling
Genetic experiments in Caenorhabditis elegans, Drosophila and mice clearly implicate phosphatidylinositol 3-kinase (PI3K) as a key component of the Ins/IGF-1 signaling pathway that regulates metabolism, cell growth and aging. Class IA, PI3K, is a heterodimer composed of one of three p85 regulatory subunits (p85α, p85β, and p85γ); the p85α subunit binds to IRS-1 and IRS-2. The PI3K heterodimers also consist of the p110 catalytic subunits that bind to the downstream AKT/PKB in the process of signal transduction (Engelman et al., 2006). The p85 regulatory subunits that complex with the p110 catalytic subunits to form the PI3K heterodimer play a unique role in the multiplicity and diversity of the insulin signaling pathway: (a) they serve as a bridge between the catalytic p110 isoforms and tyrosine-phosphorylated IRS proteins (Figs. 1 and 2); (b) they regulate p110 catalytic activity which transduces the IR signal to the downstream AKT/PKB protein; (c) as monomers, they serve to negatively regulate PI3K by competing with p85–p110 for binding to IRS proteins, and (d) the regulatory p85 subunit can down-regulate Ins/IGF-1 signaling by complexing to IRS-1/IRS-2 as a monomer (Ueki et al., 2002). Thus, the magnitude of PI3K-dependent signaling is determined by the balance between positive signals derived from p85–p110–IRS complex formation which transduces the signal to its downstream substrate (Akt/PKB) or, the level of negative signaling derived from the p85–IRS complex which prevents transduction of the signal further downstream (Fig. 3).
Fig. 3.
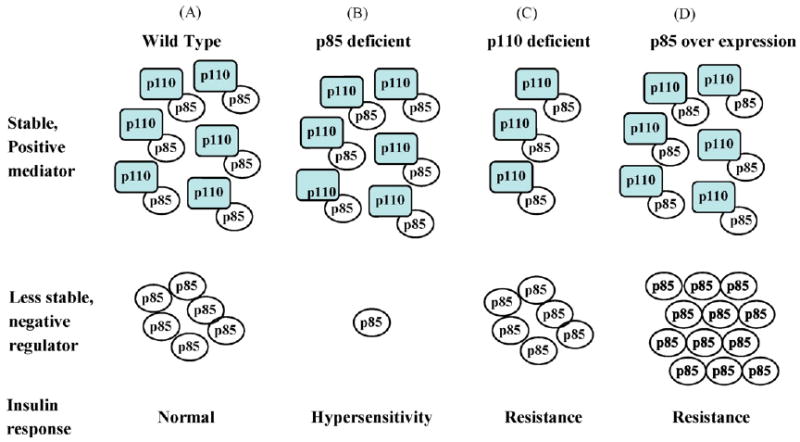
The regulation of insulin signaling by the ratio of the positive mediator, p85–p110 and the negative regulator, unbound p85. This model proposes that: (A) a given ratio between p85–p110 and monomeric p85 defines the insulin signaling pathway activity and the state of insulin sensitivity in the wild-type background. (B) In p85α(−/−) and p85β(−/−) mice, free p85 is preferentially decreased and the balance between p85–p110 and free p85 is shifted toward the positive mediator, p85–p110. This causes increased insulin sensitivity in the mutant mice. (C) In contrast, in p110α (+/−) and p110β(+/−) mice, the free p85 pool level is preferentially decreased (over free p85). This shifts the balance toward the higher levels of free p85, negative regulator, which inactivates IRS-1 and IRS-2 (free p85), resulting in decreased insulin signaling and insulin resistance. (D) Over expression of p85 causes an increase in the p85 pool which shifts the balance toward the negative regulator, resulting in decreased insulin sensitivity (This figure is a modification from Brachmann et al., 2005.)
The p85 monomer plays a novel role as a critical cross-roads site downstream of IRS-1/IRS-2, by providing both the positive (PI3K activation) and negative (p85 feedback) regulation critical in insulin signaling. These differential roles are supported by the improved insulin response that occurs by deletion of p85 or by p85 deficiency which results in insulin hypersensitivity (Fig. 3B). Monomeric p85 inhibits PIP3 production by competing with the PI3K heterodimer for binding to IRS proteins or by altering the subcellular localization of the PI3K signaling complexes. This mechanism prevents the localization of PI3K where its substrate, PI-4,5-P2, resides. Either directly or indirectly, p85 regulates the activity of PTEN in vivo in the liver. Although it is not clear how the regulation occurs, the likely mechanism is that p85α enhances PTEN function by modulating some post-translational modification or altering its subcellular localization (Luo et al., 2005; Taniguchi et al., 2006a,b).
5.1. The negative regulation of IRS-1/IRS-2 by p85
The ROS-mediated differential phosphorylation of serine residues is part of the mechanism of regulation of IRS-1/IRS-2 signaling. Another mechanism of regulation of the IRS signaling is mediated by binding of the p85 monomeric subunit to IRS-1. This complex blocks the activation of the downstream pathway, i.e., Akt/PKB, since activation of PI3K requires the binding of p85–p110 complexes to IRS-1/IRS-2 (Fig. 3). In this mechanism it is the abundance of p85 regulatory monomers compared to the levels of the p110 catalytic monomers that confer the negative regulatory function of p85. These monomers, upon binding to IRS proteins interfere with the binding of p85–p110 heterodimers to phosphorylated IRS proteins (Mauvais-Jarvis et al., 2002; Ueki et al., 2002). This negative regulatory function inhibits insulin action and the physiological processes that decrease the monomeric p85 pool level thus enhancing insulin signaling (Fig. 3). For example, mice lacking either the p85α or p85β regulatory subunits exhibit increased insulin sensitivity (Brachmann et al., 2005). Thus, in the insulin sensitive tissues of the p85α−/− animal there are decreased p110α and p110β catalytic subunits and in p110α+/− and p110β+/− heterozygotes there is ∼50% decrease in p85 expression in liver and muscle. The knockdown of p110 by RNA interference also resulted in loss of p85 proteins and is attributed to the decreased stability of the monomeric proteins. These studies suggest that insulin sensitivity is regulated by a balance between p85 and p110 monomeric pool levels and the level of the heterodimeric p85–p110 pool. Thus, p85 subunits play a negative role in insulin signaling that is independent of their p110 subunit activation, and the ratio of p85 to p110 plays a critical role in insulin sensitivity in vivo (Fig. 3). It is interesting that the ROS-stimulation of phosphorylation of serine residues inactivate IRS-1/IRS-2, thus raising the question of whether this serine modification and p85 binding to IRS-1/IRS-2 are interrelated. Furthermore, the levels of monomeric p85 and dimeric p85–p110 in animals that age normally and in the long-lived Snell, Ames and IGF-1R mutants, that are resistant to oxidative stress, may play a key role in the level of activity of the Ins/IGF-1 pathways.
5.2. p85-mediated down-regulation of JNK activity
Another important regulatory function of the monomeric p85 is its p85-dependent, but PI3K-independent signaling pathway that attenuates insulin-induced JNK1 activity in adipocytes and liver (Fig. 4; Taniguchi et al., 2006a,b, 2007; Terauchi et al., 1999; Ueki et al., 2002). This mechanism involves the p85 activation of NAD(P)H oxidase-cdc42, which is an ROS producing function. JNK1 is activated in p85−/− cells by expression of p85α and p85β independently of levels of PI3K activity. Furthermore, the expression of p85α restores insulin-induced JNK activity. Thus, the mechanism of insulin induced activation of JNK occurs via the p85-mediated activation of NAD(P)H oxidase-cdc42, without PI3K involvement. This function is localized to the N-terminal region of p85. Although JNK1 down-regulates insulin sensitivity by phosphorylation of Ser307 in IRS-1, decreased JNK1 activity does not increase or prolong tyrosine phosphorylation of IRS-1 (Aguirre et al., 2002; Lee et al., 2003; Hirosumi et al., 2002).
Fig. 4.
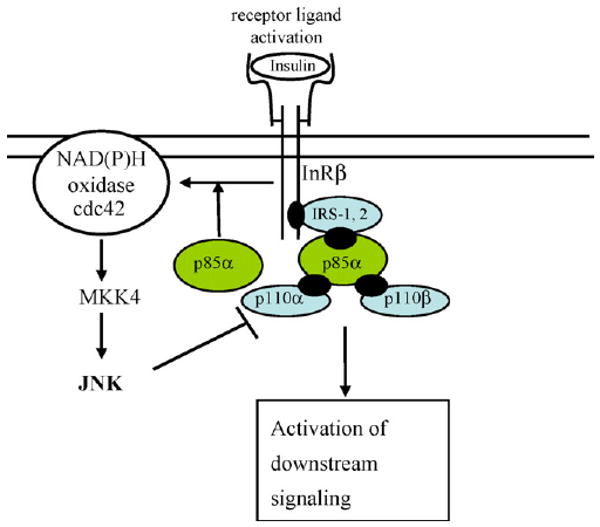
Molecular mechanism and pathway of JNK activation by p85. The full-length p85 regulatory subunit activates JNK through the insulin/cdc42/MKK pathway. Activation of JNK results in negative feedback on the insulin signaling pathway. Thus, the loss of p85 improves insulin sensitivity, in part by diminishing this JNK-mediated negative regulation. (This figure is a modification from Taniguchi et al., 2007.)
The mechanism of regulation of JNK1 activation by p85 does not involve direct interaction between these two proteins. This cascade does, however, lie downstream of Ras since a dominant negative form of Ras completely inhibits insulin-induced JNK activity. Alternatively, p85 negatively regulates a dual specificity phosphatase activity for JNK, and a reduction of p85 decreases this negative regulation, thus increasing inhibition of JNK activity.
5.3. p85–p110 in the Snell dwarf mutant
Studies of the Ins/IGF-1 pathway in Snell dwarf and in the long-lived, Age-1, C. elegans (Age-1) mutant have indicated that the PI3K (p85–p110) is a significant site of down-regulation of the pathway associated with longevity. A series of striking changes in the components of the Ins/IGF-1 signaling pathway that affect its activity, occur in the Snell dwarf mouse liver. These involve a dramatic decrease in the pool level of IRS-2, hyper-phosphorylation of its tyrosine residues accompanied by decreased docking of p85α, and a severely decreased PI3K activity. These observations suggest that these changes may be associated with the mechanism that decreases insulin signaling at that site in the dwarf (Hsieh et al., 2002a). Thus, the decreased PI3K activity correlates with the decreased Ins/IGF-1 signaling in these long-lived animals. However, the increased phosphorylation of IRS-2 does not correlate with IRS-2–p85α docking or an associated decrease in PI3K activity. On the other hand, in normal aging mice, PI3K activity associated with IRS-2 is also decreased even though the pool level increases. This suggests that age-associated attenuation of PI3K may be attributed to a decreased docking ability of interacting components. The changes in pool level and tyrosine phosphorylation of aged control vs. dwarf suggest a decrease of IRS-2 activity, but by different mechanisms. In the aged control group the high pool level and low phosphorylation are consistent with decreased protein signaling activity; in the dwarf, although the phosphorylation level is high, the low pool level may impose a decreased signaling activity. Interestingly, although a similar dramatic decrease of IRS-2 pool level has been observed in insulin-resistant lipodystrophic and ob/ob mice (Shinomura et al., 2000) and IRS-2 knockout mice (Burkes et al., 2000), the long-lived Snell dwarf mice do not develop the acute pathological characteristics of diabetes associated with decreased IRS-2 pool level. The low IRS-2 pool and high phosphorylation levels are characteristics of the Pit-1 mutation and decreased signaling at this level of the IRS-1 (Ogawa et al., 1998) and IRS-2 pathways (Sun et al., 1991; White and Kahn, 1994).
5.4. The signaling specificity of p110α and p110β
Although phosphorylation and pool levels are important factors in PI3K regulation, our studies have indicated a preferential binding of p85α–p110α to IRS-2 in aged Snell dwarfs and p85α–p110β to IRS-2 in aged controls (Hsieh et al., 2002a,b). Such specificity is seen in insulin-mediated induction of heterodimer formation (p85α–p110α and p85α–p110β) that results in marked enhancement of p110β kinase and only a very small effect on p110α kinase activity (Asano et al., 2000). Furthermore, evidence of signaling specificity of p110α and p110β subunits is suggested by the overexpression of p110β, which has no effect on basal glucose transport but leads to an increase in insulin-induced glucose uptake, whereas overexpression of p110α increased both basal and insulin-stimulated glucose uptake. Microinjection of anti-p110β but not anti-p110α completely abolished insulin-induced translocation of GLUT4 to the cell surface. Furthermore, p85α–p110α is preferred over p85α–p110β for the differential stimulation of DNA synthesis by various growth factors (Roche et al., 1994, 1998). These specific combinations of protein-protein interactions within the components of the Ins/IGF-1 signaling pathway may target genes that play a role in longevity determination. Further identification of the proteins specifically targeted by these heterodimers would provide more detailed understanding of the molecular and genetic factors of longevity. Thus, the p85α–p110α heterodimer is the primary insulin-responsive PI3K (in cultured cells) whereas p85α–p110β is dispensable. The p110α is the critical lipid kinase required for insulin signaling in adipocytes and myocytes while p110β and p110δ play a secondary role in insulin signaling in these cells (Knight et al., 2006).
The complexity of the IRS-1/IRS-2 regulatory and signaling processes is indicated by the number of amino acid sequences that define specific binding sites for a variety of signaling pathways including cross-talk with the second messenger ROS-mediated redox pathway. Thus, PI3K (p85–p110) links to the activation of Akt (PKB) and then to FOX O expression; Brb2 links IRS-1 phosphorylation to the Ras pathway (GTPase) and stimulation of ROS via Nox4 and p21ras. Insulin-stimulated phosphorylation of p66Shc enables it to bind to Grb2 and the activation of Ras (GTPase), which couples insulin signaling to MKKs and the stress response MAPKinase pathway (Sun et al., 1991). A major signaling protein that docks to IRS-1, and -2, PI3K belongs to a key family of “cross-roads” proteins that regulate the multiple metabolic insulin pathways. Signaling proceeds further downstream to Akt (PKB), a serine–threonine kinase which targets such sites as glycogen synthase kinase 3, p70S6K and protein kinase-Cξ (Shepherd et al., 1998). These signaling proteins mediate insulin-induced glucose transport, GLUT-4 translocation, glycogen and protein synthesis (Srivastava, 2005).
In summary, p85α and p85β have multiple common functions in insulin signaling, including stabilization of p110, bridging between p110 and phosphorylated IRS proteins, competing with p85–p110 for binding to IRS proteins, and down-regulating PIP3 levels, independent of regulation of PI3K. Thus, biological responses to Ins/IGF-1 mediated by PI3K is determined by a balance between the positive effect of p85–p110–IRS complexes and the negative effects derived from monomeric p85 (PI3K-dependent inhibition) and total p85 (PI3K-independent inhibition). The significance of these regulatory events in aging and longevity determination remains to be demonstrated.
6. Second messengers and mimickers of insulin
Oxidants can mimic insulin stimulation of its target cells, and mutations affecting aging and longevity also confer resistance to oxidative stress, suggesting that a balance of ROS production may play a role in the development of aging and longevity characteristics.
The interplay between insulin-induced ROS serves as a second messenger that facilitates insulin signaling. The signaling proteins that participate in this function are potentially susceptible to reversible inhibition by biochemical oxidation, e.g., the protein-tyrosine phosphatases (PTPs), are sensitive to biological oxidation and reduction and are critical components of the regulatory cross-talk between Ins/IGF-1- and ROS-generating pathways. The age-associated increase in endogenous ROS production causes a stabilized, chronic exposure to ROS that may cause reduction-of-function by PTPs, resulting in an inability to turn off signaling.
High endogenous levels of ROS play a key role in accelerating the development of senescence characteristics (Finkel, 2003). Interestingly, low endogenous levels of superoxide and H2O2 generated by insulin stimulation are important factors in regulating normal insulin signaling (Fig. 2). The mechanism of insulin-stimulated ROS production involves the NADPH oxidase system (NOX 4), the protein tyrosine phosphatases (PTPases) and certain enzymes whose activities depend upon the reduced state of critical thiol residues for their catalytic activity. These observations suggest that the age-associated increase in endogenous ROS generated by mitochondrial dysfunction may affect the activity of the insulin/IGF-1 signaling pathway. More specifically, is there a physiological difference between the level of Ins/IGF-1 signaling activity in young tissues compared to the signaling activity of aged tissues whose elevated endogenous ROS levels are stabilized and serve as a continuous stimulus to the ROS-sensitive components of the Ins/IGF-1 pathway. The fact that prolonged exposure to exogenous ROS factors (H2O2) causes insulin/IGF1 signaling abnormalities strongly suggests that age-associated alterations or abnormalities of this pathway may be due to the increased endogenous level of ROS production. Thus, the reduction-of-function of the Ins/IGF-1 signaling pathways in the long-lived mouse mutants may be a factor that decreases the ROS-mediated abnormalities of Ins/IGF-1 signaling, a factor that might also involve resistance to oxidative stress. We propose that: (1) while the endogenous levels of oxidants that serve as second messengers are within a range that mediates normal signaling; (2) the increased levels of endogenous oxidants and chronic overexposure to extrinsic oxidative stress may be the basis for Ins/IGF-1 signaling abnormalities that occur in aging tissues; (3) such an increase in ROS production in long-lived mouse models may be attenuated by the decreased activity of the Ins/IGF-1 pathway; (4) this suggests that the levels of second messengers that mimic insulin may play a role in the progression and determination of aging and longevity.
6.1. The ROS-mediated regulation of Ins/IGF-1 signaling via phosphoprotein phosphatases
Cellular phosphotyrosine phosphatases (PTPases) play a central role in the down-regulation of IR kinase activity and its downstream cascade (Goldstein, 2003). An important feature of this negative regulation of insulin signaling relies upon the sensitivity of the catalytic sites to the endogenous redox state of the cell. The PTPases contain a conserved ∼230 amino acid domain consisting of an 11-residue signature sequence that includes a cysteine residue that is required to catalyze the hydrolysis of protein phosphotyrosine residues (Andersen et al., 2001; Tonks and Neel, 2001). The cysteine residue of the catalytic site is oxidized in a stepwise manner by ROS, thereby decreasing their activity (Fig. 5; DeGnore et al., 1998; Finkel, 2003; Lee et al., 1998; Meng et al., 2002). The initial oxidation produces the sulfenic (−SOH) form, a modification that inactivates the enzyme but can be reversed by either enzyme activity or reducing agents (Claiborne et al., 1999; Denu and Tanner, 1998). Further oxidation to the sulfinic (−SO2H) and sulfonic (−SO3H) forms results in irreversible inactivation of PTPases (Barrett et al., 1999a,b). A reversible inactivation of PTPases can also be accomplished by forming a glutathiolated form that is reversed by a specific reductase (Barrett et al., 1999a,b). This mechanism represents the direct link of intracellular ROS levels to the insulin signaling pathway activity and raises the questions of whether age-associated and stabilized increase in endogenous ROS production plays a role in this regulation.
Fig. 5.
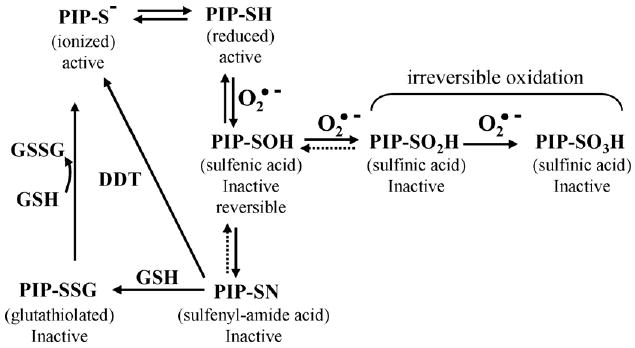
Regulation of PTPase catalytic activity by oxidation, and reduction of the catalytic cysteine residue. The regulation of signal transduction by PTPases involves the modulation of their activity by modification of the oxidation–reduction state of the catalytic center. The catalytic cysteine of the PTPases is especially reactive because of the ionized state of the cysteinyl hydrogen. The stepwise oxidation of the catalytic cysteine residue by reactive oxygen species results in inactivation of the enzyme. Oxidation of the catalytic thiol to the sulfenic (−SOH) form is reversible; oxidation to sulfinic (SO2H) and sulfonic (–SO3H) forms lead to irreversible PTPase inactivation. Mildly oxidized PTPase can undergo disulfide conjugation in the cell with glutathione. A novel sulfenyl-amide derivative of PTP1B has been shown to undergo GSH conjugation or direct biochemical reduction with DTT. Active enzyme can be regenerated from glutathiolated enzyme by cellular GSH reductases. (This figure is a modification from Goldstein et al., 2005.)
There is considerable evidence that the protein tyrosine phosphatase 1B (PTB1B) plays a major role in the negative down-regulation of the insulin signaling pathway in vivo (Elchebly et al., 1999; Galic et al., 2003; Klaman et al., 2006). This is supported by the enhancement of insulin receptor autophosphorylation in the PTP1B knockout, and insulin responsiveness in skeletal muscle and liver is indication of the absence of negative regulation by the PTB1B action.
7. The Klotho mutant–increases resistance to oxidative stress
The klotho gene encodes a single-pass transmembrane protein detected mainly in distal convoluted tubules of the kidney and choroid plexus in the brain. Over expression of the gene extends lifespan by ∼20–30% in males and 18–19% in females whereas mutation of the gene (KL−/−) leads to premature aging and death at ∼2 months of age, suggesting that this gene functions as a suppressor of aging in mammals (Kuro-o et al., 1997; Kurosu et al., 2005). Klotho is a circulating peptide hormone that suppresses insulin signaling by attenuating tyrosine phosphorylation of the insulin and IGF-1 receptors. This reduces the IRS protein association with PI3K, thereby decreasing insulin and IGF-1 signaling. The Klotho mutant (KL−/−) exhibits accelerated age-dependent loss of function in multiple age-associated traits. Thus, klotho is an aging suppressor gene whose product functions as a hormone that inhibits intracellular insulin and IGF-1 signaling (Kuro-o et al., 1997; Takahashi et al., 2000).
7.1. Klotho inhibits intracellular insulin and IGF-1 signaling
The extracellular soluble Klotho peptide, comprised of 952 amino acids, is detected in blood and cerebrospinal fluid in wild type mice but not in KL−/− mice (Imura et al., 2004; Takahashi et al., 2000). There is clear evidence that Klotho does not inhibit ligand binding to IR and that its action involves suppression of the ligand stimulated autophosphorylation of IR and IGF-1R. Consequently, downstream signaling events, i.e., activation of phosphorylation of IRS-1– and IRS-2–p85 docking sites are also attenuated; previously activated IR is also inactivated by Klotho. A similar effect occurs with IGF-1R autophosphorylation. In general, klotho inhibits activation of IR and IGF-1R and represses activated receptors suggests that the cross-talk between Klotho and IR may also regulate the redox activities of these proteins.
7.2. Inhibition of insulin and IGF-1 signaling rescues KL−/− phenotypes
Since inhibition of insulin and IGF-1 signaling by Klotho retards senescence, independent inhibition of these signaling pathways may ameliorate some of the aging-like phenotypes of KL−/− mice. Thus, survival is improved in KL−/− mice heterozygous for IRS-1 null allele (KL−/−/IRS-1+/−) relative to the KL−/− control. These studies suggest that the potential decrease in oxidative stress due to reduction-of-signaling activity of Ins/IGF-1 may be a factor in increased longevity of these animals due to their resistance to oxidative stress.
The fact that Klotho-induced inhibition of insulin/IGF-1 signaling is associated with resistance to oxidative stress emphasizes the redox sensitivity of its anti-aging processes. The mechanism of this resistance involves (1) inhibition of FOXO phosphorylation, which (2) promotes FOXO nuclear translocation; (3) FOXO binds to MnSOD promoter, up-regulates its expression to facilitate removal of ROS and confer resistance to oxidative stress (Yamamoto et al., 2005). Thus, Klotho protein suppresses aging via two distinct anti-aging mechanisms that are evolutionarily conserved: (1) the inhibition of insulin-like signaling; (2) increase resistance to oxidative stress.
The decreased activity of IR and IGF-1R is part of the mechanism of longevity determination of Klotho and dwarf mice (Brown-Borg et al., 1996; Coschigano et al., 2000; Dominici et al., 2002; Flurkey et al., 2001; Hsieh et al., 2002a,b; Tatar et al., 2003). Some of the important (basic) determining factors of the longevity of these mutants that are linked to ROS cross-talk are (1) enhanced insulin sensitivity; (2) reduced insulin secretion and the resulting alteration of insulin signaling to its downstream targets, p85-p110 specificity of the p110 targets in response to insulin; (3) decreased glucose tolerance (Coschigano et al., 2000; Dominici et al., 2002; Hsieh et al., 2002a,b); (4) divergent modification of insulin action in different organs as well as to the various p110 pathways (Coschigano et al., 2000; Dominici et al., 2002; Hsieh et al., 2002a,b); (5) overlapping effects of insulin and IGF-1 on activation of IRS-1 and IRS-2. Involvement of both insulin and IGF-1 signaling in the control of aging and longevity is supported by increased longevity of mice heterozygous for IGF-1R mutation (Holzenberger et al., 2003) and mice with adipose specific IR knockout (Bluher et al., 2003).
Increased longevity in Klotho overexpressing transgenic mice provides a link to the interaction and cross-talk between insulin and IGF-1 signaling and longevity determination (Kurosu et al., 2005).
8. The p66shc mutant–a redox link to longevity determination
The protein p66Shc is a 66 kDa alternatively spliced isoform of the growth factor adapter Shc that is phosphorylated in response to oxidative stress (H2O2) (Migliaccio et al., 1999; Nemoto et al., 2006; Orsini et al., 2004; Pinton et al., 2007). It is a genetic determinant of lifespan in mammals; ablation of the gene extends lifespan with no pathological consequences (Migliaccio et al., 1999). Its function involves the regulation of ROS metabolism and apoptosis and is one of several mammalian genes associated with longevity determination, i.e., the knockout results in extended lifespan of about 30% (Migliaccio et al., 1999). These p66shc knockouts exhibit resistance to exogenous oxidative stress in vivo, and their cells in culture show lower levels of ROS production (Francia et al., 2004; Migliaccio et al., 1999; Napoli et al., 2003; Nemoto and Finkel, 2002; Trinei et al., 2002). A fraction of p66Shc localizes to the mitochondrial intermembrane space where it binds to cytochrome c and acts as ROS generating oxidoreductase, leading to mitochondrial dysfunction and cell death (Giorgio et al., 2005; Nemoto et al., 2006; Orsini et al., 2004). This process involves the use of reducing equivalents of the mitochondrial ETC through the oxidation of cytochrome c. Thus, the p66Shc system provides an alternative redox process associated with mitochondrial electron transport chain activity that generates proapoptitic ROS in response to stress signals. Phosphorylation of Shc Ser36 occurs in the cytoplasm and is necessary for Shc activation and translocation to the mitochondrial intermembrane space. This signaling route links an oxidative challenge to the activation of p66Shc, to the recruitment of mitochondria in apoptosis and its role in the integration of signaling pathways that control aging, longevity and cell death (Hajnoczky and Hoek, 2007; Pinton et al., 2007). The long-lived mice lacking p66shc exhibit decreased mitochondrial function including mitochondrial generated ROS (Giorgio et al., 2005; Nemoto et al., 2006).
The decreased oxidative capacity in p66Shc(−/−) cells is partially offset by increased levels of glycolysis. This switch to aerobic glycolysis results in increased lactate production and requirement of glucose to maintain ATP levels. These data suggest that p66Shc regulates mitochondrial oxidative capacity and that its ability to extend lifespan may reside in the partitioning of metabolic energy conversion away from oxidative and toward glycolytic pathways (Johnson et al., 2001; Nemoto et al., 2006), This affects the level of ROS production and raises the question of the role of decreased ROS levels in the regulation of Ins/IGF-1 pathway activities in p66Shc(−/−) animals.
The translocation of p66Sshc to the mitochondrial intermembrane space in response to H2O2 is promoted by the activation of PKCβ which induces p66Sshc phosphorylation and binding of p66shc to Pin1 (Pinton et al., 2007). In this interaction, Pin1, a prolyl isomerase that induces cis–trans isomerization of phosphorylated Ser-Pro (e.g., as in p66Shc), confers phosphorylation-dependant conformational changes upon Pin1 targets. Thus, conformational changes induced by the binding of phosphorylated p66Shc to Pin1 may expose a hidden sequence that targets p66Shc to mitochondria (Pinton et al., 2007). The imported p66Shc causes alteration of mitochondrial Ca2+ responses and three-dimensional structure thus inducing apoptosis. This signaling route activates an apoptotic inducer shortening the lifespan.
The mechanism of mitochondrial p66Shc activity involves (a) phosphorylation of Ser36 of p66Shc by PKCβ; (b) interaction of p66Shc with Pin1 induces cis–trans isomerization and translocation to inter-mitochondrial space; (c) p66Shc in inter-mitochondrial space interacts with cytochrome c to produce H2O2; (d) this promotes opening of mitochondrial permeability transition core; (e) mitochondrial generated H2O2 is required for pore opening; (f) this is proposed to be p66Shc-mediated ROS generation.
In this review although we discussed the mechanism of p66shc stimulated ROS production, activation of p52Shc a splice variant of p66shc is also involved in intracellular ROS production via signaling from activated tyrosine kinases to Ras (Pelicci et al., 1992). This is in contrast to the ROS-mediated phosphorylation and activation of p66shc. Interestingly, the sites of ROS production by p66shc (mitochondrial) and p52Shc (cytoplasmic) clearly point to the diversity and complexity of the hormone vs. the ROS-mediated activation of the signaling pathways that play an important role in the determination of aging and longevity.
9. Discussion
A number of long-lived mouse mutants modulate the insulin/IGF-1/GH signaling pathway in a manner similar to the long-lived C. elegans and Drosophila mutants. The Ames and Snell mouse mutants are two of the most extensively studied strains. These long-lived mice exhibit such characteristics as modulation of the insulin/IGF-1 signaling as well as resistance to oxidative stress and increased anti-oxidant scavenging activity (Balaban et al., 2005), which suggest that the mechanisms of aging and longevity may involve cross-talk between the ligand–receptor and the ROS signaling pathways.
Although the free radical theory of aging hypothesizes that accumulation of oxidatively modified proteins is a mediator of the aging processes, ROS also act as second messengers that regulate cellular functions such as signaling pathways (Balaban et al., 2005; Barrett et al., 1999a,b; Harman, 1956; Humphries et al., 1998; Thomas et al., 1995; Thomas and Mallis, 2001). In general, signaling processes that are regulated in response to changes in redox-balance represent a safety net of regulation residing between reversible and irreversible redox-mediated protein modifications. Through these reactions, redox-modified proteins regulate cell signaling, transcriptional control and metabolism (Droge, 2002; Ghezzi et al., 2005; Rhee, 1999; Shelton et al., 2005). The irreversible oxidation of enzymes and signaling proteins potentially play a critical role in aging and in the progression of age-associated diseases (Beal, 1995; Schapira, 1999.)
In the mutants used to study lifespan, the level of resistance to oxidative stress is a common characteristic associated with longevity, while increased sensitivity to oxidative stress correlates with decreased lifespan. Thus, elucidation of how aging may affect the mechanisms of reversible oxidative processes, the function of components involved and the metabolic consequences may provide a more in depth understanding of the molecular mechanisms of aging and the development of biochemical characteristics of aging (Humphries et al., 2007). This raises a number of questions: (1) what are the redox-levels that are critical to the regulation (positive effects), i.e., the positive functional boundaries; (2) what are the mechanisms of regulation of the pathways and their targets; (3) what are the beneficial regulatory functions of the intracellular signaling responses to free radicals and what are the damaging effects of redox-imbalance either hyper- or hypo-oxidation; (4) how/what is the contribution or role of ROS generated by mitochondrial dysfunction; (5) how does this relate to free radical theory of aging. Importantly, although free radicals can promote reversible responses as well as damaging effects, is there a window of demarcation to these responses.
Acknowledgments
This publication was supported by USPHS grant 1P01 AG021832-04 awarded by the National Institute on Aging, and the USPHS grant 1 P30 AG024832-03, the Claude D. Pepper Older Americans Independence Center grant awarded by the National Institute on Aging, and by the UTMB Sealy Center on Aging.
References
- Aguirre V, Wener ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser(307) in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatases domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T, Kanda Q, Katagiri H, Nawano M, Ogihara T, Inakai K, Anai M, Fukushima Y, Yazaki Y, Kikuchi M, Hooshmand-Rad R, Heldin CH, Oka Y, Funaki M. p110β is upregulated during differentiation of 3T3-L1 cells and contributes to the highly insulin-responsive glucose transport activity. J Biol Chem. 2000;275:17671–17676. doi: 10.1074/jbc.M910391199. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of super oxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatases 1B. J Biol Chem. 1999a;274:34543–345436. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999b;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- Bartke A, Brown-Borg HM, Bode AM, Carlson J, Hunter WS, Bronson RT. Does growth hormone prevent or accelerate aging. Exp Gerontol. 1998;33:675–687. doi: 10.1016/s0531-5565(98)00032-1. [DOI] [PubMed] [Google Scholar]
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- Berhanu P, Anderson C, Hickman M, Ciaraldi TP. Insulin signal transduction by a mutant human insulin receptor lacking the NPEY sequence. Evidence for an alternate mitogenic signaling pathway that is independent of Shc phosphorylation. J Biol Chem. 1997;272:22884–22890. doi: 10.1074/jbc.272.36.22884. [DOI] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluher M, Kahn B, Kahn C. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol. 2005;25:1596–1607. doi: 10.1128/MCB.25.5.1596-1607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Burkes D, Font de Mora J, Schubert M, Withers DJ, Myers MG, Towery HH, Altamuro SL, Flint CL, White MF. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000;4079:377–382. doi: 10.1038/35030105. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Izivion G, Nasrin N, Ogg S, Dore JH, Ruvkun G, Alexander-Bridges M. Phosphatidylinositol 3-kinase signaling inhibits DAF16 DNA binding and function via 14-3-3-dependent and 14-3-3 independent pathways. J Biol Chem. 2001;276:13402–13410. doi: 10.1074/jbc.M010042200. [DOI] [PubMed] [Google Scholar]
- Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein–sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- Clancy D, Gems D, Harshman LG, Oldham S, Hafen E, Leevers SJ, Partridge L. Extension of life span by loss of chico, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology. 2000;141:2608–2613. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- DeGnore JP, Konig S, Barrett WC, Chock PB, Fales HM. Identification of the oxidation states of the active site cysteine in a recombinant protein tyrosine phosphatases by electrospray mass spectrometry using on-line desalting. Rapid Commun Mass Spectrom. 1998;12:1457–1462. doi: 10.1002/(SICI)1097-0231(19981030)12:20<1457::AID-RCM346>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide—evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Dominici FP, Hauck S, Agentino DP, Bartke A, Turyn D. Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS-1) and (IRS-2) in liver of Ames dwarf mice. J Endocrinol. 2002;173:81–94. doi: 10.1677/joe.0.1730081. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Droge W. Oxidative enhancement of insulin receptor signaling experimental findings and clinical implications. Antioxid Redox Signal. 2005a;7:1071–1077. doi: 10.1089/ars.2005.7.1071. [DOI] [PubMed] [Google Scholar]
- Droge W. Oxidative aging and insulin receptor signaling. J Gerontol A: Biol Sci Med Sci. 2005b;60:1378–1385. doi: 10.1093/gerona/60.11.1378. [DOI] [PubMed] [Google Scholar]
- DuPont J, Derouet M, Simm J, Taouis M. Nutritional state regulates insulin receptor and IRS-1 phosphorylation and expression in chicken. Am J Physiol. 1998;274:E309–316. doi: 10.1152/ajpendo.1998.274.2.E309. [DOI] [PubMed] [Google Scholar]
- Erbina Y, Ellis L, Jarnagin K, Edery M, Graf L, Ou JH, Masiarz F, Kan YW, Goldfine ID. Human insulin receptor cDNA: the structural basis for hormone activated trans-membrane signaling. Cell. 1985;40:747–758. doi: 10.1016/0092-8674(85)90334-4. [DOI] [PubMed] [Google Scholar]
- Egawa K, Nakashima N, Sharma PM, Maegawa H, Nagar Y, Kashiwagi A, Kikkawa R, Olefsky JM. Persistent activation of phosphatidylinositol 3-kinase causes insulin resistance due to accelerated insulin-induced insulin receptor substrate-1 degradation in 3T3-L1 adipocytes. Endocrinology. 2000;141:1930–1935. doi: 10.1210/endo.141.6.7516. [DOI] [PubMed] [Google Scholar]
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatases-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Lujo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Harrison DE. The Snell dwarf mutation Pit1 (dw) can increase life span in mice. Mech Ageing Dev. 2002;123:121–130. doi: 10.1016/s0047-6374(01)00339-6. [DOI] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia P, deli Gatti C, Bachschmid MK, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;110:2889–2895. doi: 10.1161/01.CIR.0000147731.24444.4D. [DOI] [PubMed] [Google Scholar]
- Furukawa N, Ongusaha P, Jahung WJ, Araki K, Choi CS, Kim HJ, Lee YH, Kaibuchi K, Kahn BB, Masuzaki H, Kim JK, Lee SW, Kim YB. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab. 2005;2:119–129. doi: 10.1016/j.cmet.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK, Tiganis T. Regulation of insulin receptor signaling by the protein tyrosine phosphatases TCPTP. Mol Cell Biol. 2003;23:2096–2108. doi: 10.1128/MCB.23.6.2096-2108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DD, Eguchi S, Reynolds CM, Eguchi K, Frank GD, Motley ED. Hydrogen peroxide inhibits insulin signaling in vascular smooth muscle cells. Exp Biol Med (Maywood) 2003;228:836–842. doi: 10.1177/15353702-0322807-09. [DOI] [PubMed] [Google Scholar]
- Ghezzi P, Bonetto B, Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizuto R, Bernardi P, Paollucci F, Pellici PB, et al. Electron transfer between cytochrome c and p66shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Goldstein BJ. Protein-tyrosine phosphatases and the regulation of insulin action. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus: A fundamental and Clinical TGext. Vol. 3. Lippincott; Philadelphia: 2003. pp. 255–268. [Google Scholar]
- Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson TA, He W, Craparo A, Schaub CD, O'Neill TJ. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol Cell Biol. 1995;15:2500–2508. doi: 10.1128/mcb.15.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Hoek JB. Mitochondrial longevity pathways. Science. 2007;315:60–X. doi: 10.1126/science.1138825. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Hayes GR, Lockwood DH. Role of insulin receptor phosphorylation in the insulinomimetic effects of hydrogen peroxide. Proc Natl Acad Sci U S A. 1987;84:8115–8119. doi: 10.1073/pnas.84.22.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang LF, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even P, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Hsieh CC, DeFord JH, Flurkey K, Harrison DE, Papaconstantinou J. Implications of the insulin signaling pathway in Snell dwarf mouse longevity: a similarity with the C. elegans longevity paradigm. Mech Ageing Dev. 2002a;123:1229–1244. doi: 10.1016/s0047-6374(02)00036-2. [DOI] [PubMed] [Google Scholar]
- Hsieh CC, DeFord JH, Flurkey K, Harrison DE, Papaconstantinou J. Effects of the Pit 1 mutation on the insulin signaling pathway: implications on the longevity of the long-lived Snell Dwarf mouse. Mech Ageing Dev. 2002b;123:1245–1255. doi: 10.1016/s0047-6374(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries KM, Sweda PA, Szweda LI. Aging: a shift from redox regulation to oxidative damage. Free Radic Res. 2007;40:1239–1243. doi: 10.1080/10715760600913184. [DOI] [PubMed] [Google Scholar]
- Humphries KM, Yoo Y, Szweda LI. Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry. 1998;37:552–557. doi: 10.1021/bi971958i. [DOI] [PubMed] [Google Scholar]
- Imura A, Iwano A, Tohtyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujmimori T, Nabeshima Y. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565:143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Keyse SM. Protein phosphatases and the regulation of MAP kinase activity. Semin Cell Dev Biol. 1998;9:143–152. doi: 10.1006/scdb.1997.0219. [DOI] [PubMed] [Google Scholar]
- Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O'Brien WE, Littman DR, Shulman GI. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of PI3K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe d, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of PI3K family defines a role for p110a in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, De Ruiter ND, DeVries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloons I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema HH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kusari AB, Byon J, Bandyopadhyay D, Kenner KA, Kusari J. Insulin-induced mitogen-activated protein (MAP) kinase phosphatase-1 (MKP-1) attenuates insulin-stimulated MAP kinase activity: a mechanism for the feedback inhibition of insulin signaling. Mol Endocrinol. 1997;11:1532–1543. doi: 10.1210/mend.11.10.9998. [DOI] [PubMed] [Google Scholar]
- Lawrence JC, Jr, Larner J. Activation of glycogen synthase in rat adipocytes by insulin and glucose involves increased glucose transport and phosphorylation. J Biol Chem. 1978;253:2104–2113. [PubMed] [Google Scholar]
- Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. 2003;278:2896–2902. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatases 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- Li J, DeFea K, Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J Biol Chem. 1999;274:9351–9356. doi: 10.1074/jbc.274.14.9351. [DOI] [PubMed] [Google Scholar]
- Little SA, de Haen C. Effects of hydrogen peroxide on basal and hormone-stimulated lipolysis in perfused rat fat cells in relation to the mechanism of action of insulin. J Biol Chem. 1980;255:10888–10895. [PubMed] [Google Scholar]
- Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinosotide-3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol. 2005;170:455–464. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem. 2001a;276:48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001b;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141–149. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JM, de Haen C. The insulin-like effect of hydrogen peroxide on pathways of lipid synthesis in rat adipocytes. J Biol Chem. 1979;254:9017–9021. [PubMed] [Google Scholar]
- Meng TC, Fukada T, Tonk NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, DiFiore PP, Lanfrancone L, Pelicci PG. Opposite effects of p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase fos signaling pathway. EMBO J. 1997;16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeschel K, Beck A, Weigert C, Lammers R, Kalbacher H, Voelter W, Schleicher ED, Haring HU, Lehmann R. Protein kinase C-zeta-induced phosphorylation of Ser(318) in insulin receptor substrate-1 (IRS-1) attenuates the interaction with the insulin receptor and the tyrosine phosphorylation of IRS-1. J Biol Chem. 2004;279:25157–25163. doi: 10.1074/jbc.M402477200. [DOI] [PubMed] [Google Scholar]
- Mothe I, Van Obberghen E. Phosphorylation of insulin receptor substrate-1 on multiple serine residues 612, 632, 662, and 731, modulates insulin action. J Biol Chem. 1996;271:11222–11227. doi: 10.1074/jbc.271.19.11222. [DOI] [PubMed] [Google Scholar]
- Nakae J, Park B, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274:15982–15985. doi: 10.1074/jbc.274.23.15982. [DOI] [PubMed] [Google Scholar]
- Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci U S A. 2003;100:2112–2116. doi: 10.1073/pnas.0336359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Combs CA, French S, Ahn BH, Fergusson MM, Balaban RS, Finkel T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J Biol Chem. 2006;281:10555–10560. doi: 10.1074/jbc.M511626200. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T. Redox regulation of Forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- Ogawa W, Matozaki T, Kasuga M. Role of binding proteins to IRS-1 in insulin signaling. Mol Cell Biochem. 1998;182:13–22. [PubMed] [Google Scholar]
- Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, Puri C, Tacchetti C, Ferrini M, Mannucci R, Nicoletti I, Lanfrancone L, Giorgio M, Pelicci PG. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Grignani F, Pawson T, Pelicci PG. A novel transforming protein (SCH) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowskiu MR, Del Sal G, Pelicci PG, Rizzuto R. Rprotyein kinase Cβ and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. 2007;315:659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- Potashnik R, Bloch-Damati A, Boshan N, Rudich A. IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia. 2003;46:639–648. doi: 10.1007/s00125-003-1097-5. [DOI] [PubMed] [Google Scholar]
- Ren JM, Li PM, Zhang WR, Sweet LJ, Cline G, Shulman GI, Livingston JN, Goldstein BJ. Transgenic mice deficient in the LAR protein-tyrosine phosphatase exhibit profound defects in glucose homeostasis. Diabetes. 1998;47:493–497. doi: 10.2337/diabetes.47.3.493. [DOI] [PubMed] [Google Scholar]
- Rena G, Guo S, Cichy SC, Uterman TG, Cohen P. Phosphorylation of the transcription factor Forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- Roche S, Koegl M, Courtneidge SA. The phosphatidylinositol-kinase alpha is required for DNA synthesis induced by some but not all growth factors. Proc Natl Acad Sci U S A. 1994;91:9185–9189. doi: 10.1073/pnas.91.19.9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S, Downward J, Raynal P, Courtneidge AA. A function of phosphatidylinositol 3-kinase beta (p85alpha-p110 beta) in fibroblasts during mitogenesis: requirement for insulin and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol. 1998;18:7119–7129. doi: 10.1128/mcb.18.12.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes. 1998;47:1562–1569. doi: 10.2337/diabetes.47.10.1562. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Rose DW, Jhun BH, Saltiel AR, Draznin B, Olefsky JM. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem. 1994a;269:13689–13694. [PubMed] [Google Scholar]
- Sasaoka T, Drazin B, Leitner JW, Langlois WJ, Olefsky JM. Shc is the predominant signaling molecule coupling insulin receptors to activation of guanine nucleotide releasing factor and p21 ras-STP formation. J Biol Chem. 1994b;269:10734–10738. [PubMed] [Google Scholar]
- Sattar AA, Berhanu C, Gebreselassie S, Berhanu P. Human insulin receptor juxtamembrane domain independent insulin signaling. Cell Biol Int. 2007;31:815–824. doi: 10.1016/j.cellbi.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Schapira HH. Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- Schmid E, El Benna J, Galter D, Klein G, Droge W. Redox priming of the insulin receptor beta-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation. FASEB J. 1998;12:863–870. doi: 10.1096/fasebj.12.10.863. [DOI] [PubMed] [Google Scholar]
- Schmid E, Hotz-Wagenblatt A, Hacj V, Droge W. Phosphorylation of the insulin receptor kinase by phosphocreatine in combination with hydrogen peroxide: the structural basis of redox priming. FASEB J. 1999;13:1491–1500. doi: 10.1096/fasebj.13.12.1491. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signaling. Biochem J. 1998;333(Pt 3):471–490. doi: 10.1042/bj3330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1C lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- Srivastava AK. Redox regulation of insulin action and signaling. Antioxid Redox Signal. 2005;7:1011–1013. doi: 10.1089/ars.2005.7.1011. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Rothenberg P, Kahn CR, Backer JM, Arakai E, Wilden PA, Cahill DA, Goldstein BJ, White MF. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG, Jr, Glasheen E, Lane WS, Pierce JH, White MF. Role of IRS-2 in insulin and cytokine signaling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Tebbey PW, Granner DK. Oxidative and chemical stress mimic insulin by selectively inhibiting the expression of phosphoenolpyruvate carboxykinase in hepatoma cells. Diabetes. 1997;46:17–22. doi: 10.2337/diab.46.1.17. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Kuro-O M, Ishikawa F. Aging mechanisms. Proc Natl Acad Sci U S A. 2000;97:12407–12408. doi: 10.1073/pnas.210382097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Aleman JO, Ueki K, Luo J, Asano T, Kaneto H, Stephanopoulos G, Cantley LC, Kahn CR. The p85α regulatory subunit of phosphoinositide 3-kinsae potentiates c-Jun N-terminal kinase-mediated insulin resistance. Mol Cell Biol. 2007;27:2830–2840. doi: 10.1128/MCB.00079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes of signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006a;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Tran TT, Kondo T, Luo J, Ueki K, Cantley LC, Kahn CR. Phosphoinositide 3-kinase regualtory subunit p85alpha suppresses insulin action via positive regulation of PTEN. Proc Natl Acad Sci U S A. 2006b;103:12093–12097. doi: 10.1073/pnas.0604628103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A, Inukai K, Asano T, Kaburagi Y, Ueki K, Nakajima H, Hanafusa T, Matsuzawa Y, Sekihara H, Yin Y, Barrett JC, Oda H, Ishikawa T, Akanuma Y, Komuro I, Suzuki M, Yamamura K, Kodama T, Suzuki H, Yamamura K, Kodama T, Suzuki H, Koyasu S, Aizawa S, Tobe K, Fukui Y, Yazaki Y, Kadowaki T. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21:230–235. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Mallis RJ. Aging and oxidation of reactive protein sulfhydryls. Exp Gerontol. 2001;36:1519–1526. doi: 10.1016/s0531-5565(01)00137-1. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys. 1995;319:1–9. doi: 10.1006/abbi.1995.1261. [DOI] [PubMed] [Google Scholar]
- Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine posphatases. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG. A p53–p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22:965–977. doi: 10.1128/MCB.22.3.965-977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem. 2004;279:35298–35305. doi: 10.1074/jbc.M405203200. [DOI] [PubMed] [Google Scholar]
- White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- Whitehead JP, Clark SF, Urso B, James DE. Signaling through the insulin receptor. Curr Opin Cell Biol. 2000;12:222–228. doi: 10.1016/s0955-0674(99)00079-4. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, Mityoshi M, Ogawa Y, Castrillon DH, Rosenblatt KP, Kuro-o M. Regulation of oxidative stress by the anti-aging hormone Klotho. J Biol Chem. 2005;280:38029–38034. doi: 10.1074/jbc.M509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zhong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]